

Overexpression of Glutathione S-Transferases in Human Diseases: Drug Targets and Therapeutic Implications
Abstract
:1. Introduction
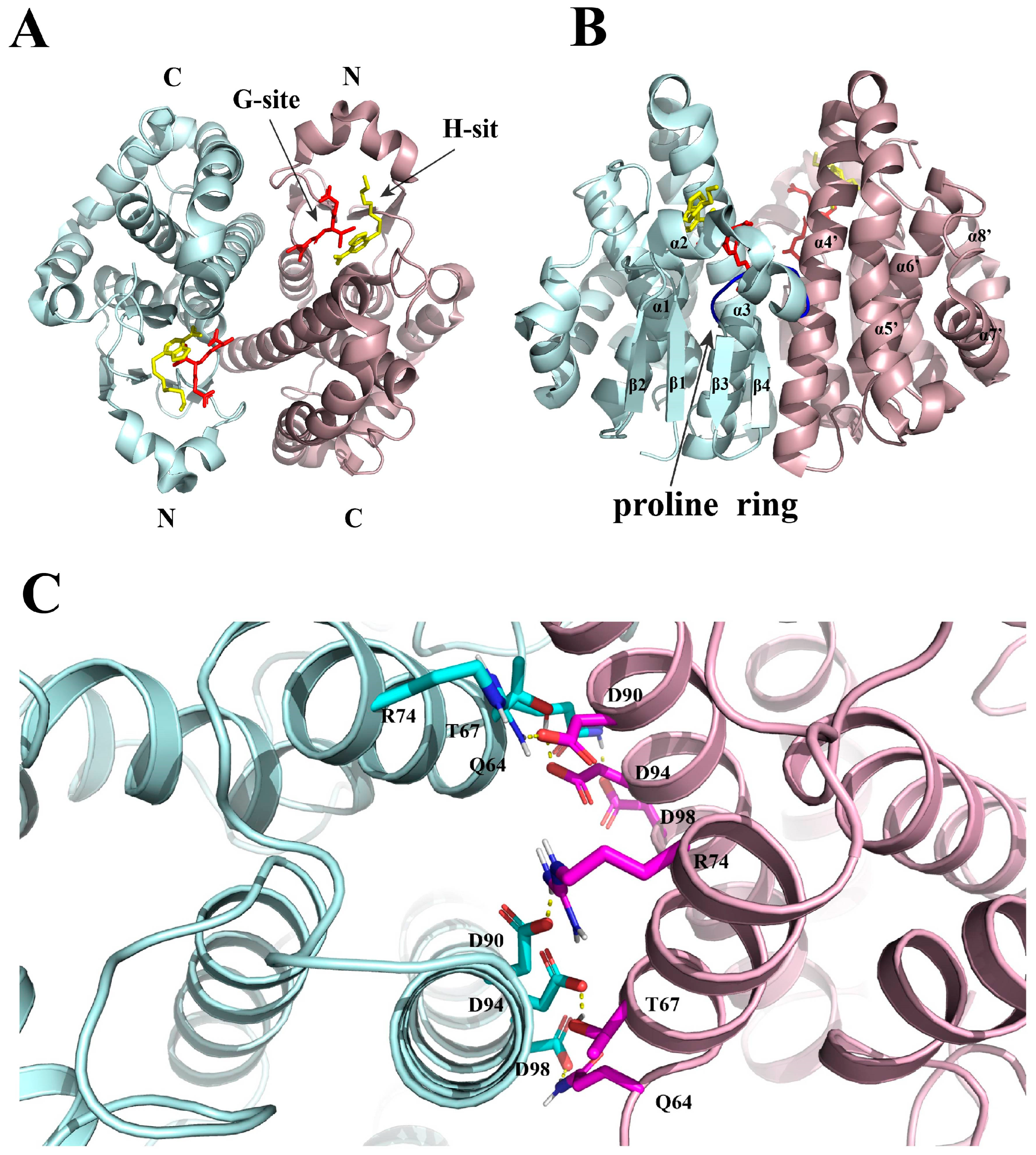
