
Heme Oxygenase-1 and Its Role in Colorectal Cancer

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction to HO-1 and Its Role in Physiology and Pathophysiology
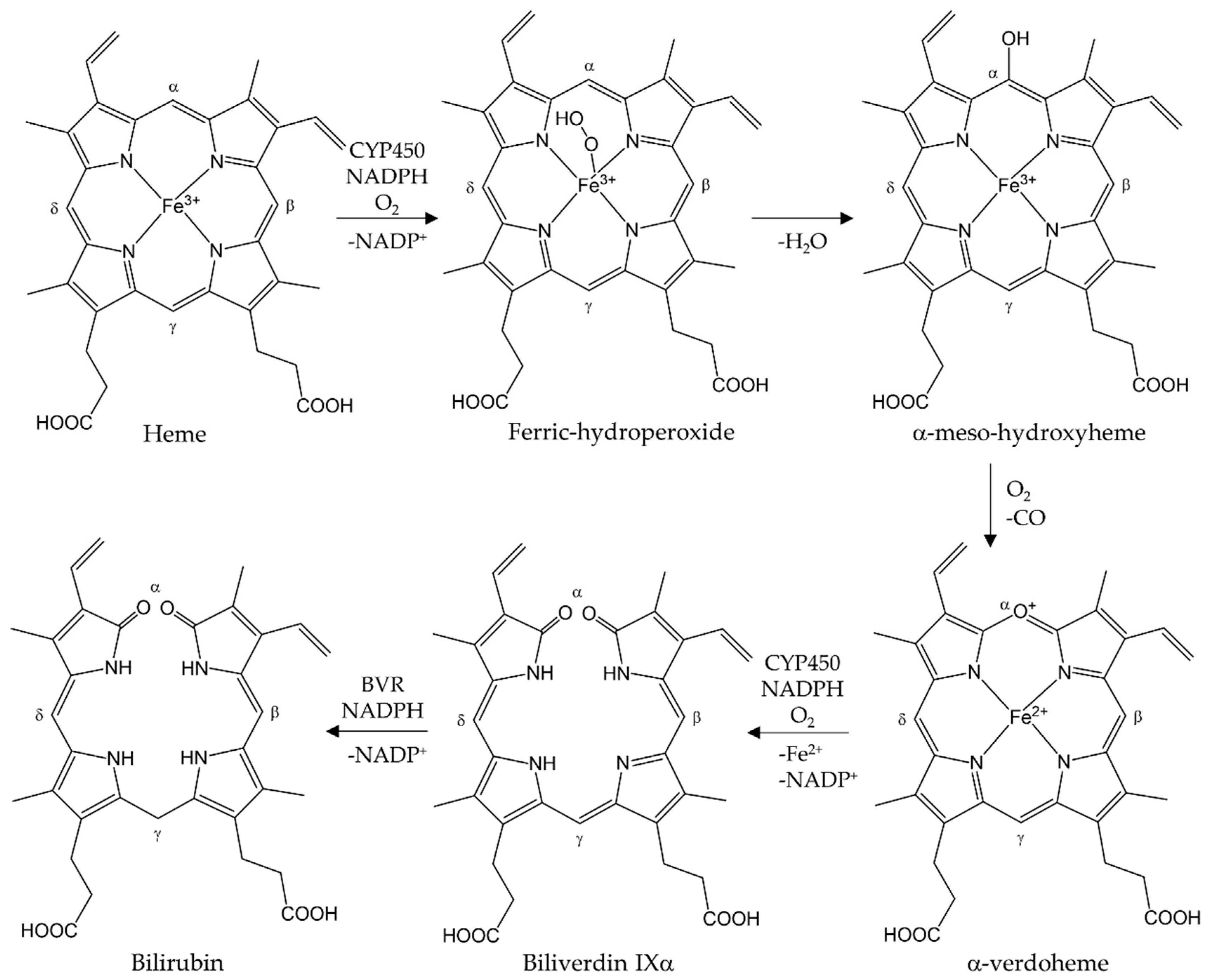
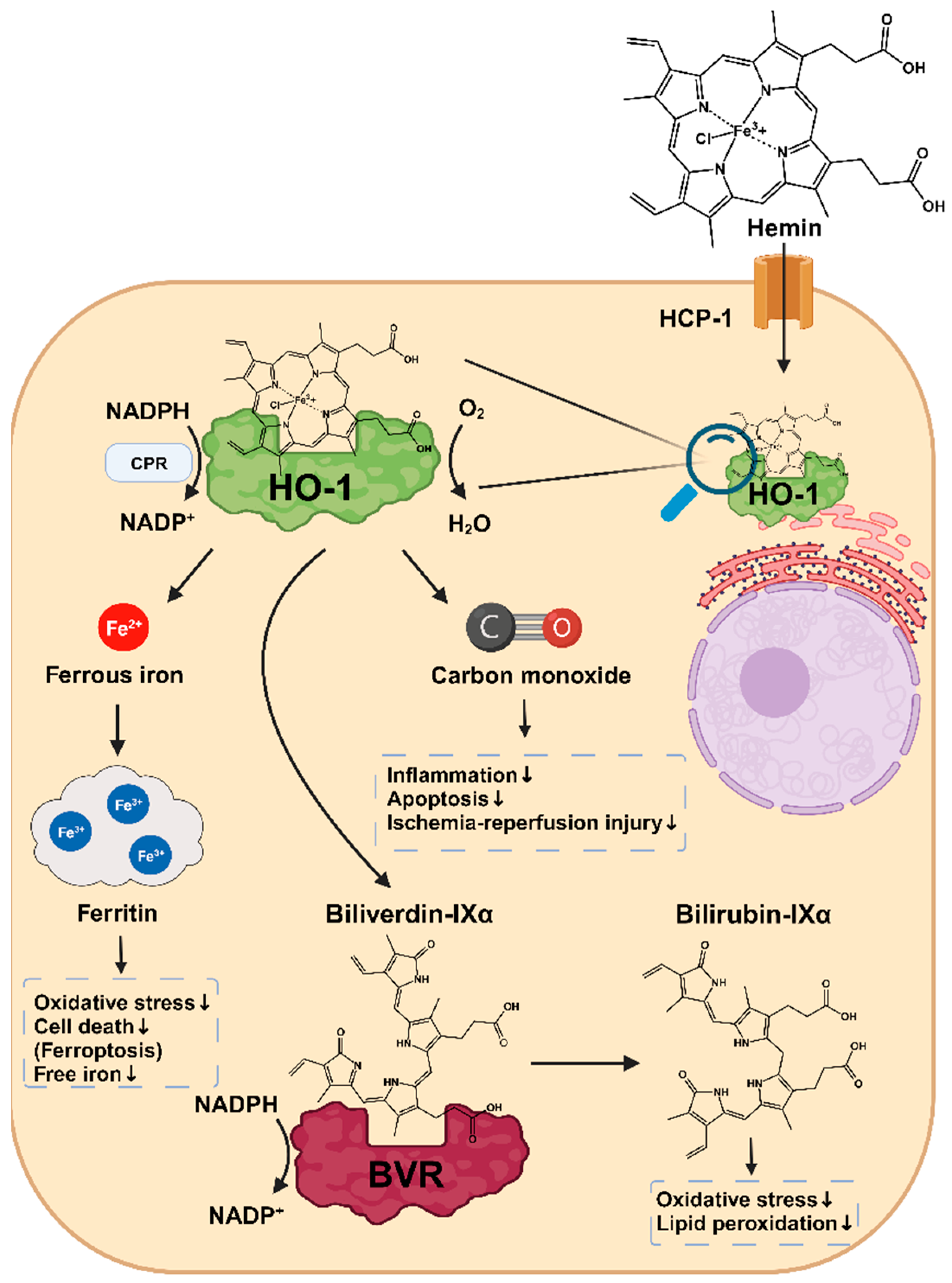
2. Structure and Function of HO-1
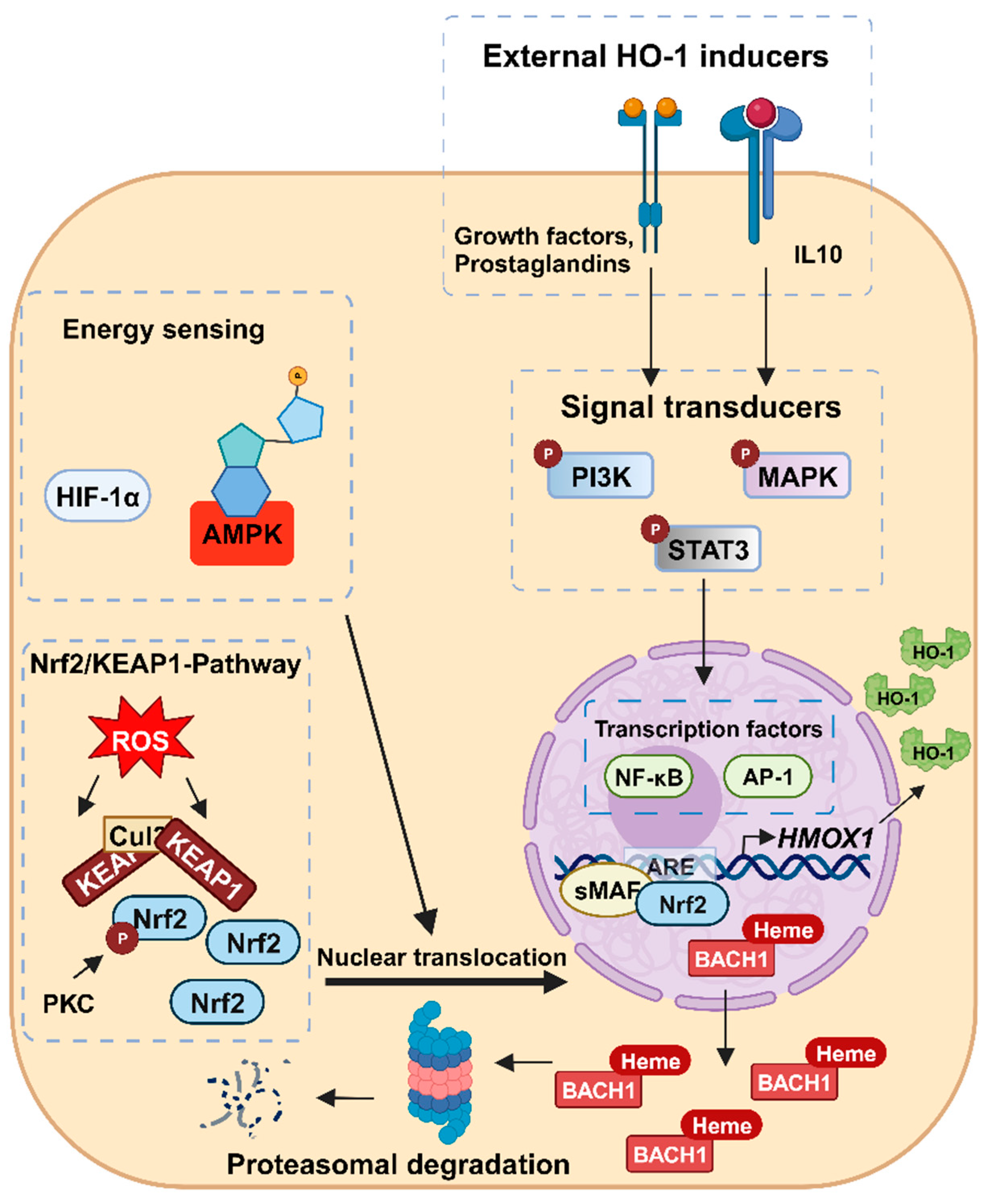
3. Regulation of HO-1 Expression
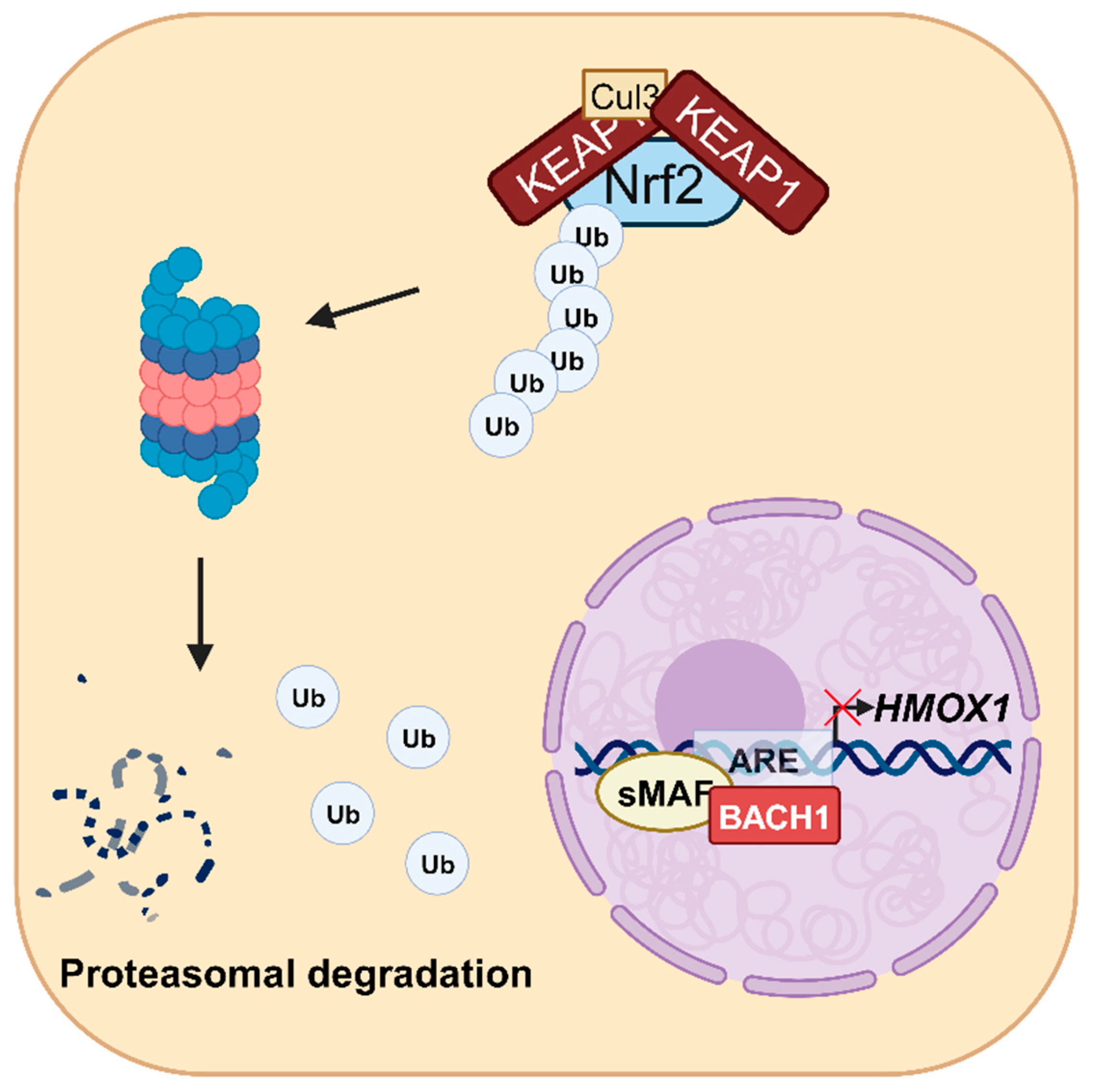
3.1. Transcriptional Regulation of HMOX1
3.2. Regulation of HMOX1 Expression by Signaling Pathways and Post-Transcriptional Mechanisms
4. Other HO Isoforms
5. Cytoprotective Effects of HO-1
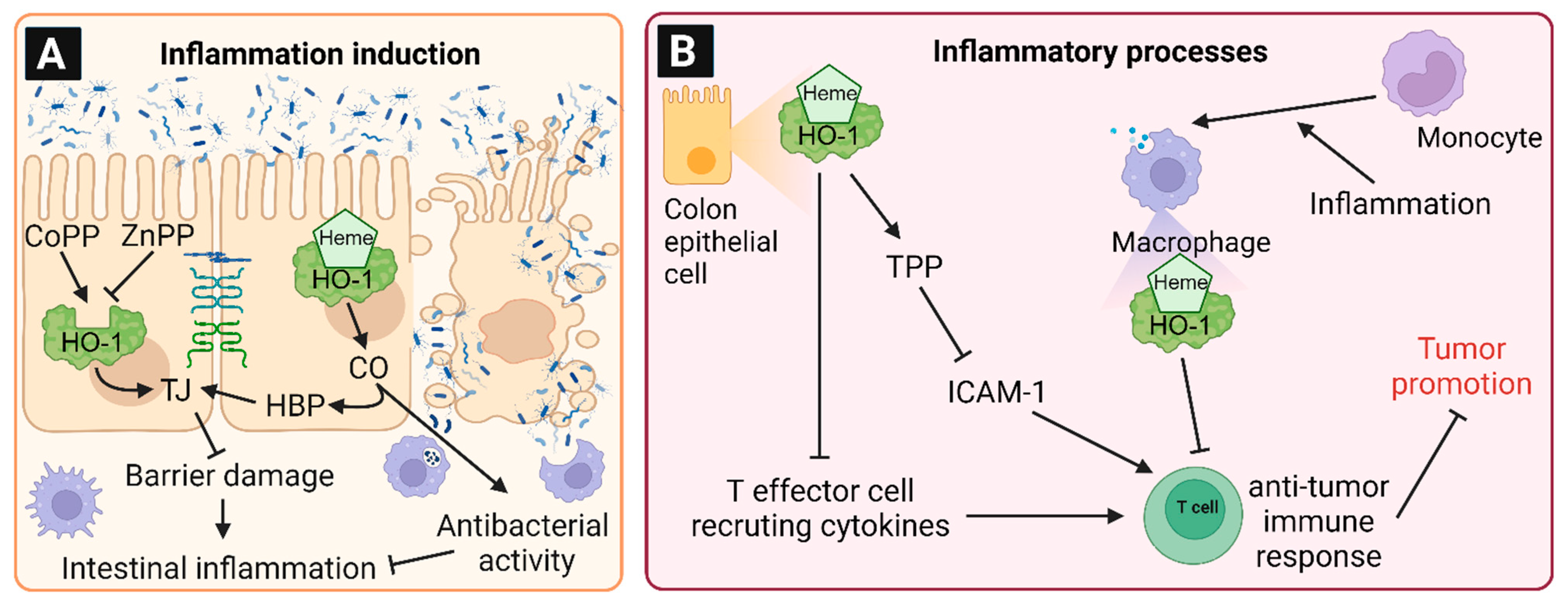
6. Involvement of HO-1 in Intestinal Inflammation and Chronic Diseases
7. Colorectal Carcinogenesis and the Role of HO-1
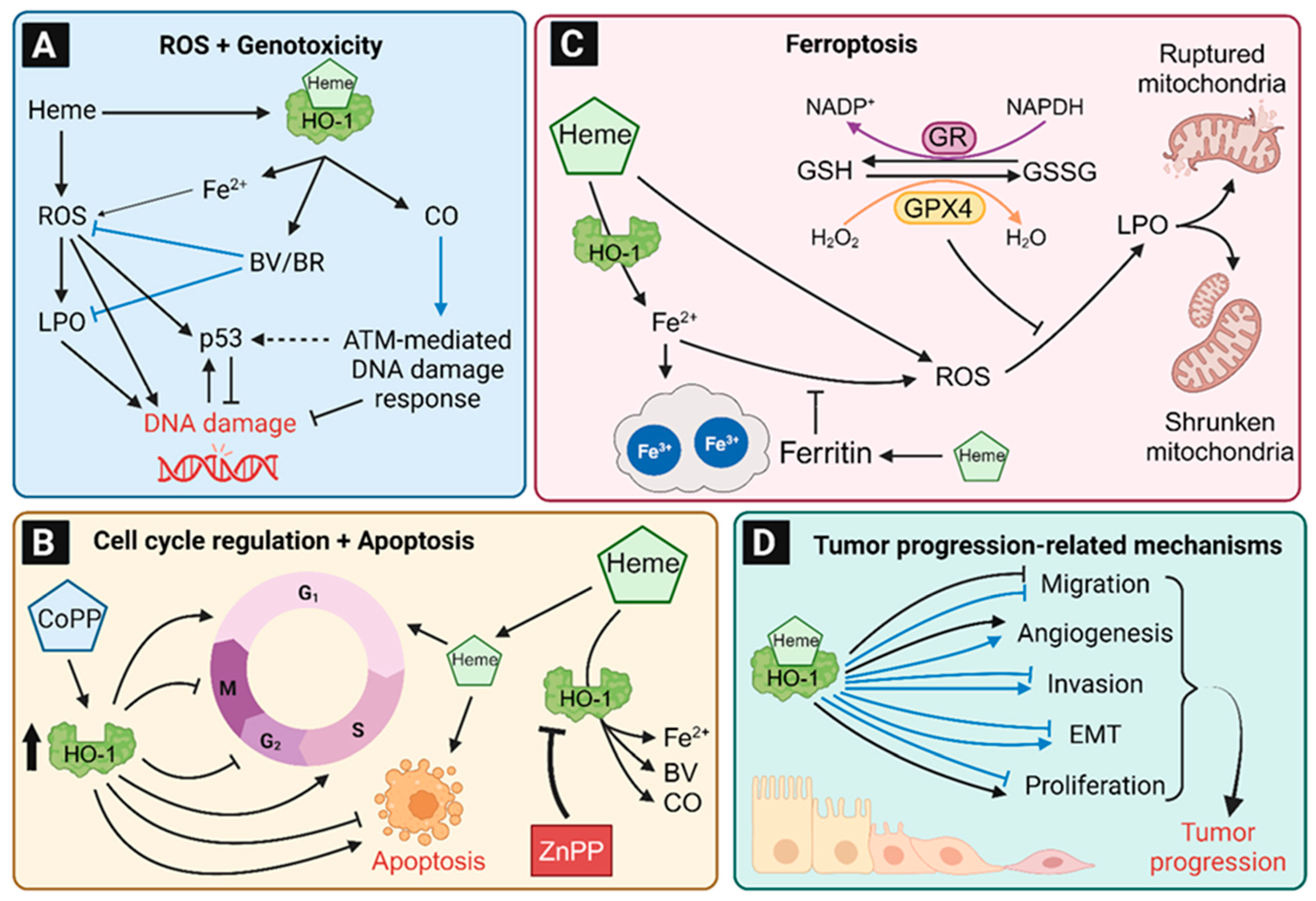
8. Role of HO-1 in Tumor Induction
8.1. HO-1-Dependent Protection against ROS and Lipid Peroxidation
8.2. Role of HO-1 in Genotoxicity and DNA Damage Response
8.3. Regulation of the Cell Cycle, Apoptosis and Cell Viability by HO-1
8.4. The Contribution of HO-1 to Ferroptosis
8.5. Role of HO-1 in Tumor Initiation in Non-Colon Tissues
9. Role of HO-1 in Tumor Progression
10. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| γ-GCS | γ-glutamylcysteine synthetase |
| ACSL4 | Long-chain-fatty-acid-CoA ligase 4 |
| AKI | Acute kidney injury |
| AMPK | AMP-activated protein kinase |
| AP-1 | Activator protein-1 |
| AP-2 | Activator protein-2 |
| ARE | Antioxidant response element |
| BACH1 | BTB and CNC homology 1 |
| BK | Calcium-sensitive potassium |
| BR | Bilirubin |
| BV | Biliverdin-IXα |
| BVR | Biliverdin reductase |
| bZIP | Basic-region leucine zipper |
| Caco-2 | Human epithelial colorectal adenocarcinoma cells |
| CD | Crohn’s disease |
| CK2 | Casein kinase 2 |
| CNC | Cap’n’collar |
| CoPP | Cobalt protoporphyrin IX |
| CRC | Colorectal cancer |
| Cul3 | Cullin 3 |
| DDS | Dextran sodium sulphate |
| DEN | Diethylnitrosamine |
| ECE-1 | Endothelin-converting enzyme-1 |
| Egr-1 | Early growth response protein 1 |
| EMT | Epithelial-mesenchymal transition |
| ER | Endoplasmatic reticulum |
| ERK | Extracellular signal-regulated kinase |
| FPN | Ferroportin |
| FtH | Ferritin heavy chain |
| FtL | Ferritin light chain |
| GPX4 | Glutathione peroxidase 4 |
| GRP78 | Glucose-related protein 78 |
| GSH | Glutathione |
| GSSG | Glutathione disulfide |
| HCEC | Human colonic epithelial cells |
| HIF-1α | Hypoxia-inducible factor-1 |
| HMEC-1 | Human microvascular endothelial cell line |
| HO-1 | Heme oxygenase-1 |
| HO-2 | Heme oxygenase-2 |
| HO-3 | Heme oxygenase-3 |
| HRMs | Heme-regulatory motifs |
| HUVEC | Human umbilical vein endothelial cells |
| IBD | Inflammatory bowel disease |
| ICAM-1 | Intercellular adhesion molecule 1 |
| ICH | Intracerebral haemorrhage |
| IFN-γ | Interferon-γ |
| IL-1β | Interleukin-1β |
| IL-6 | Interleukin 6 |
| IR | Ischemia-reperfusion |
| IRI | Ischemia-reperfusion injury |
| JNK | C-Jun N-terminal kinase |
| KEAP-1 | Kelch-like ECH-associated protein 1 |
| KRAS | Kirsten-ras |
| LPS | Lipopolysaccharide |
| MAPK | Mitogen-activated protein kinase |
| MIP-1β | Macrophage inflammatory protein-1β |
| miRs | MicroRNAs |
| MMP-9 | Matrix metallopeptidase 9 |
| MPO | Myeloperoxidase |
| MSCs | Mesenchymal stem cells |
| NAMs | New approached methodologies |
| NF-κB | Nuclear factor-κB |
| NQO1 | NAD(P)H quinone oxidoreductase 1 |
| Nrf2 | Nuclear factor erythroid 2-related factor 2 |
| PI3K | Phasphatidylinositol 3-kinase |
| PKC | Protein kinase C |
| PPARγ | Peroxisome proliferator-activated receptor-γ |
| PTGS2 | Prostaglandin-endoperoxide synthase 2 |
| ROS | Reactive oxygen species |
| sMAF | Small musculoaponeurotic fibrosarcoma |
| SnPP | Tin protoporphyrin IX |
| SPF | Specific pathogen-free |
| STAT | Signal Transducer and Activator of Transcription |
| TBARS | Thiobarbituric acid reactive substances |
| TCR -α | T cell receptor-alpha |
| TJ | Tight junction |
| TLR | Toll-like receptor |
| TNFα | Tumor necrosis factor-α |
| TPP | Tristetraprolin |
| UC | Ulcerative colitis |
| VCAM-1 | Vascular cell adhesion protein 1 |
| ZnPP | Zinc protoporphyrine IX |
References
- Tenhunen, R.; Amarver, H.S.; Schmid, R. The enzymatic conversion of heme to bilirubin by microsomal heme oxygenase. Biochemistry 1968, 61, 748–755. [Google Scholar] [CrossRef]
- Ryter, S.W. Heme Oxgenase-1, a Cardinal Modulator of Regulated Cell Death and Inflammation. Cells 2021, 10, 515. [Google Scholar] [CrossRef]
- Rucker, H.; Amslinger, S. Identification of heme oxygenase-1 stimulators by a convenient ELISA-based bilirubin quantification assay. Free Radic. Biol. Med. 2015, 78, 135–146. [Google Scholar] [CrossRef]
- Huber, W.J., 3rd; Backes, W.L. Expression and Characterization of Full-Length Human Heme Oxygenase-1: The Presence of Intact Membrane-Binding Region Leads to Increased Binding Affinity for NADPH Cytochrome P450 Reductase. Biochemistry 2007, 46, 12212–12219. [Google Scholar] [CrossRef]
- Prawan, A.; Kundu, J.K.; Surh, Y.J. Molecular basis of heme oxygenase-1 induction: Implications for chemoprevention and chemoprotection. Antioxid. Redox Signal. 2005, 7, 1688–1703. [Google Scholar] [CrossRef]
- Maines, M.D.; Trakshel, G.M.; Kutty, R.K. Characterization of two constitutive forms of rat liver microsomal heme oxygenase. Only one molecular species of the enzyme is inducible. J. Biol. Chem. 1986, 261, 411–419. [Google Scholar] [CrossRef]
- Ryter, S.W. Heme Oxygenase-1: An Anti-Inflammatory Effector in Cardiovascular, Lung, and Related Metabolic Disorders. Antioxidants 2022, 11, 555. [Google Scholar] [CrossRef]
- Kim, H.P.; Wang, X.; Galbiati, F.; Ryter, S.W.; Choi, A.M. Caveolae compartmentalization of heme oxygenase-1 in endothelial cells. FASEB J. 2004, 18, 1080–1089. [Google Scholar] [CrossRef]
- Lin, Q.; Weis, S.; Yang, G.; Weng, Y.H.; Helston, R.; Rish, K.; Smith, A.; Bordner, J.; Polte, T.; Gaunitz, F.; et al. Heme oxygenase-1 protein localizes to the nucleus and activates transcription factors important in oxidative stress. J. Biol. Chem. 2007, 282, 20621–20633. [Google Scholar] [CrossRef]
- Biswas, C.; Shah, N.; Muthu, M.; La, P.; Fernando, A.P.; Sengupta, S.; Yang, G.; Dennery, P.A. Nuclear heme oxygenase-1 (HO-1) modulates subcellular distribution and activation of Nrf2, impacting metabolic and anti-oxidant defenses. J. Biol. Chem. 2014, 289, 26882–26894. [Google Scholar] [CrossRef]
- Yang, G.; Biswasa, C.; Lin, Q.S.; La, P.; Namba, F.; Zhuang, T.; Muthu, M.; Dennery, P.A. Heme oxygenase-1 regulates postnatal lung repair after hyperoxia: Role of beta-catenin/hnRNPK signaling. Redox Biol. 2013, 1, 234–243. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, P.; Pandey, V.C. Mitochondrial Heme Oxygenase of lMastomys couclza. Int. J. Biochem. Cell Biol. 1996, 28, 1071–1077. [Google Scholar] [CrossRef] [PubMed]
- Consoli, V.; Sorrenti, V.; Grosso, S.; Vanella, L. Heme Oxygenase-1 Signaling and Redox Homeostasis in Physiopathological Conditions. Biomolecules 2021, 11, 589. [Google Scholar] [CrossRef] [PubMed]
- Yachie, A.; Niida, Y.; Wada, T.; Igarashi, N.; Kaneda, H.; Toma, T.; Ohta, K.; Kasahara, Y.; Koizumi, S. Oxidative stress causes enhanced endothelial cell injury in human heme oxygenase-1 deficiency. J. Clin. Investig. 1999, 103, 129–135. [Google Scholar] [CrossRef]
- Poss, K.D.; Tonegawa, S. Reduced stress defense in heme oxygenase 1-deficient cells. Proc. Natl. Acad. Sci. USA 1997, 94, 10925–10930. [Google Scholar] [CrossRef]
- Poss, K.D.; Tonegawa, S. Heme oxygenase 1 is required for mammalian iron reutilization. Proc. Natl. Acad. Sci. USA 1997, 94, 10919–10924. [Google Scholar] [CrossRef]
- Fujii, H.; Takahashi, T.; Nakahira, K.; Uehara, K.; Shimizu, H.; Matsumi, M.; Morita, K.; Hirakawa, M.; Akagi, R.; Sassa, S. Protective role of heme oxygenase-1 in the intestinal tissue injury in an experimental model of sepsis. Crit. Care Med. 2003, 31, 893–902. [Google Scholar] [CrossRef]
- Akagi, R.; Akagi, M.; Hatori, Y.; Inouye, S. Prevention of Barrier Disruption by Heme Oxygenase-1 in Intestinal Bleeding Model. Biol. Pharm. Bull. 2016, 39, 1007–1012. [Google Scholar] [CrossRef]
- Hegazi, R.A.; Rao, K.N.; Mayle, A.; Sepulveda, A.R.; Otterbein, L.E.; Plevy, S.E. Carbon monoxide ameliorates chronic murine colitis through a heme oxygenase 1-dependent pathway. J. Exp. Med. 2005, 202, 1703–1713. [Google Scholar] [CrossRef]