2. Structure
3. Physiological Function
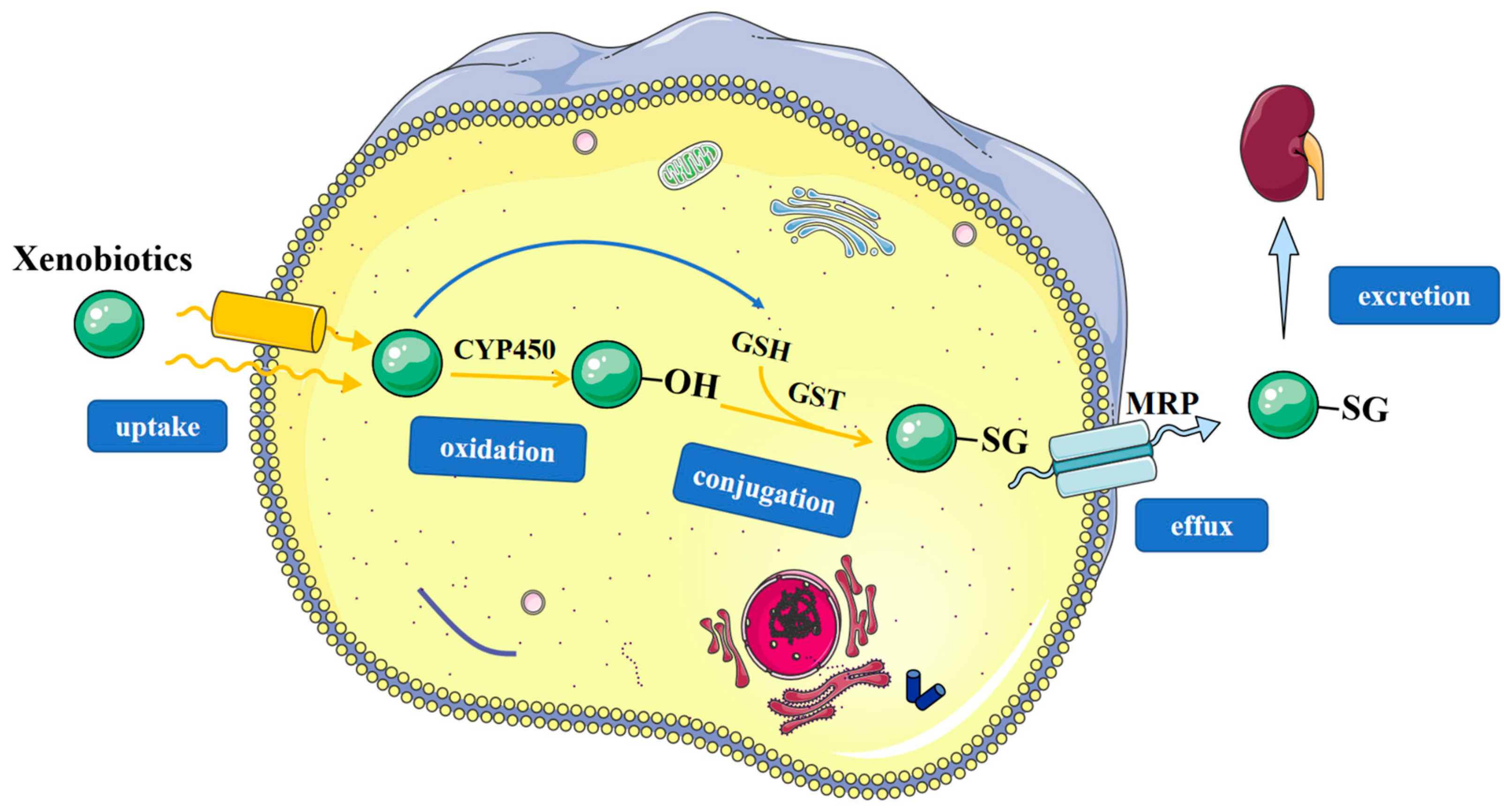
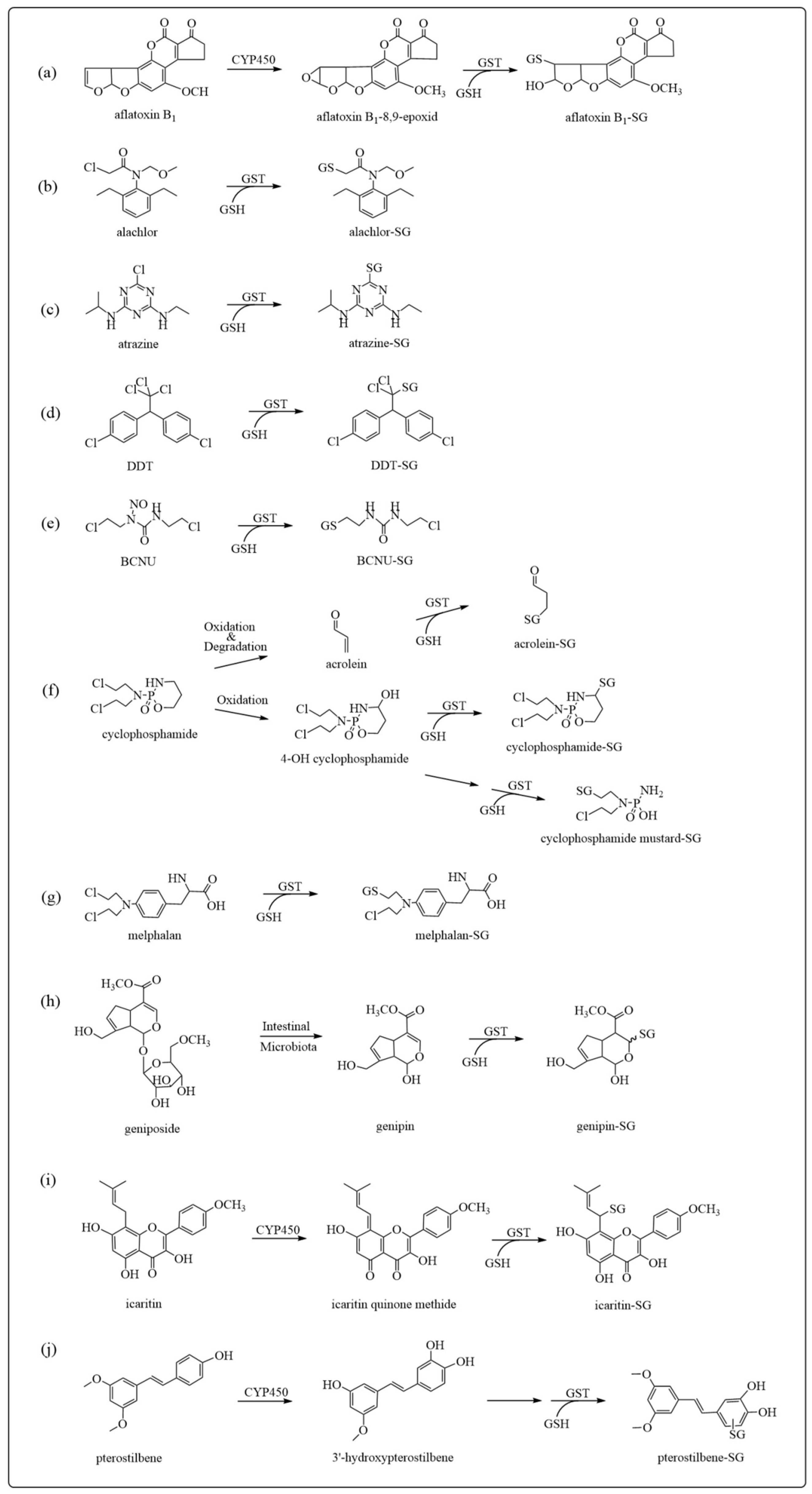
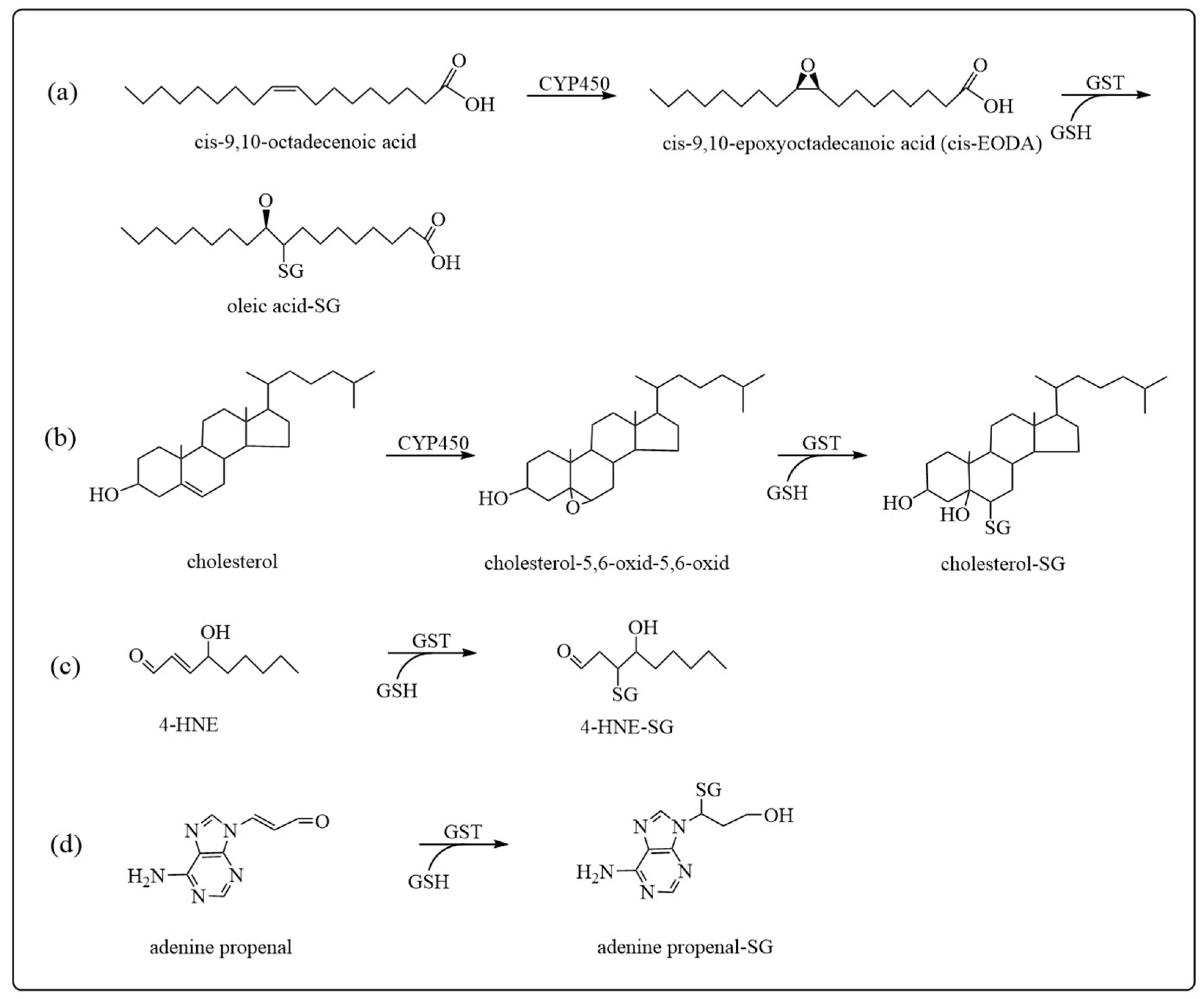
3.1. Detoxification