- Wegiel, B.; Larsen, R.; Gallo, D.; Chin, B.Y.; Harris, C.; Mannam, P.; Kaczmarek, E.; Lee, P.J.; Zuckerbraun, B.S.; Flavell, R.; et al. Macrophages sense and kill bacteria through carbon monoxide-dependent inflammasome activation. J. Clin. Investig. 2014, 124, 4926–4940. [Google Scholar] [CrossRef]
- Seiwert, N.; Wecklein, S.; Demuth, P.; Hasselwander, S.; Kemper, T.A.; Schwerdtle, T.; Brunner, T.; Fahrer, J. Heme oxygenase 1 protects human colonocytes against ROS formation, oxidative DNA damage and cytotoxicity induced by heme iron, but not inorganic iron. Cell Death Dis. 2020, 11, 787. [Google Scholar] [CrossRef] [PubMed]
- Vanella, L.; Kim, D.H.; Asprinio, D.; Peterson, S.J.; Barbagallo, I.; Vanella, A.; Goldstein, D.; Ikehara, S.; Kappas, A.; Abraham, N.G. HO-1 expression increases mesenchymal stem cell-derived osteoblasts but decreases adipocyte lineage. Bone 2010, 46, 236–243. [Google Scholar] [CrossRef] [PubMed]
- Zwerina, J.; Tzima, S.; Hayer, S.; Redlich, K.; Hoffmann, O.; Hanslik-Schnabel, B.; Smolen, J.S.; Kollias, G.; Schett, G. Heme oxygenase 1 (HO-1) regulates osteoclastogenesis and bone resorption. FASEB J. 2005, 19, 2011–2013. [Google Scholar] [CrossRef] [PubMed]
- Kutty, R.K.; Kutty, G.; Rodriguez, I.R.; Chader, G.J.; Wiggert, B. Chromosomal localization of the human heme oxygenase genes: Heme oxygenase-1 (HMOX1) maps to chromosome 22q12 and heme oxygenase-2 (HMOX2) maps to chromosome 16p13.3. Genomics 1994, 20, 513–516. [Google Scholar] [CrossRef]
- Montellano, P.R. The mechanism of heme oxygenase. Curr. Opin. Chem. Biol. 2000, 4, 221–227. [Google Scholar] [CrossRef]
- Gottlieb, Y.; Truman, M.; Cohen, L.A.; Leichtmann-Bardoogo, Y.; Meyron-Holtz, E.G. Endoplasmic reticulum anchored heme-oxygenase 1 faces the cytosol. Haematologica 2012, 97, 1489–1493. [Google Scholar] [CrossRef]
- Zhu, Y.; Silverman, R.B. Revisiting Heme Mechanisms. A Perspective on the Mechanisms of Nitric Oxide Synthase (NOS), Heme Oxygenase (HO), and Cytochrome P450s (CYP450s). Biochemistry 2008, 47, 2231–2243. [Google Scholar] [CrossRef]
- Zhang, X.; Fujii, H.; Matera, K.M.; Migita, C.T.; Sun, D.; Sato, M.; Ikeda-Saito, M.; Yoshida, T. Stereoselectivity of each of the three steps of the heme oxygenase reaction: Hemin to meso-hydroxyhemin, meso-hydroxyhemin to verdoheme, and verdoheme to biliverdin. Biochemistry 2003, 42, 7418–7426. [Google Scholar] [CrossRef]
- Campbell, N.K.; Fitzgerald, H.K.; Dunne, A. Regulation of inflammation by the antioxidant haem oxygenase 1. Nat. Rev. Immunol. 2021, 21, 411–425. [Google Scholar] [CrossRef]
- Ferrándiz, M.L.; Devesa, I. Inducers of Heme Oxygenase-1. Curr. Pharm. Des. 2008, 14, 473–486. [Google Scholar] [CrossRef]
- Zhang, A.; Suzuki, T.; Adachi, S.; Naganuma, E.; Suzuki, N.; Hosoya, T.; Itoh, K.; Sporn, M.B.; Yamamoto, M. Distinct Regulations of HO-1 Gene Expression for Stress Response and Substrate Induction. Mol. Cell. Biol. 2021, 41, e0023621. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Igarashi, K.; Hayashi, N.; Nishizawa, M.; Yamamoto, M. Cloning and characterization of a novel erythroid cell-derived CNC family transcription factor heterodimerizing with the small Maf family proteins. Mol. Cell. Biol. 1995, 15, 4184–4193. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, K.; Sun, J.; Taketani, S.; Nakajima, O.; Nishitani, C.; Sassa, S.; Hayashi, N.; Yamamoto, M.; Shibahara, S.; Fujita, H.; et al. Heme mediates derepression of Maf recognition element through direct binding to transcription repressor Bach1. EMBO J. 2001, 20, 2835–2843. [Google Scholar] [CrossRef] [PubMed]
- Thanas, C.; Ziros, P.G.; Chartoumpekis, D.V.; Renaud, C.O.; Sykiotis, G.P. The Keap1/Nrf2 Signaling Pathway in the Thyroid-2020 Update. Antioxidants 2020, 9, 1082. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I.; et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 1997, 236, 312–322. [Google Scholar] [CrossRef] [PubMed]
- Katsuoka, F.; Motohashi, H.; Ishii, T.; Aburatani, H.; Engel, J.D.; Yamamoto, M. Genetic evidence that small maf proteins are essential for the activation of antioxidant response element-dependent genes. Mol. Cell. Biol. 2005, 25, 8044–8051. [Google Scholar] [CrossRef]
- Sun, J.; Brand, M.; Zenke, Y.; Tashiro, S.; Groudine, M.; Igarashi, K. Heme regulates the dynamic exchange of Bach1 and NF-E2-related factors in the Maf transcription factor network. Proc. Natl. Acad. Sci. USA 2004, 101, 1461–1466. [Google Scholar] [CrossRef]
- Suzuki, H.; Tashiro, S.; Hira, S.; Sun, J.; Yamazaki, C.; Zenke, Y.; Ikeda-Saito, M.; Yoshida, M.; Igarashi, K. Heme regulates gene expression by triggering Crm1-dependent nuclear export of Bach1. EMBO J. 2004, 23, 2544–2553. [Google Scholar] [CrossRef]
- Zenke-Kawasaki, Y.; Dohi, Y.; Katoh, Y.; Ikura, T.; Ikura, M.; Asahara, T.; Tokunaga, F.; Iwai, K.; Igarashi, K. Heme induces ubiquitination and degradation of the transcription factor Bach1. Mol. Cell. Biol. 2007, 27, 6962–6971. [Google Scholar] [CrossRef]
- Wiel, C.; Le Gal, K.; Ibrahim, M.X.; Jahangir, C.A.; Kashif, M.; Yao, H.; Ziegler, D.V.; Xu, X.; Ghosh, T.; Mondal, T.; et al. BACH1 Stabilization by Antioxidants Stimulates Lung Cancer Metastasis. Cell 2019, 178, 330–345.e322. [Google Scholar] [CrossRef]
- Wright, M.M.; Kim, J.; Hock, T.D.; Leitinger, N.; Freeman, B.A.; Agarwal, A. Human haem oxygenase-1 induction by nitro-linoleic acid is mediated by cAMP, AP-1 and E-box response element interactions. Biochem. J. 2009, 422, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.Y.; Yang, Y.C.; Li, C.C.; Liu, K.L.; Lii, C.K.; Chen, H.W. Andrographolide inhibits TNFalpha-induced ICAM-1 expression via suppression of NADPH oxidase activation and induction of HO-1 and GCLM expression through the PI3K/Akt/Nrf2 and PI3K/Akt/AP-1 pathways in human endothelial cells. Biochem. Pharmacol. 2014, 91, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Ockaili, R.; Natarajan, R.; Salloum, F.; Fisher, B.J.; Jones, D.; Fowler, A.A., 3rd; Kukreja, R.C. HIF-1 activation attenuates postischemic myocardial injury: Role for heme oxygenase-1 in modulating microvascular chemokine generation. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, H542–H548. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Guo, Y.; Ou, Q.; Cui, C.; Wu, W.J.; Tan, W.; Zhu, X.; Lanceta, L.B.; Sanganalmath, S.K.; Dawn, B.; et al. Gene transfer of inducible nitric oxide synthase affords cardioprotection by upregulating heme oxygenase-1 via a nuclear factor-kappaB-dependent pathway. Circulation 2009, 120, 1222–1230. [Google Scholar] [CrossRef]
- Yang, C.M.; Lin, C.C.; Yang, C.C.; Cho, R.L.; Hsiao, L.D. Mevastatin-Induced AP-1-Dependent HO-1 Expression Suppresses Vascular Cell Adhesion Molecule-1 Expression and Monocyte Adhesion on Human Pulmonary Alveolar Epithelial Cells Challenged with TNF-alpha. Biomolecules 2020, 10, 381. [Google Scholar] [CrossRef]
- Shen, H.H.; Wang, C.J.; Zhang, X.Y.; Sheng, Y.R.; Yang, S.L.; Zheng, Z.M.; Shi, J.L.; Qiu, X.M.; Xie, F.; Li, M.Q. HIF1A-induced heme oxygenase 1 promotes the survival of decidual stromal cells against excess heme-mediated oxidative stress. Reproduction 2021, 163, 33–43. [Google Scholar] [CrossRef]
- Wu, G.; Marin-Garcia, J.; Rogers, T.B.; Lakatta, E.G.; Long, X. Phosphorylation and hypoxia-induced heme oxygenase-1 gene expression in cardiomyocytes. J. Card. Fail. 2004, 10, 519–526. [Google Scholar] [CrossRef]
- Gong, P.; Hu, B.; Cederbaum, A.I. Diallyl sulfide induces heme oxygenase-1 through MAPK pathway. Arch. Biochem. Biophys. 2004, 432, 252–260. [Google Scholar] [CrossRef]
- Niu, T.; Fu, G.; Zhou, J.; Han, H.; Chen, J.; Wu, W.; Chen, H. Floridoside Exhibits Antioxidant Properties by Activating HO-1 Expression via p38/ERK MAPK Pathway. Mar. Drugs 2020, 18, 105. [Google Scholar] [CrossRef]
- Kietzmann, T.; Samoylenko, A.; Immenschuh, S. Transcriptional regulation of heme oxygenase-1 gene expression by MAP kinases of the JNK and p38 pathways in primary cultures of rat hepatocytes. J. Biol. Chem. 2003, 278, 17927–17936. [Google Scholar] [CrossRef]
- Alam, J.; Cook, J.L. How many transcription factors does it take to turn on the heme oxygenase-1 gene? Am. J. Respir. Cell Mol. Biol. 2007, 36, 166–174. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.C.; Nguyen, T.; Pickett, C.B. Phosphorylation of Nrf2 at Ser-40 by protein kinase C regulates antioxidant response element-mediated transcription. J. Biol. Chem. 2002, 277, 42769–42774. [Google Scholar] [CrossRef] [PubMed]