3.2. Cellular Signaling Regulation
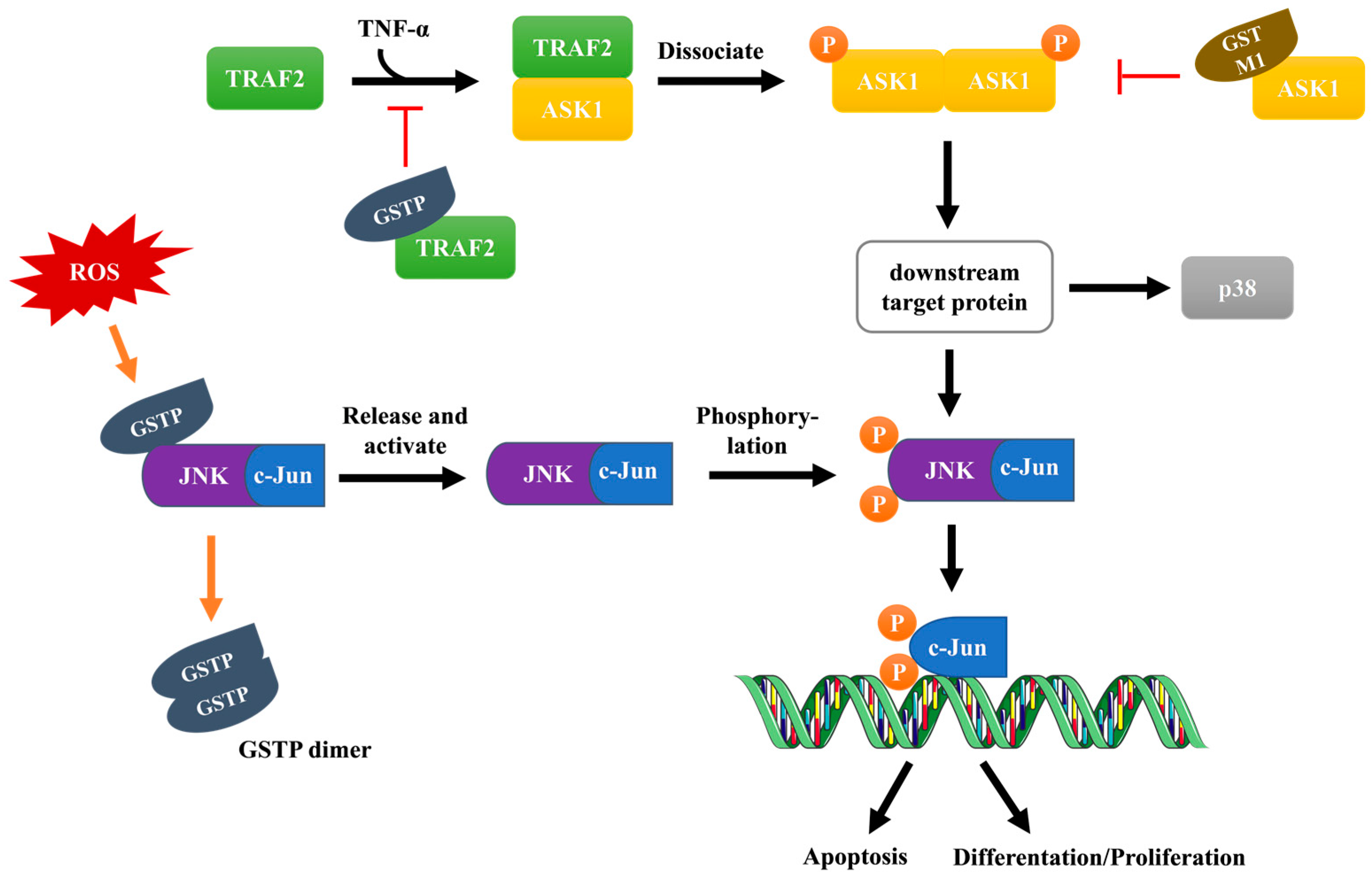
3.2.1. JNK Signaling Pathway
3.2.2. ASK1 Signaling Pathway
3.2.3. Other Signaling Pathways