- Ricchetti, G.A.; Williams, L.M.; Foxwell, B.M. Heme oxygenase 1 expression induced by IL-10 requires STAT-3 and phosphoinositol-3 kinase and is inhibited by lipopolysaccharide. J. Leukoc. Biol. 2004, 76, 719–726. [Google Scholar] [CrossRef] [PubMed]
- Xi, Y.D.; Yu, H.L.; Ding, J.; Ma, W.W.; Yuan, L.H.; Feng, J.F.; Xiao, X.Y.; Xiao, R. Flavonoids protect cerebrovascular endothelial cells through Nrf2 and PI3K from β-amyloid peptide-induced oxidative damage. Curr. Neurovasc. Res. 2012, 9, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.M.; Peyton, K.J.; Shebib, A.R.; Wang, H.; Korthuis, R.J.; Durante, W. Activation of AMPK stimulates heme oxygenase-1 gene expression and human endothelial cell survival. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, H84–H93. [Google Scholar] [CrossRef]
- Mo, C.; Wang, L.; Zhang, J.; Numazawa, S.; Tang, H.; Tang, X.; Han, X.; Li, J.; Yang, M.; Wang, Z.; et al. The crosstalk between Nrf2 and AMPK signal pathways is important for the anti-inflammatory effect of berberine in LPS-stimulated macrophages and endotoxin-shocked mice. Antioxid. Redox Signal. 2014, 20, 574–588. [Google Scholar] [CrossRef]
- Piras, S.; Furfaro, A.L.; Caggiano, R.; Brondolo, L.; Garibaldi, S.; Ivaldo, C.; Marinari, U.M.; Pronzato, M.A.; Faraonio, R.; Nitti, M. microRNA-494 Favors HO-1 Expression in Neuroblastoma Cells Exposed to Oxidative Stress in a Bach1-Independent Way. Front. Oncol. 2018, 8, 199. [Google Scholar] [CrossRef]
- Stachurska, A.; Ciesla, M.; Kozakowska, M.; Wolffram, S.; Boesch-Saadatmandi, C.; Rimbach, G.; Jozkowicz, A.; Dulak, J.; Loboda, A. Cross-talk between microRNAs, nuclear factor E2-related factor 2, and heme oxygenase-1 in ochratoxin A-induced toxic effects in renal proximal tubular epithelial cells. Mol. Nutr. Food Res. 2013, 57, 504–515. [Google Scholar] [CrossRef]
- Hou, W.; Zhu, X.; Liu, J.; Ma, J. Inhibition of miR-153 ameliorates ischemia/reperfusion-induced cardiomyocytes apoptosis by regulating Nrf2/HO-1 signaling in rats. Biomed. Eng. Online 2020, 19, 15. [Google Scholar] [CrossRef]
- Pulkkinen, K.H.; Yla-Herttuala, S.; Levonen, A.L. Heme oxygenase 1 is induced by miR-155 via reduced BACH1 translation in endothelial cells. Free Radic. Biol. Med. 2011, 51, 2124–2131. [Google Scholar] [CrossRef]
- Eades, G.; Yang, M.; Yao, Y.; Zhang, Y.; Zhou, Q. miR-200a regulates Nrf2 activation by targeting Keap1 mRNA in breast cancer cells. J. Biol. Chem. 2011, 286, 40725–40733. [Google Scholar] [CrossRef] [PubMed]
- Fiedler, J.; Stohr, A.; Gupta, S.K.; Hartmann, D.; Holzmann, A.; Just, A.; Hansen, A.; Hilfiker-Kleiner, D.; Eschenhagen, T.; Thum, T. Functional microRNA library screening identifies the hypoxamir miR-24 as a potent regulator of smooth muscle cell proliferation and vascularization. Antioxid. Redox Signal. 2014, 21, 1167–1176. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Song, N.Y.; Kim, E.H.; Na, H.K.; Joe, Y.; Chung, H.T.; Surh, Y.J. 15-deoxy-Delta12,14-prostaglandin J(2) induces p53 expression through Nrf2-mediated upregulation of heme oxygenase-1 in human breast cancer cells. Free Radic. Res. 2014, 48, 1018–1027. [Google Scholar] [CrossRef]
- Liu, L.; Dumbrepatil, A.B.; Fleischhacker, A.S.; Marsh, E.N.G.; Ragsdale, S.W. Heme oxygenase-2 is post-translationally regulated by heme occupancy in the catalytic site. J. Biol. Chem. 2020, 295, 17227–17240. [Google Scholar] [CrossRef] [PubMed]
- Fleischhacker, A.S.; Gunawan, A.L.; Kochert, B.A.; Liu, L.; Wales, T.E.; Borowy, M.C.; Engen, J.R.; Ragsdale, S.W. The heme-regulatory motifs of heme oxygenase-2 contribute to the transfer of heme to the catalytic site for degradation. J. Biol. Chem. 2020, 295, 5177–5191. [Google Scholar] [CrossRef]
- Boehning, D.; Moon, C.; Sharma, S.; Hurt, K.J.; Hester, L.D.; Ronnett, G.V.; Shugar, D.; Snyder, S.H. Carbon Monoxide Neurotransmission Activated by CK2 Phosphorylation of Heme Oxygenase-2. Neuron 2003, 40, 129–137. [Google Scholar] [CrossRef]
- Vukomanovic, D.; McLaughlin, B.E.; Rahman, M.N.; Szarek, W.A.; Brien, J.F.; Jia, Z.; Nakatsu, K. Selective activation of heme oxygenase-2 by menadione. Can. J. Physiol. Pharmacol. 2011, 89, 861–864. [Google Scholar] [CrossRef]
- Burnetti, A.L.; Johns, D.G.; Kriegsfeld, L.J.; Klein, S.L.; Calvin, D.C.; Demas, G.E.; Schramm, L.P.; Tonegawa, S.; Nelson, R.J.; Synder, S.H.; et al. Ejaculatory abnormalities in mice with targeted disruption of the gene for heme oxygenase-2. Nat. Med. 1998, 4, 84–87. [Google Scholar] [CrossRef]
- Stocker, R.; Yamamoto, Y.; McDonagh, A.F.; Glazer, A.N.; Ames, B.N. Bilirubin Is an Antioxidant of Possible Physiological Importance. Science 1987, 235, 1043–1046. [Google Scholar] [CrossRef]
- Wang, J.; Zhuang, H.; Dore, S. Heme oxygenase 2 is neuroprotective against intracerebral hemorrhage. Neurobiol. Dis. 2006, 22, 473–476. [Google Scholar] [CrossRef]
- Qu, Y.; Chen-Roetling, J.; Benvenisti-Zarom, L.; Regan, R.F. Attenuation of oxidative injury after induction of experimental intracerebral hemorrhage in heme oxygenase–2 knockout mice. J. Neurosurg. 2007, 106, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Wang, Z.; Chen, Q.; Zhang, T.; Zhu, Y.; Xian, X.; Han, X. Heme oxygenase-2 suppresses acute inflammation and improves the survival of skin allografts. Int. Immunopharmacol. 2018, 63, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Goodman, A.I.; Chander, P.N.; Rezzani, R.; Schwartzman, M.L.; Regan, R.F.; Rodella, L.; Turkseven, S.; Lianos, E.A.; Dennery, P.A.; Abraham, N.G. Heme oxygenase-2 deficiency contributes to diabetes-mediated increase in superoxide anion and renal dysfunction. J. Am. Soc. Nephrol. 2006, 17, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Xing, S.; Shen, D.; Chen, C.; Wang, J.; Yu, Z. Early induction of oxidative stress in a mouse model of Alzheimer’s disease with heme oxygenase activity. Mol. Med. Rep. 2014, 10, 599–604. [Google Scholar] [CrossRef]
- Ayuso, P.; Martinez, C.; Lorenzo-Betancor, O.; Pastor, P.; Luengo, A.; Jimenez-Jimenez, F.J.; Alonso-Navarro, H.; Villalba, M.T.; Agundez, J.A.; Garcia-Martin, E. A polymorphism located at an ATG transcription start site of the heme oxygenase-2 gene is associated with classical Parkinson’s disease. Pharmacogeneti. Genom. 2011, 21, 565–571. [Google Scholar] [CrossRef]
- Hayashi, S.; Omata, Y.; Sakamoto, H.; Higashimoto, Y.; Hara, T.; Sagara, Y.; Noguchi, M. Characterization of rat heme oxygenase-3 gene. Implication of processed pseudogenes derived from heme oxygenase-2 gene. Gene 2004, 336, 241–250. [Google Scholar] [CrossRef]
- Dutt, S.; Hamza, I.; Bartnikas, T.B. Molecular Mechanisms of Iron and Heme Metabolism. Annu. Rev. Nutr. 2022, 42, 311–335. [Google Scholar] [CrossRef]
- Chambers, I.G.; Willoughby, M.M.; Hamza, I.; Reddi, A.R. One ring to bring them all and in the darkness bind them: The trafficking of heme without deliverers. Biochim. Biophys. Acta Mol. Cell Res. 2021, 1868, 118881. [Google Scholar] [CrossRef]
- Korolnek, T.; Hamza, I. Like iron in the blood of the people: The requirement for heme trafficking in iron metabolism. Front. Pharmacol. 2014, 5, 126. [Google Scholar] [CrossRef]
- Kumar, A.; Ganini, D.; Deterding, L.J.; Ehrenshaft, M.; Chatterjee, S.; Mason, R.P. Immuno-spin trapping of heme-induced protein radicals: Implications for heme oxygenase-1 induction and heme degradation. Free Radic. Biol. Med. 2013, 61, 265–272. [Google Scholar] [CrossRef]
- Seiwert, N.; Adam, J.; Steinberg, P.; Wirtz, S.; Schwerdtle, T.; Adams-Quack, P.; Hövelmeyer, N.; Kaina, B.; Foersch, S.; Fahrer, J. Chronic intestinal inflammation drives colorectal tumor formation triggered by dietary heme iron in vivo. Arch. Toxicol. 2021, 95, 2507–2522. [Google Scholar] [CrossRef] [PubMed]
- Jeney, V.; Balla, J.; Yachie, A.; Varga, Z.; Vercellotti, G.M.; Eaton, J.W.; Balla, G. Pro-oxidant and cytotoxic effects of circulating heme. Blood 2002, 100, 879–887. [Google Scholar] [CrossRef] [PubMed]
- Yachie, A. Heme Oxygenase-1 Deficiency and Oxidative Stress: A Review of 9 Independent Human Cases and Animal Models. Int. J. Mol. Sci. 2021, 22, 1514. [Google Scholar] [CrossRef] [PubMed]
- Kovtunovych, G.; Ghosh, M.C.; Ollivierre, W.; Weitzel, R.P.; Eckhaus, M.A.; Tisdale, J.F.; Yachie, A.; Rouault, T.A. Wild-type macrophages reverse disease in heme oxygenase 1-deficient mice. Blood 2014, 124, 1522–1530. [Google Scholar] [CrossRef] [PubMed]
- Baranano, D.E.; Rao, M.; Ferris, C.D.; Snyder, S.H. Biliverdin reductase: A major physiologic cytoprotectant. Proc. Natl. Acad. Sci. USA 2002, 99, 16093–16098. [Google Scholar] [CrossRef]
- Suh, S.; Cho, Y.R.; Park, M.K.; Kim, D.K.; Cho, N.H.; Lee, M.K. Relationship between serum bilirubin levels and cardiovascular disease. PLoS ONE 2018, 13, e0193041. [Google Scholar] [CrossRef]
- Ziberna, L.; Martelanc, M.; Franko, M.; Passamonti, S. Bilirubin is an Endogenous Antioxidant in Human Vascular Endothelial Cells. Sci. Rep. 2016, 6, 29240. [Google Scholar] [CrossRef]
- Sedlak, T.W.; Saleh, M.; Higginson, D.S.; Paul, B.D.; Juluri, K.R.; Snyder, S.H. Bilirubin and glutathione have complementary antioxidant and cytoprotective roles. Proc. Natl. Acad. Sci. USA 2009, 106, 5171–5176. [Google Scholar] [CrossRef]
- Zuckerbraun, B.S.; Chin, B.Y.; Bilban, M.; d’Avila, J.C.; Rao, J.; Billiar, T.R.; Otterbein, L.E. Carbon monoxide signals via inhibition of cytochrome c oxidase and generation of mitochondrial reactive oxygen species. FASEB J. 2007, 21, 1099–1106. [Google Scholar] [CrossRef]
- Takagi, T.; Naito, Y.; Mizushima, K.; Hirai, Y.; Harusato, A.; Okayama, T.; Katada, K.; Kamada, K.; Uchiyama, K.; Handa, O.; et al. Heme oxygenase-1 prevents murine intstinal inflammation. J. Clin. Biochem. Nutr. 2018, 63, 169–174. [Google Scholar] [CrossRef]
- Kim, H.S.; Loughran, P.A.; Rao, J.; Billiar, T.R.; Zuckerbraun, B.S. Carbon monoxide activates NF-kappaB via ROS generation and Akt pathways to protect against cell death of hepatocytes. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 295, G146–G152. [Google Scholar] [CrossRef] [PubMed]
- Taille, C.; El-Benna, J.; Lanone, S.; Boczkowski, J.; Motterlini, R. Mitochondrial respiratory chain and NAD(P)H oxidase are targets for the antiproliferative effect of carbon monoxide in human airway smooth muscle. J. Biol. Chem. 2005, 280, 25350–25360. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Huang, S.; Zeng, L.; Ma, J.; Sun, S.; Zeng, F.; Kong, F.; Cheng, X. HMOX-1 inhibits TGF-beta-induced epithelial-mesenchymal transition in the MCF-7 breast cancer cell line. Int. J. Mol. Med. 2017, 40, 411–417. [Google Scholar] [CrossRef] [PubMed]
- Vogel, M.E.; Idelman, G.; Konaniah, E.S.; Zucker, S.D. Bilirubin Prevents Atherosclerotic Lesion Formation in Low-Density Lipoprotein Receptor-Deficient Mice by Inhibiting Endothelial VCAM-1 and ICAM-1 Signaling. J. Am. Heart Assoc. 2017, 6, e004820. [Google Scholar] [CrossRef]
- Zhou, Z.X.; Chen, J.K.; Hong, Y.Y.; Zhou, R.; Zhou, D.M.; Sun, L.Y.; Qin, W.L.; Wang, T.C. Relationship between the Serum Total Bilirubin and Inflammation in Patients with Psoriasis Vulgaris. J. Clin. Lab. Anal. 2016, 30, 768–775. [Google Scholar] [CrossRef]
- Akamatsu, Y.; Haga, M.; Tyagi, S.; Yamashita, K.; Graca-Souza, A.V.; Ollinger, R.; Czismadia, E.; May, G.A.; Ifedigbo, E.; Otterbein, L.E.; et al. Heme oxygenase-1-derived carbon monoxide protects hearts from transplant associated ischemia reperfusion injury. FASEB J. 2004, 18, 771–772. [Google Scholar] [CrossRef]
- Verma, A.; Hirsch, D.J.; Glatt, C.E.; Ronnett, G.V.; Snyder, S.H. Carbon monoxide: A putative neural messenger. Science 1993, 259, 381–384. [Google Scholar] [CrossRef]
- Nikolic, I.; Saksida, T.; Mangano, K.; Vujicic, M.; Stojanovic, I.; Nicoletti, F.; Stosic-Grujicic, S. Pharmacological application of carbon monoxide ameliorates islet-directed autoimmunity in mice via anti-inflammatory and anti-apoptotic effects. Diabetologia 2014, 57, 980–990. [Google Scholar] [CrossRef]
- Wu, L.; Wang, R. Carbon monoxide: Endogenous production, physiological functions, and pharmacological applications. Pharmacol. Rev. 2005, 57, 585–630. [Google Scholar] [CrossRef]
- Sylvester, J.T.; McGowan, C. The effects of agents that bind to cytochrome P-450 on hypoxic pulmonary vasoconstriction. Circ. Res. 1978, 43, 429–437. [Google Scholar] [CrossRef]
- Wang, R.; Wang, Z.; Wu, L. Carbon monoxide-induced vasorelaxation and the underlying mechanisms. Br. J. Pharmacol. 1997, 121, 927–934. [Google Scholar] [CrossRef] [PubMed]
- Brouard, S.; Otterbein, L.E.; Anrather, J.; Tobiasch, E.; Bach, F.H.; Choi, A.M.; Soares, M.P. Carbon monoxide generated by heme oxygenase 1 suppresses endothelial cell apoptosis. J. Exp. Med. 2000, 192, 1015–1026. [Google Scholar] [CrossRef] [PubMed]
- Morita, T.; Kourembanas, S. Endothelial cell expression of vasoconstrictors and growth factors is regulated by smooth muscle cell-derived carbon monoxide. J. Clin. Investig. 1995, 96, 2676–2682. [Google Scholar] [CrossRef] [PubMed]
- Nakahira, K.; Kim, H.P.; Geng, X.H.; Nakao, A.; Wang, X.; Murase, N.; Drain, P.F.; Wang, X.; Sasidhar, M.; Nabel, E.G.; et al. Carbon monoxide differentially inhibits TLR signaling pathways by regulating ROS-induced trafficking of TLRs to lipid rafts. J. Exp. Med. 2006, 203, 2377–2389. [Google Scholar] [CrossRef]
- Bilban, M.; Bach, F.H.; Otterbein, S.L.; Ifedigbo, E.; d’Avila, J.C.; Esterbauer, H.; Chin, B.Y.; Usheva, A.; Robson, S.C.; Wagner, O.; et al. Carbon monoxide orchestrates a protective response through PPARgamma. Immunity 2006, 24, 601–610. [Google Scholar] [CrossRef]
- Otterbein, L.E.; Bach, F.H.; Alam, J.; Soares, M.; Lu, H.T.; Wysk, M.; Davis, R.J.; Flavell, R.A.; Choi, A.M. Carbon monoxide has anti-inflammatory effects involving the mitogen-activated protein kinase pathway. Nat. Med. 2000, 6, 422–428. [Google Scholar] [CrossRef]
- Takagi, T.; Naito, Y.; Mizushima, K.; Akagiri, S.; Suzuki, T.; Hirata, I.; Omatsu, T.; Handa, O.; Kokura, S.; Ichikawa, H.; et al. Inhalation of carbon monoxide ameliorates TNBS-induced colitis in mice through the inhibition of TNF-alpha expression. Dig. Dis. Sci. 2010, 55, 2797–2804. [Google Scholar] [CrossRef]
- Zhang, X.; Shan, P.; Alam, J.; Davis, R.J.; Flavell, R.A.; Lee, P.J. Carbon monoxide modulates Fas/Fas ligand, caspases, and Bcl-2 family proteins via the p38alpha mitogen-activated protein kinase pathway during ischemia-reperfusion lung injury. J. Biol. Chem. 2003, 278, 22061–22070. [Google Scholar] [CrossRef]
- Zhang, X.; Shan, P.; Alam, J.; Fu, X.Y.; Lee, P.J. Carbon monoxide differentially modulates STAT1 and STAT3 and inhibits apoptosis via a phosphatidylinositol 3-kinase/Akt and p38 kinase-dependent STAT3 pathway during anoxia-reoxygenation injury. J. Biol. Chem. 2005, 280, 8714–8721. [Google Scholar] [CrossRef]
- Chin, B.Y.; Jiang, G.; Wegiel, B.; Wang, H.J.; MacDonald, T.; Zhang, X.C.; Gallo, D.; Cszimadia, E.; Bach, F.H.; Lee, P.J.; et al. Hypoxia-inducible factor 1α stabilization by carbon monoxide results in cytoprotective preconditioning. Proc. Natl. Acad. Sci. USA 2007, 104, 5109–5114. [Google Scholar] [CrossRef]
- Hu, Q.S.; Chen, Y.X.; Huang, Q.S.; Deng, B.Q.; Xie, S.L.; Wang, J.F.; Nie, R.Q. Carbon Monoxide Releasing Molecule Accelerates Reendothelialization after Carotid Artery Balloon Injury in Rat. Biomed. Environ. Sci. 2015, 28, 253–262. [Google Scholar] [CrossRef]
- Sheftel, A.D.; Kim, S.F.; Ponka, P. Non-heme Induction of Heme Oxygenase-1 Does Not Alter Cellular Iron Metabolism. J. Biol. Chem. 2007, 282, 10480–10486. [Google Scholar] [CrossRef]
- Eisenstein, R.S.; Garcia-Mayol, D.; Pettingell, W.; Munro, H.N. Regulation of ferritin and heme oxygenase synthesis in rat fibroblasts by different forms of iron. Proc. Natl. Acad. Sci. USA 1991, 88, 688–692. [Google Scholar] [CrossRef]
- Koorts, A.M.; Viljoen, M. Ferritin and ferritin isoforms I: Structure-function relationships, synthesis, degradation and secretion. Arch. Physiol. Biochem. 2007, 113, 30–54. [Google Scholar] [CrossRef] [PubMed]
- Vanoaica, L.; Darshan, D.; Richman, L.; Schumann, K.; Kuhn, L.C. Intestinal ferritin H is required for an accurate control of iron absorption. Cell Metab. 2010, 12, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Zarjou, A.; Bolisetty, S.; Joseph, R.; Traylor, A.; Apostolov, E.O.; Arosio, P.; Balla, J.; Verlander, J.; Darshan, D.; Kuhn, L.C.; et al. Proximal tubule H-ferritin mediates iron trafficking in acute kidney injury. J. Clin. Investig. 2013, 123, 4423–4434. [Google Scholar] [CrossRef] [PubMed]
- Pham, C.G.; Bubici, C.; Zazzeroni, F.; Papa, S.; Jones, J.; Alvarez, K.; Jayawardena, S.; De Smaele, E.; Cong, R.; Beaumont, C.; et al. Ferritin heavy chain upregulation by NF-kappaB inhibits TNFalpha-induced apoptosis by suppressing reactive oxygen species. Cell 2004, 119, 529–542. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhang, L.; Zhang, Q.; Liu, B.; Li, F.; Xin, Y.; Duan, Z. HO-1/CO Maintains Intestinal Barrier Integrity through NF-kappaB/MLCK Pathway in Intestinal HO-1(−/−) Mice. Oxid. Med. Cell Longev. 2021, 2021, 6620873. [Google Scholar] [CrossRef]
- Akagi, R. Role of Heme Oxygenase in Gastrointestinal Epithelial Cells. Antioxidants 2022, 11, 1323. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, Z.; Liu, B.; Jin, Y.; Tian, Y.; Xin, Y.; Duan, Z. The Protective Effect of Heme Oxygenase-1 against Intestinal Barrier Dysfunction in Cholestatic Liver Injury Is Associated with NF-κB Inhibition. Mol. Med. 2017, 23, 215–224. [Google Scholar] [CrossRef]
- Glassner, K.L.; Abraham, B.P.; Quigley, E.M.M. The microbiome and inflammatory bowel disease. J. Allergy Clin. Immunol. 2020, 145, 16–27. [Google Scholar] [CrossRef]
- Kaplan, G.G. The global burden of IBD: From 2015 to 2025. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 720–727. [Google Scholar] [CrossRef]
- Takagi, T.; Naito, Y.; Mizushima, K.; Nukigi, Y.; Okada, H.; Suzuki, T.; Hirata, I.; Omatsu, T.; Okayama, T.; Handa, O.; et al. Increased intestinal expression of heme oxygenase-1 and its localization in patients with ulcerative colitis. J. Gastroenterol. Hepatol. 2008, 23 (Suppl. S2), S229–S233. [Google Scholar] [CrossRef]
- Sheikh, S.Z.; Hegazi, R.A.; Kobayashi, T.; Onyiah, J.C.; Russo, S.M.; Matsuoka, K.; Sepulveda, A.R.; Li, F.; Otterbein, L.E.; Plevy, S.E. An anti-inflammatory role for carbon monoxide and heme oxygenase-1 in chronic Th2-mediated murine colitis. J. Immunol. 2011, 186, 5506–5513. [Google Scholar] [CrossRef]
- Kapturczak, M.H.; Wasserfall, C.; Brusko, T.; Campbell-Thompson, M.; Ellis, T.M.; Atkinson, M.A.; Agarwal, A. Heme Oxygenase-1 Modulates Early Inflammatory Responses. Am. J. Pathol. 2004, 165, 1045–1053. [Google Scholar] [CrossRef]
- Greil, J.; Verga-Falzacappa, M.V.; Echner, N.E.; Behnisch, W.; Bandapalli, O.R.; Pechanska, P.; Immenschuh, S.; Vijayan, V.; Balla, J.; Tsukahara, H.; et al. Mutating heme oxygenase-1 into a peroxidase causes a defect in bilirubin synthesis associated with microcytic anemia and severe hyperinflammation. Haematologica 2016, 101, e437. [Google Scholar] [CrossRef] [PubMed]
- Gionchetti, P.; Rizzello, F.; Helwig, U.; Venturi, A.; Lammers, K.M.; Brigidi, P.; Vitali, B.; Poggioli, G.; Miglioli, M.; Campieri, M. Prophylaxis of pouchitis onset with probiotic therapy: A double-blind, placebo-controlled trial. Gastroenterology 2003, 124, 1202–1209. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, L.; Singleton, J.; Sessions, J.; Hanauer, S.; Krawitt, E.; Rankin, G.; Summers, R.; Mekhjian, H.; Greenberger, N.; Kelly, M. Double blind, placebo controlled trial of metronidazole in Crohn’s disease. Gut 1991, 32, 1071–1075. [Google Scholar] [CrossRef] [PubMed]
- Onyiah, J.C.; Sheikh, S.Z.; Maharshak, N.; Steinbach, E.C.; Russo, S.M.; Kobayashi, T.; Mackey, L.C.; Hansen, J.J.; Moeser, A.J.; Rawls, J.F.; et al. Carbon monoxide and heme oxygenase-1 prevent intestinal inflammation in mice by promoting bacterial clearance. Gastroenterology 2013, 144, 789–798. [Google Scholar] [CrossRef]
- Paul, G.; Bataille, F.; Obermeier, F.; Bock, J.; Klebl, F.; Strauch, U.; Lochbaum, D.; Rummele, P.; Farkas, S.; Scholmerich, J.; et al. Analysis of intestinal haem-oxygenase-1 (HO-1) in clinical and experimental colitis. Clin. Exp. Immunol. 2005, 140, 547–555. [Google Scholar] [CrossRef]
- Seo, G.S.; Jiang, W.-Y.; Chi, J.H.; Jin, H.; Park, W.-C.; Sohn, D.H.; Park, P.-H.; Lee, S.H. Heme oxygenase-1 promotes tumor progression and metastasis of colorectal carcinoma cells by inhibiting antitumor immunity. Oncotarget 2015, 6, 19792–19806. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer Immunoediting: Integrating Immunity’s Roles in Cancer Suppression and Promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef] [PubMed]
- Lim, E.J.; Kang, J.H.; Kim, Y.J.; Kim, S.; Lee, S.J. ICAM-1 promotes cancer progression by regulating SRC activity as an adapter protein in colorectal cancer. Cell Death Dis. 2022, 13, 417. [Google Scholar] [CrossRef] [PubMed]
- Spaander, M.C.W.; Zauber, A.G.; Syngal, S.; Blaser, M.J.; Sung, J.J.; You, Y.N.; Kuipers, E.J. Young-onset colorectal cancer. Nat. Rev. Dis. Primers 2023, 9, 21. [Google Scholar] [CrossRef] [PubMed]
- Keum, N.; Giovannucci, E. Global burden of colorectal cancer: Emerging trends, risk factors and prevention strategies. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 713–732. [Google Scholar] [CrossRef] [PubMed]
- Murphy, N.; Moreno, V.; Hughes, D.J.; Vodicka, L.; Vodicka, P.; Aglago, E.K.; Gunter, M.J.; Jenab, M. Lifestyle and dietary environmental factors in colorectal cancer susceptibility. Mol. Asp. Med. 2019, 69, 2–9. [Google Scholar] [CrossRef]
- Seiwert, N.; Heylmann, D.; Hasselwander, S.; Fahrer, J. Mechanism of colorectal carcinogenesis triggered by heme iron from red meat. Biochim. Biophys. Acta. Rev. Cancer 2020, 1873, 188334. [Google Scholar] [CrossRef]
- Dörsam, B.; Seiwert, N.; Foersch, S.; Stroh, S.; Nagel, G.; Begaliew, D.; Diehl, E.; Kraus, A.; McKeague, M.; Minneker, V.; et al. PARP-1 protects against colorectal tumor induction, but promotes inflammation-driven colorectal tumor progression. Proc. Natl. Acad. Sci. USA 2018, 115, E4061–E4070. [Google Scholar] [CrossRef]
- Fearon, E.R.; Vogelstein, B.; Fearon, E.R.; Vogelstein, B. A Genetic Model for Colorectal Tumorigenesis. Cell 1990, 61, 759–767. [Google Scholar] [CrossRef]
- Vargas, A.J.; Thompson, P.A.; Vargas, A.J.; Thompson, P.A. Diet and Nutrient Factors in Colorectal Cancer Risk. Nutr. Clin. Pract. 2012, 27, 613–623. [Google Scholar] [CrossRef]
- Kostka, T.; Fohrer, J.; Guigas, C.; Briviba, K.; Seiwert, N.; Fahrer, J.; Steinberg, P.; Empl, M.T. Synthesis and in vitro characterization of the genotoxic, mutagenic and cell-transforming potential of nitrosylated heme. Arch. Toxicol. 2020, 94, 3911–3927. [Google Scholar] [CrossRef]
- Fahrer, J.; Christmann, M. DNA Alkylation Damage by Nitrosamines and Relevant DNA Repair Pathways. Int. J. Mol. Sci. 2023, 24, 4684. [Google Scholar] [CrossRef]
- Mimmler, M.; Peter, S.; Kraus, A.; Stroh, S.; Nikolova, T.; Seiwert, N.; Hasselwander, S.; Neitzel, C.; Haub, J.; Monien, B.H.; et al. DNA damage response curtails detrimental replication stress and chromosomal instability induced by the dietary carcinogen PhIP. Nucleic Acids Res. 2016, 44, 10259–10276. [Google Scholar] [CrossRef] [PubMed]
- Lynch, H.T.; Lynch, J.F.; Lynch, P.M.; Attard, T. Hereditary colorectal cancer syndromes: Molecular genetics, genetic counseling, diagnosis and management. Fam. Cancer 2008, 7, 27–39. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.; Emmerson, E.; Maynard, J.; Best, J.M.; Jordan, S.; Williams, G.T.; Sampson, J.R.; Cheadle, J.P. Biallelic germline mutations in MYH predispose to multiple colorectal adenoma and somatic G:C-->T:A mutations. Hum. Mol. Genet. 2002, 11, 2961–2967. [Google Scholar] [CrossRef] [PubMed]
- Fahrer, J.; Kaina, B. O6-methylguanine-DNA methyltransferase in the defense against N-nitroso compounds and colorectal cancer. Carcinogenesis 2013, 34, 2435–2442. [Google Scholar] [CrossRef] [PubMed]
- Markowitz, S.D.; Bertagnolli, M.M. Molecular origins of cancer: Molecular basis of colorectal cancer. N. Engl. J. Med. 2009, 361, 2449–2460. [Google Scholar] [CrossRef]
- Kinzler, K.W.; Vogelstein, B.; Kinzler, K.W.; Vogelstein, B. Lessons from Hereditary Colorectal Cancer. Cell 1996, 87, 159–170. [Google Scholar] [CrossRef]
- Tomasetti, C.; Marchionni, L.; Nowak, M.A.; Parmigiani, G.; Vogelstein, B.; Tomasetti, C.; Marchionni, L.; Nowak, M.A.; Parmigiani, G.; Vogelstein, B. Only three driver gene mutations are required for the development of lung and colorectal cancers. Proc. Natl. Acad. Sci. USA 2015, 112, 118–123. [Google Scholar] [CrossRef]
- Harlozinska, A. Progress in Molecular Mechanisms of Tumor Metastasis and Angiogenesis. Anticancer Res. 2005, 25, 3327–3334. [Google Scholar]
- Andres, N.C.; Fermento, M.E.; Gandini, N.A.; Romero, A.L.; Ferro, A.; Donna, L.G.; Curino, A.C.; Facchinetti, M.M. Heme oxygenase-1 has antitumoral effects in colorectal cancer: Involvement of p53. Exp. Mol. Pathol. 2014, 97, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Kang, K.A.; Maeng, Y.H.; Zhang, R.; Yang, Y.R.; Piao, M.J.; Kim, K.C.; Kim, G.Y.; Kim, Y.R.; Koh, Y.S.; Kang, H.K.; et al. Involvement of heme oxygenase-1 in Korean colon cancer. Tumour Biol. 2012, 33, 1031–1038. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Fang, J.; Liao, L.; Maeda, H.; Su, Q. Upregulation of heme oxygenase-1 in colorectal cancer patients with increased circulation carbon monoxide levels, potentially affects chemotherapeutic sensitivity. BMC Cancer 2014, 14, 436. [Google Scholar] [CrossRef] [PubMed]
- Timney, B.L.; Raveh, B.; Mironska, R.; Trivedi, J.M.; Kim, S.J.; Russel, D.; Wente, S.R.; Sali, A.; Rout, M.P. Simple rules for passive diffusion through the nuclear pore complex. J. Cell Biol. 2016, 215, 57–76. [Google Scholar] [CrossRef]
- Ma, J.; Goryaynov, A.; Sarma, A.; Yang, W. Self-regulated viscous channel in the nuclear pore complex. Proc. Natl. Acad. Sci. USA 2012, 109, 7326–7331. [Google Scholar] [CrossRef]
- Vanella, L.; Barbagallo, I.; Tibullo, D.; Forte, S.; Zappalà, A.; Volti, G.L. The non-canonical functions of the heme oxygenases. Oncotarget 2016, 7, 69075–69086. [Google Scholar] [CrossRef]
- Becker, J.C.; Fukui, H.; Imai, Y.; Sekikawa, A.; Kimura, T.; Yamagishi, H.; Yoshitake, N.; Pohle, T.; Domschke, W.; Fujimori, T. Colonic expression of heme oxygenase-1 is associated with a better long-term survival in patients with colorectal cancer. Scand. J. Gastroenterol. 2007, 42, 852–858. [Google Scholar] [CrossRef]
- Kimura, S.; Aung, N.Y.; Ohe, R.; Yano, M.; Hashimoto, T.; Fujishima, T.; Kimura, W.; Yamakawa, M. Increasing Heme Oxygenase-1-Expressing Macrophages Indicates a Tendency of Poor Prognosis in Advanced Colorectal Cancer. Digestion 2020, 101, 401–410. [Google Scholar] [CrossRef]
- Alaluf, E.; Vokaer, B.; Detavernier, A.; Azouz, A.; Splittgerber, M.; Carrette, A.; Boon, L.; Libert, F.; Soares, M.; Le Moine, A.; et al. Heme oxygenase-1 orchestrates the immunosuppressive program of tumor-associated macrophages. JCI Insight 2020, 5, e133929. [Google Scholar] [CrossRef]
- Jiang, Z.; Tao, G.; Zhu, Y. The Expression and Clinical Significance of miRNA-135a and Bach1 in Colorectal Cancer. Int. Surg. 2021, 105, 649–654. [Google Scholar] [CrossRef]
- Zhou, W.; Li, X.; Liu, F.; Xiao, Z.; He, M.; Shen, S.; Liu, S. MiR-135a promotes growth and invasion of colorectal cancer via metastasis suppressor 1 in vitro. Acta Biochim. Biophys. Sin. 2012, 44, 838–846. [Google Scholar] [CrossRef] [PubMed]
- Novikov, N.M.; Zolotaryova, S.Y.; Gautreau, A.M.; Denisov, E.V. Mutational drivers of cancer cell migration and invasion. Br. J. Cancer 2021, 124, 102–114. [Google Scholar] [CrossRef] [PubMed]
- Rios-Arrabal, S.; Puentes-Pardo, J.D.; Moreno-SanJuan, S.; Szuba, A.; Casado, J.; Garcia-Costela, M.; Escudero-Feliu, J.; Verbeni, M.; Cano, C.; Gonzalez-Puga, C.; et al. Endothelin-1 as a Mediator of Heme Oxygenase-1-Induced Stemness in Colorectal Cancer: Influence of p53. J. Pers. Med. 2021, 11, 509. [Google Scholar] [CrossRef] [PubMed]
- Perez-Moreno, P.; Indo, S.; Niechi, I.; Huerta, H.; Cabello, P.; Jara, L.; Aguayo, F.; Varas-Godoy, M.; Burzio, V.A.; Tapia, J.C. Endothelin-converting enzyme-1c promotes stem cell traits and aggressiveness in colorectal cancer cells. Mol. Oncol. 2020, 14, 347–362. [Google Scholar] [CrossRef]
- Huang, E.H.; Hynes, M.J.; Zhang, T.; Ginestier, C.; Dontu, G.; Appelman, H.; Fields, J.Z.; Wicha, M.S.; Boman, B.M. Aldehyde dehydrogenase 1 is a marker for normal and malignant human colonic stem cells (SC) and tracks SC overpopulation during colon tumorigenesis. Cancer Res. 2009, 69, 3382–3389. [Google Scholar] [CrossRef]
- Du, L.; Wang, H.; He, L.; Zhang, J.; Ni, B.; Wang, X.; Jin, H.; Cahuzac, N.; Mehrpour, M.; Lu, Y.; et al. CD44 is of functional importance for colorectal cancer stem cells. Clin. Cancer Res. 2008, 14, 6751–6760. [Google Scholar] [CrossRef]
- Todaro, M.; Gaggianesi, M.; Catalano, V.; Benfante, A.; Iovino, F.; Biffoni, M.; Apuzzo, T.; Sperduti, I.; Volpe, S.; Cocorullo, G.; et al. CD44v6 is a marker of constitutive and reprogrammed cancer stem cells driving colon cancer metastasis. Cell Stem Cell 2014, 14, 342–356. [Google Scholar] [CrossRef]
- Tavassolifar, M.J.; Vodjgani, M.; Salehi, Z.; Izad, M. The Influence of Reactive Oxygen Species in the Immune System and Pathogenesis of Multiple Sclerosis. Autoimmune Dis. 2020, 2020, 5793817. [Google Scholar] [CrossRef]
- Lien, G.S.; Wu, M.S.; Bien, M.Y.; Chen, C.H.; Lin, C.H.; Chen, B.C. Epidermal growth factor stimulates nuclear factor-kappaB activation and heme oxygenase-1 expression via c-Src, NADPH oxidase, PI3K, and Akt in human colon cancer cells. PLoS ONE 2014, 9, e104891. [Google Scholar] [CrossRef]
- Foresti, R.; Goatly, H.; Green, C.J.; Motterlini, R. Role of heme oxygenase-1 in hypoxia-reoxygenation: Requirement of substrate heme to promote cardioprotection. Am. J. Physiol.—Heart C 2001, 281, H1976–H1984. [Google Scholar] [CrossRef]
- Cuypers, H.T.M.; Ter Haar, E.M.; Jansen, P.L. Microsomal conjugation and oxidation of bilirubin. Biochim. Biophys. Acta 1983, 758, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Wang, H. Oxaliplatin induces ferroptosis and oxidative stress in HT29 colorectal cancer cells by inhibiting the Nrf2 signaling pathway. Exp. Ther. Med. 2022, 23, 394. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Sawa, T.; Akaike, T.; Akuta, T.; Sahoo, S.K.; Khaled, G.; Hamada, A.; Maeda, H. In Vivo Antitumor Activity of Pegylated Zinc Protoporphyrin: Targeted Inhibition of Heme Oxygenase in Solid Tumor. Cancer Res. 2003, 63, 3567–3574. [Google Scholar]
- Nowis, D.; Bugajski, M.; Winiarska, M.; Bil, J.; Szokalska, A.; Salwa, P.; Issat, T.; Was, H.; Jozkowicz, A.; Dulak, J.; et al. Zinc protoporphyrin IX, a heme oxygenase-1 inhibitor, demonstrates potent antitumor effects but is unable to potentiate antitumor effects of chemotherapeutics in mice. BMC Cancer 2008, 8, 197. [Google Scholar] [CrossRef] [PubMed]
- Olejniczak, A.; Szarynska, M.; Kmiec, Z. In vitro characterization of spheres derived from colorectal cancer cell lines. Int. J. Oncol. 2018, 52, 599–612. [Google Scholar] [CrossRef] [PubMed]
- Luk, I.Y.; Jenkins, L.J.; Schoffer, K.L.; Ng, I.; Tse, J.W.T.; Mouradov, D.; Kaczmarczyk, S.; Nightingale, R.; Burrows, A.D.; Anderson, R.L.; et al. Epithelial de-differentiation triggered by co-ordinate epigenetic inactivation of the EHF and CDX1 transcription factors drives colorectal cancer progression. Cell Death Differ. 2022, 29, 2288–2302. [Google Scholar] [CrossRef]
- Wu, M.S.; Chien, C.C.; Chang, J.; Chen, Y.C. Pro-apoptotic effect of haem oxygenase-1 in human colorectal carcinoma cells via endoplasmic reticular stress. J. Cell Mol. Med. 2019, 23, 5692–5704. [Google Scholar] [CrossRef]
- Pierre, F.; Peiro, G.; Tache, S.; Cross, A.J.; Bingham, S.A.; Gasc, N.; Gottardi, G.; Corpet, D.E.; Gueraud, F.; Pierre, F.; et al. New marker of colon cancer risk associated with heme intake: 1,4-dihydroxynonane mercapturic acid. Cancer Epidemiol. Biomark. Prev. 2006, 15, 2274–2279. [Google Scholar] [CrossRef]
- Ijssennagger, N.; Rijnierse, A.; Wit, N.J.W.d.; Boekschoten, M.V.; Dekker, J.; Schonewille, A.; Müller, M.; van der Meer, R. Dietary heme induces acute oxidative stress, but delayed cytotoxicity and compensatory hyperproliferation in mouse colon. Carcinogenesis 2013, 34, 1628–1635. [Google Scholar] [CrossRef]
- Cheng, Z.; Li, Y. What Is Responsible for the Initiating Chemistry of Iron-Mediated Lipid Peroxidation: An Update. Chem. Rev. 2007, 107, 748–766. [Google Scholar] [CrossRef]
- Gaschler, M.M.; Stockwell, B.R. Lipid peroxidation in cell death. Biochem. Biophys. Res. Commun. 2017, 482, 419–425. [Google Scholar] [CrossRef]
- Guéraud, F.; Atalay, M.; Bresgen, N.; Cipak, A.; Eckl, P.M.; Huc, L.; Jouanin, I.; Siems, W.; Uchida, K. Chemistry and biochemistry of lipid peroxidation products. Free Radic. Res. 2010, 44, 1098–1124. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Xia, T.; Yang, G.; Yang, H.; Zhang, J. Zinc protoporphyrin IX improves the sensitivity of colorectal cancer cells to paclitaxel by inactivating AKT/mTOR pathway via HO-1. Trop. J. Pharm. Res. 2022, 21, 2249–2253. [Google Scholar] [CrossRef]
- Otterbein, L.E.; Hedblom, A.; Harris, C.; Csizmadia, E.; Gallo, D.; Wegiel, B. Heme oxygenase-1 and carbon monoxide modulate DNA repair through ataxia-telangiectasia mutated (ATM) protein. Proc. Natl. Acad. Sci. USA 2011, 108, 14491–14496. [Google Scholar] [CrossRef]
- Nikolova, T.; Dvorak, M.; Jung, F.; Adam, I.; Kramer, E.; Gerhold-Ay, A.; Kaina, B. The gammaH2AX assay for genotoxic and nongenotoxic agents: Comparison of H2AX phosphorylation with cell death response. Toxicol. Sci. 2014, 140, 103–117. [Google Scholar] [CrossRef] [PubMed]
- Fahrer, J.; Frisch, J.; Nagel, G.; Kraus, A.; Dörsam, B.; Thomas, A.D.; Reißig, S.; Waisman, A.; Kaina, B. DNA repair by MGMT, but not AAG, causes a threshold in alkylation-induced colorectal carcinogenesis. Carcinogenesis 2015, 36, 1235–1244. [Google Scholar] [CrossRef]
- Ayala, A.; Munoz, M.F.; Arguelles, S.; Ayala, A.; Muñoz, M.F.; Argüelles, S. Lipid Peroxidation: Production, Metabolism, and Signaling Mechanisms of Malondialdehyde and 4-Hydroxy-2-Nonenal. Oxid. Med. Cell Longev. 2014, 2014, 360438. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.D.; Fahrer, J.; Johnson, G.E.; Kaina, B. Theoretical considerations for thresholds in chemical carcinogenesis. Mutat. Res. 2015, 765, 56–67. [Google Scholar] [CrossRef]
- Lowe, S.W.; Cepero, E.; Evan, G. Intrinsic tumour suppression. Nature 2004, 432, 307–315. [Google Scholar] [CrossRef]
- Busserolles, J.; Megias, J.; Terencio, M.C.; Alcaraz, M.J. Heme oxygenase-1 inhibits apoptosis in Caco-2 cells via activation of Akt pathway. Int. J. Biochem. Cell Biol. 2006, 38, 1510–1517. [Google Scholar] [CrossRef]
- Nowis, D.; Legat, M.; Grzela, T.; Niderla, J.; Wilczek, E.; Wilczynski, G.M.; Glodkowska, E.; Mrowka, P.; Issat, T.; Dulak, J.; et al. Heme oxygenase-1 protects tumor cells against photodynamic therapy-mediated cytotoxicity. Oncogene 2006, 25, 3365–3374. [Google Scholar] [CrossRef] [PubMed]
- Uc, A.; Britigan, B.E. Does Heme Oxygenase-1 Have a Role in Caco-2 Cell Cycle Progression? Exp. Biol. Med. 2003, 228, 590–595. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.-K.; Chen, C.-C.; Panyod, S.; Chen, R.-A.; Wu, M.-S.; Sheen, L.-Y.; Chang, S.-C. Optimization of fecal sample processing for microbiome study—The journey from bathroom to bench. J. Formos. Med. Assoc.=Taiwan. Yi Zhi 2019, 118, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Li, J.; Cao, F.; Yin, H.L.; Huang, Z.J.; Lin, Z.T.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, present and future. Cell Death Dis. 2020, 11, 88. [Google Scholar] [CrossRef]
- Zukor, H.; Song, W.; Liberman, A.; Mui, J.; Vali, H.; Fillebeen, C.; Pantopoulos, K.; Wu, T.D.; Guerquin-Kern, J.L.; Schipper, H.M. HO-1-mediated macroautophagy: A mechanism for unregulated iron deposition in aging and degenerating neural tissues. J. Neurochem. 2009, 109, 776–791. [Google Scholar] [CrossRef]
- Ryan, S.K.; Ugalde, C.L.; Rolland, A.S.; Skidmore, J.; Devos, D.; Hammond, T.R. Therapeutic inhibition of ferroptosis in neurodegenerative disease. Trends Pharmacol. Sci. 2023, 44, 674–688. [Google Scholar] [CrossRef]
- Friedmann Angeli, J.P.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.A.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E.; et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 2014, 16, 1180–1191. [Google Scholar] [CrossRef]
- Yan, B.; Ai, Y.; Sun, Q.; Ma, Y.; Cao, Y.; Wang, J.; Zhang, Z.; Wang, X. Membrane Damage during Ferroptosis Is Caused by Oxidation of Phospholipids Catalyzed by the Oxidoreductases POR and CYB5R1. Mol. Cell 2021, 81, 355–369.e310. [Google Scholar] [CrossRef]
- Macias-Rodriguez, R.U.; Inzaugarat, M.E.; Ruiz-Margain, A.; Nelson, L.J.; Trautwein, C.; Cubero, F.J. Reclassifying Hepatic Cell Death during Liver Damage: Ferroptosis-A Novel Form of Non-Apoptotic Cell Death? Int. J. Mol. Sci. 2020, 21, 1651. [Google Scholar] [CrossRef]
- Song, Y.Q.; Yan, X.D.; Wang, Y.; Wang, Z.Z.; Mao, X.L.; Ye, L.P.; Li, S.W. Role of ferroptosis in colorectal cancer. World J. Gastrointest. Oncol. 2023, 15, 225–239. [Google Scholar] [CrossRef] [PubMed]
- Miotto, G.; Rossetto, M.; Di Paolo, M.L.; Orian, L.; Venerando, R.; Roveri, A.; Vuckovic, A.M.; Bosello Travain, V.; Zaccarin, M.; Zennaro, L.; et al. Insight into the mechanism of ferroptosis inhibition by ferrostatin-1. Redox Biol. 2020, 28, 101328. [Google Scholar] [CrossRef] [PubMed]
- Dang, D.; Meng, Z.; Zhang, C.; Li, Z.; Wei, J.; Wu, H. Heme induces intestinal epithelial cell ferroptosis via mitochondrial dysfunction in transfusion-associated necrotizing enterocolitis. FASEB J. 2022, 36, e22649. [Google Scholar] [CrossRef]
- Kwon, M.-Y.; Park, E.; Lee, S.-J.; Chung, S.W. Heme oxygenase-1 accelerates erastin-induced ferroptotic cell death. Oncotarget 2015, 6, 24393–24403. [Google Scholar] [CrossRef] [PubMed]
- Hino, K.; Yanatori, I.; Hara, Y.; Nishina, S. Iron and liver cancer: An inseparable connection. FEBS J. 2022, 289, 7810–7829. [Google Scholar] [CrossRef] [PubMed]
- Ichimura, Y.; Waguri, S.; Sou, Y.S.; Kageyama, S.; Hasegawa, J.; Ishimura, R.; Saito, T.; Yang, Y.; Kouno, T.; Fukutomi, T.; et al. Phosphorylation of p62 activates the Keap1-Nrf2 pathway during selective autophagy. Mol. Cell 2013, 51, 618–631. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, M.; Kurokawa, H.; Waguri, S.; Taguchi, K.; Kobayashi, A.; Ichimura, Y.; Sou, Y.-S.; Ueno, I.; Sakamoto, A.; Tong, K.I.; et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 2010, 12, 213–223. [Google Scholar] [CrossRef]
- Sun, X.; Ou, Z.; Chen, R.; Niu, X.; Chen, D.; Kang, R.; Tang, D. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology 2016, 63, 173–184. [Google Scholar] [CrossRef]
- Nishizawa, H.; Yamanaka, M.; Igarashi, K. Ferroptosis: Regulation by competition between NRF2 and BACH1 and propagation of the death signal. FEBS J. 2023, 290, 1688–1704. [Google Scholar] [CrossRef]
- Chen, E.I.; Hewel, J.; Krueger, J.S.; Tiraby, C.; Weber, M.R.; Kralli, A.; Becker, K.; Yates, J.R., 3rd; Felding-Habermann, B. Adaptation of energy metabolism in breast cancer brain metastases. Cancer Res. 2007, 67, 1472–1486. [Google Scholar] [CrossRef]
- Trachootham, D.; Alexandre, J.; Huang, P. Targeting cancer cells by ROS-mediated mechanisms: A radical therapeutic approach? Nat. Rev. Drug Discov. 2009, 8, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Brigelius-Flohe, R.; Maiorino, M. Glutathione peroxidases. Biochim. Biophys. Acta 2013, 1830, 3289–3303. [Google Scholar] [CrossRef] [PubMed]
- Singhal, R.; Mitta, S.R.; Das, N.K.; Kerk, S.A.; Sajjakulnukit, P.; Solanki, S.; Andren, A.; Kumar, R.; Olive, K.P.; Banerjee, R.; et al. HIF-2alpha activation potentiates oxidative cell death in colorectal cancers by increasing cellular iron. J. Clin. Investig. 2021, 131, e143691. [Google Scholar] [CrossRef] [PubMed]
- Almahi, W.A.; Yu, K.N.; Mohammed, F.; Kong, P.; Han, W. Hemin enhances radiosensitivity of lung cancer cells through ferroptosis. Exp. Cell Res. 2022, 410, 112946. [Google Scholar] [CrossRef]
- Gao, Z.; Zhang, Z.; Gu, D.; Li, Y.; Zhang, K.; Dong, X.; Liu, L.; Zhang, J.; Chen, J.; Wu, D.; et al. Hemin mitigates contrast-induced nephropathy by inhibiting ferroptosis via HO-1/Nrf2/GPX4 pathway. Clin. Exp. Pharmacol. Physiol. 2022, 49, 858–870. [Google Scholar] [CrossRef] [PubMed]
- Malfa, G.A.; Tomasello, B.; Acquaviva, R.; Genovese, C.; La Mantia, A.; Cammarata, F.P.; Ragusa, M.; Renis, M.; Di Giacomo, C. Betula etnensis Raf. (Betulaceae) Extract Induced HO-1 Expression and Ferroptosis Cell Death in Human Colon Cancer Cells. Int. J. Mol. Sci. 2019, 20, 2723. [Google Scholar] [CrossRef]
- Jin, J.; Wang, D.; Xiao, H.; Wei, H.; Matunda, C.; Zhang, H.; Li, X.; Wang, C.; Zou, C.; Gao, X.; et al. Enhancement of DEN-induced liver tumorigenesis in heme oxygenase-1 G143H mutant transgenic mice. Biochem. Biophys. Res. Commun. 2016, 481, 169–175. [Google Scholar] [CrossRef]
- Barikbin, R.; Berkhout, L.; Bolik, J.; Schmidt-Arras, D.; Ernst, T.; Ittrich, H.; Adam, G.; Parplys, A.; Casar, C.; Krech, T.; et al. Early heme oxygenase 1 induction delays tumour initiation and enhances DNA damage repair in liver macrophages of Mdr2(−/−) mice. Sci. Rep. 2018, 8, 16238. [Google Scholar] [CrossRef]
- Hedblom, A.; Hejazi, S.M.; Canesin, G.; Choudhury, R.; Hanafy, K.A.; Csizmadia, E.; Persson, J.L.; Wegiel, B. Heme detoxification by heme oxygenase-1 reinstates proliferative and immune balances upon genotoxic tissue injury. Cell Death Dis. 2019, 10, 72. [Google Scholar] [CrossRef]
- Lang, D.; Reuter, S.; Buzescu, T.; August, C.; Heidenreich, S. Heme-induced heme oxygenase-1 (HO-1) in human monocytes inhibits apoptosis despite caspase-3 up-regulation. Int. Immunol. 2005, 17, 155–165. [Google Scholar] [CrossRef]
- Chen, G.G.; Liu, Z.M.; Vlantis, A.C.; Tse, G.M.; Leung, B.C.; van Hasselt, C.A. Heme oxygenase-1 protects against apoptosis induced by tumor necrosis factor-alpha and cycloheximide in papillary thyroid carcinoma cells. J. Cell Biochem. 2004, 92, 1246–1256. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.M.; Chen, G.G.; Ng, E.K.; Leung, W.K.; Sung, J.J.; Chung, S.C. Upregulation of heme oxygenase-1 and p21 confers resistance to apoptosis in human gastric cancer cells. Oncogene 2004, 23, 503–513. [Google Scholar] [CrossRef] [PubMed]
- Sass, G.; Leukel, P.; Schmitz, V.; Raskopf, E.; Ocker, M.; Neureiter, D.; Meissnitzer, M.; Tasika, E.; Tannapfel, A.; Tiegs, G. Inhibition of heme oxygenase 1 expression by small interfering RNA decreases orthotopic tumor growth in livers of mice. Int. J. Cancer 2008, 123, 1269–1277. [Google Scholar] [CrossRef] [PubMed]
- Sass, G.; Soares, M.C.P.; Yamashita, K.; Seyfried, S.; Zimmermann, W.-H.; Eschenhagen, T.; Kaczmarek, E.; Ritter, T.; Volk, H.-D.; Tiegs, G. Heme oxygenase-1 and its reaction product, carbon monoxide, prevent inflammation-related apoptotic liver damage in mice. Hepatology 2003, 38, 909–918. [Google Scholar] [CrossRef] [PubMed]
- Sass, G.; Seyfried, S.; Parreira Soares, M.; Yamashita, K.; Kaczmarek, E.; Neuhuber, W.L.; Tiegs, G. Cooperative effect of biliverdin and carbon monoxide on survival of mice in immune-mediated liver injury. Hepatology 2004, 40, 1128–1135. [Google Scholar] [CrossRef]
- Tertil, M.; Golda, S.; Skrzypek, K.; Florczyk, U.; Weglarczyk, K.; Kotlinowski, J.; Maleszewska, M.; Czauderna, S.; Pichon, C.; Kieda, C.; et al. Nrf2-heme oxygenase-1 axis in mucoepidermoid carcinoma of the lung: Antitumoral effects associated with down-regulation of matrix metalloproteinases. Free Radic. Biol. Med. 2015, 89, 147–157. [Google Scholar] [CrossRef]
- Qi, Y.; Chen, X.; Chan, C.Y.; Li, D.; Yuan, C.; Yu, F.; Lin, M.C.; Yew, D.T.; Kung, H.F.; Lai, L. Two-dimensional differential gel electrophoresis/analysis of diethylnitrosamine induced rat hepatocellular carcinoma. Int. J. Cancer 2008, 122, 2682–2688. [Google Scholar] [CrossRef]
- Tolba, R.; Kraus, T.; Liedtke, C.; Schwarz, M.; Weiskirchen, R. Diethylnitrosamine (DEN)-induced carcinogenic liver injury in mice. Lab. Anim. 2015, 49, 59–69. [Google Scholar] [CrossRef]
- Verna, L.; Whysner, J.; Wiliams, G.M. N-Nitrosodiethylamine Mechanistic Data and Risk Assessment: Bioactivation, DNA-Adduct Formation, Mutagenicity, and Tumor Initiation. Pharmacol. Ther. 1996, 71, 57–81. [Google Scholar] [CrossRef]
- Gerard, C.; Goldbeter, A. The balance between cell cycle arrest and cell proliferation: Control by the extracellular matrix and by contact inhibition. Interface Focus 2014, 4, 20130075. [Google Scholar] [CrossRef]
- Cheng, C.C.; Guan, S.S.; Yang, H.J.; Chang, C.C.; Luo, T.Y.; Chang, J.; Ho, A.S. Blocking heme oxygenase-1 by zinc protoporphyrin reduces tumor hypoxia-mediated VEGF release and inhibits tumor angiogenesis as a potential therapeutic agent against colorectal cancer. J. Biomed. Sci. 2016, 23, 18. [Google Scholar] [CrossRef] [PubMed]
- Neitzel, C.; Demuth, P.; Wittmann, S.; Fahrer, J. Targeting Altered Energy Metabolism in Colorectal Cancer: Oncogenic Reprogramming, the Central Role of the TCA Cycle and Therapeutic Opportunities. Cancers 2020, 12, 1731. [Google Scholar] [CrossRef]
- Peng, J.K.; Shen, S.Q.; Wang, J.; Jiang, H.W.; Wang, Y.Q. Hypoxia-inducible factor 1-alpha promotes colon cell proliferation and migration by upregulating AMPK-related protein kinase 5 under hypoxic conditions. Oncol. Lett. 2018, 15, 3639–3645. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.J.; Chen, W.Y.; Huang, C.Y.; Liu, H.H.; Wei, P.L. Glucose-regulated protein 78 (GRP78) regulates colon cancer metastasis through EMT biomarkers and the NRF-2/HO-1 pathway. Tumour Biol. 2015, 36, 1859–1869. [Google Scholar] [CrossRef] [PubMed]
- Audebert, M.; Assmann, A.S.; Azqueta, A.; Babica, P.; Benfenati, E.; Bortoli, S.; Bouwman, P.; Braeuning, A.; Burgdorf, T.; Coumoul, X.; et al. New approach methodologies to facilitate and improve the hazard assessment of non-genotoxic carcinogens-a PARC project. Front. Toxicol. 2023, 5, 1220998. [Google Scholar] [CrossRef]
- Li, D.; Yun, Y.; Gao, R. Oxygenated Polycyclic aromatic hydrocarbons (Oxy-PAHs) facilitate lung cancer metastasis by epigenetically regulating the epithelial-to-mesenchymal transition (EMT). Environ. Pollut. 2019, 255, 113261. [Google Scholar] [CrossRef] [PubMed]
- Coló, G.P.; Schweitzer, K.; Oresti, G.M.; Alonso, E.G.; Chávez, L.F.; Mascaró, M.; Giorgi, G.; Curino, A.C.; Facchinetti, M.M. Proteomic analysis of the effect of hemin in breast cancer. Sci. Rep. 2022, 13, 10091. [Google Scholar] [CrossRef]
- Park, S.Y.; Jin, M.L.; Kim, Y.H.; Lee, S.J.; Park, G. Sanguinarine inhibits invasiveness and the MMP-9 and COX-2 expression in TPA-induced breast cancer cells by inducing HO-1 expression. Oncol. Rep. 2014, 31, 497–504. [Google Scholar] [CrossRef]
- Gueron, G.; De Siervi, A.; Ferrando, M.; Salierno, M.; De Luca, P.; Elguero, B.; Meiss, R.; Navone, N.; Vazquez, E.S. Critical role of endogenous heme oxygenase 1 as a tuner of the invasive potential of prostate cancer cells. Mol. Cancer Res. 2009, 7, 1745–1755. [Google Scholar] [CrossRef]
- Gueron, G.; Giudice, J.; Valacco, P.; Paez, A.; Elguero, B.; Toscani, M.; Jaworski, F.; Leskow, F.C.; Cotignola, J.; Marti, M.; et al. Heme-oxygenase-1 implications in cell morphology and the adhesive behavior of prostate cancer cells. Oncotarget 2014, 5, 4087. [Google Scholar] [CrossRef]
- Kwok, S.C. Zinc Protoporphyrin Upregulates Heme Oxygenase-1 in PC-3 Cells via the Stress Response Pathway. Int. J. Cell Biol. 2013, 2013, 162094. [Google Scholar] [CrossRef]
- Zhao, Z.; Zhao, J.; Xue, J.; Zhao, X.; Liu, P. Autophagy inhibition promotes epithelial-mesenchymal transition through ROS/HO-1 pathway in ovarian cancer cells. Am. J. Cancer Res. 2016, 6, 2162. [Google Scholar] [PubMed]
- Jacob, A.; Prekeris, R. The regulation of MMP targeting to invadopodia during cancer metastasis. Front. Cell Dev. Biol. 2015, 3, 4. [Google Scholar] [CrossRef] [PubMed]
- Kang, X.; Kong, F.; Wu, X.; Ren, Y.; Wu, S.; Wu, K.; Jiang, Z.; Zhang, W. High glucose promotes tumor invasion and increases metastasis-associated protein expression in human lung epithelial cells by upregulating heme oxygenase-1 via reactive oxygen species or the TGF-beta1/PI3K/Akt signaling pathway. Cell Physiol. Biochem. 2015, 35, 1008–1022. [Google Scholar] [CrossRef] [PubMed]
- Tertil, M.; Skrzypek, K.; Florczyk, U.; Weglarczyk, K.; Was, H.; Collet, G.; Guichard, A.; Gil, T.; Kuzdzal, J.; Jozkowicz, A.; et al. Regulation and novel action of thymidine phosphorylase in non-small cell lung cancer: Crosstalk with Nrf2 and HO-1. PLoS ONE 2014, 9, e97070. [Google Scholar] [CrossRef]
- Lin, P.H.; Lan, W.M.; Chau, L.Y. TRC8 suppresses tumorigenesis through targeting heme oxygenase-1 for ubiquitination and degradation. Oncogene 2013, 32, 2325–2334. [Google Scholar] [CrossRef]
- Mucha, O.; Podkalicka, P.; Czarnek, M.; Biela, A.; Mieczkowski, M.; Kachamakova-Trojanowska, N.; Stepniewski, J.; Jozkowicz, A.; Dulak, J.; Loboda, A. Pharmacological versus genetic inhibition of heme oxygenase-1—The comparison of metalloporphyrins, shRNA and CRISPR/Cas9 system. Acta Biochim. Pol. 2018, 65, 277–286. [Google Scholar] [CrossRef]
- Sunamura, M.; Duda, D.G.; Ghattas, M.H.; Lozonschi, L.; Motoi, F.; Yamauchi, J.-I.; Matsuno, S.; Shibahara, S.; Abraham, N.G. Heme oxygenase-1 accelerates tumor angiogenesis of human pancreatic cancer. Angiogenesis 2003, 6, 15–24. [Google Scholar] [CrossRef]
- Was, H.; Cichon, T.; Smolarczyk, R.; Rudnicka, D.; Stopa, M.; Chevalier, C.; Leger, J.J.; Lackowska, B.; Grochot, A.; Bojkowska, K.; et al. Overexpression of heme oxygenase-1 in murine melanoma: Increased proliferation and viability of tumor cells, decreased survival of mice. Am. J. Pathol. 2006, 169, 2181–2198. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fahrer, J.; Wittmann, S.; Wolf, A.-C.; Kostka, T. Heme Oxygenase-1 and Its Role in Colorectal Cancer. Antioxidants 2023, 12, 1989. https://doi.org/10.3390/antiox12111989
Fahrer J, Wittmann S, Wolf A-C, Kostka T. Heme Oxygenase-1 and Its Role in Colorectal Cancer. Antioxidants. 2023; 12(11):1989. https://doi.org/10.3390/antiox12111989
Chicago/Turabian StyleFahrer, Jörg, Simon Wittmann, Ann-Cathrin Wolf, and Tina Kostka. 2023. "Heme Oxygenase-1 and Its Role in Colorectal Cancer" Antioxidants 12, no. 11: 1989. https://doi.org/10.3390/antiox12111989