3.3. Protein S-Glutathionylation
4. Roles of GST Overexpression in Human Diseases
4.1. GSTs and Tumor Multidrug Resistance
4.1.1. Nuclear Localization of GSTP1
4.1.2. Effects of GSTs on Glycolysis
4.1.3. Effects of GSTs on DNA Repair
4.1.4. Effects of GSTs on Autophagy
4.1.5. Effects of GSTs on Ferroptosis
4.2. GSTs and Parkinson’s Disease
4.3. GSTs and Epilepsy
4.4. GSTs and Idiopathic Pulmonary Fibrosis
5. GST Inhibition in Disease Therapeutics
5.1. GSTP Inhibitors
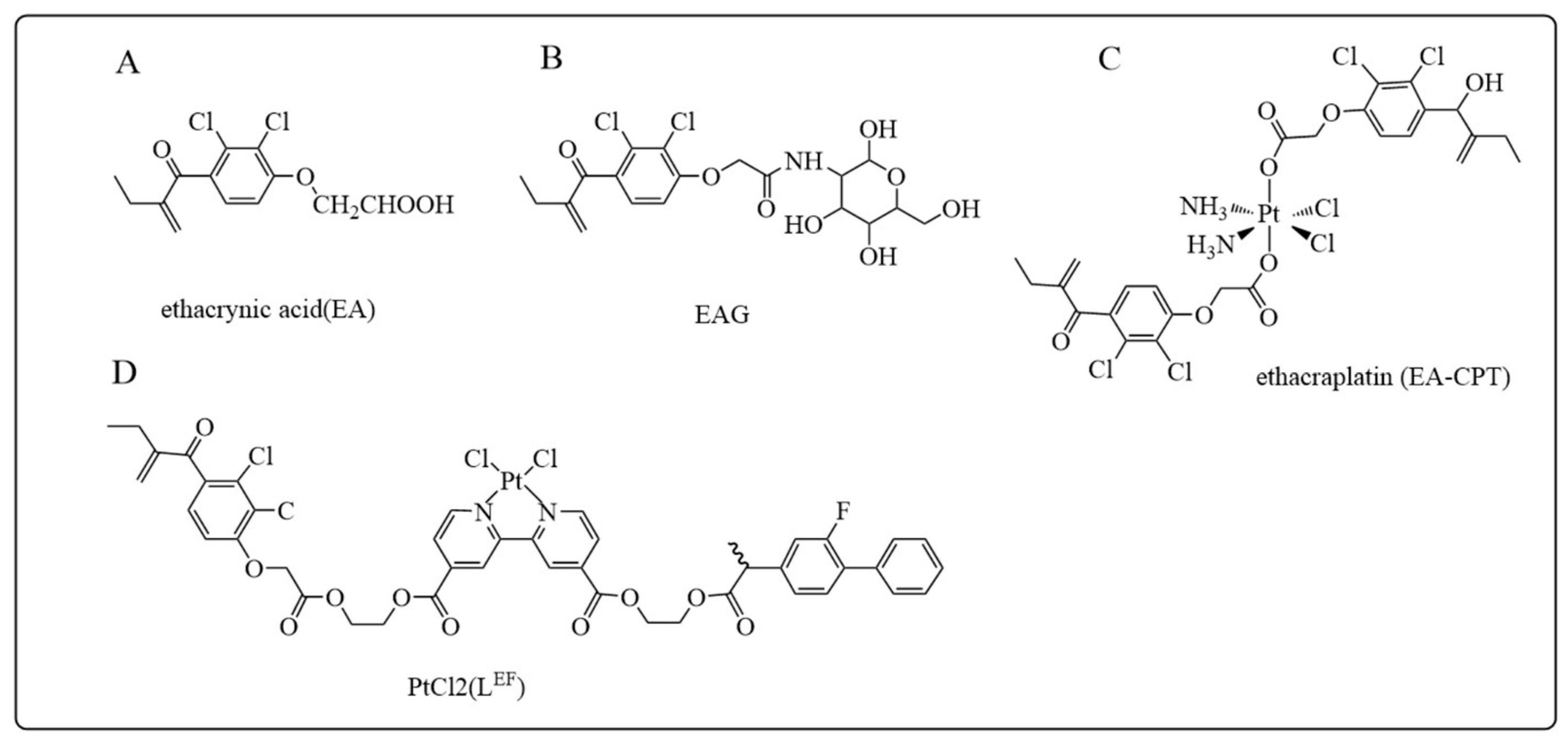
5.1.1. Ethacrynic Acid and Its Derivatives
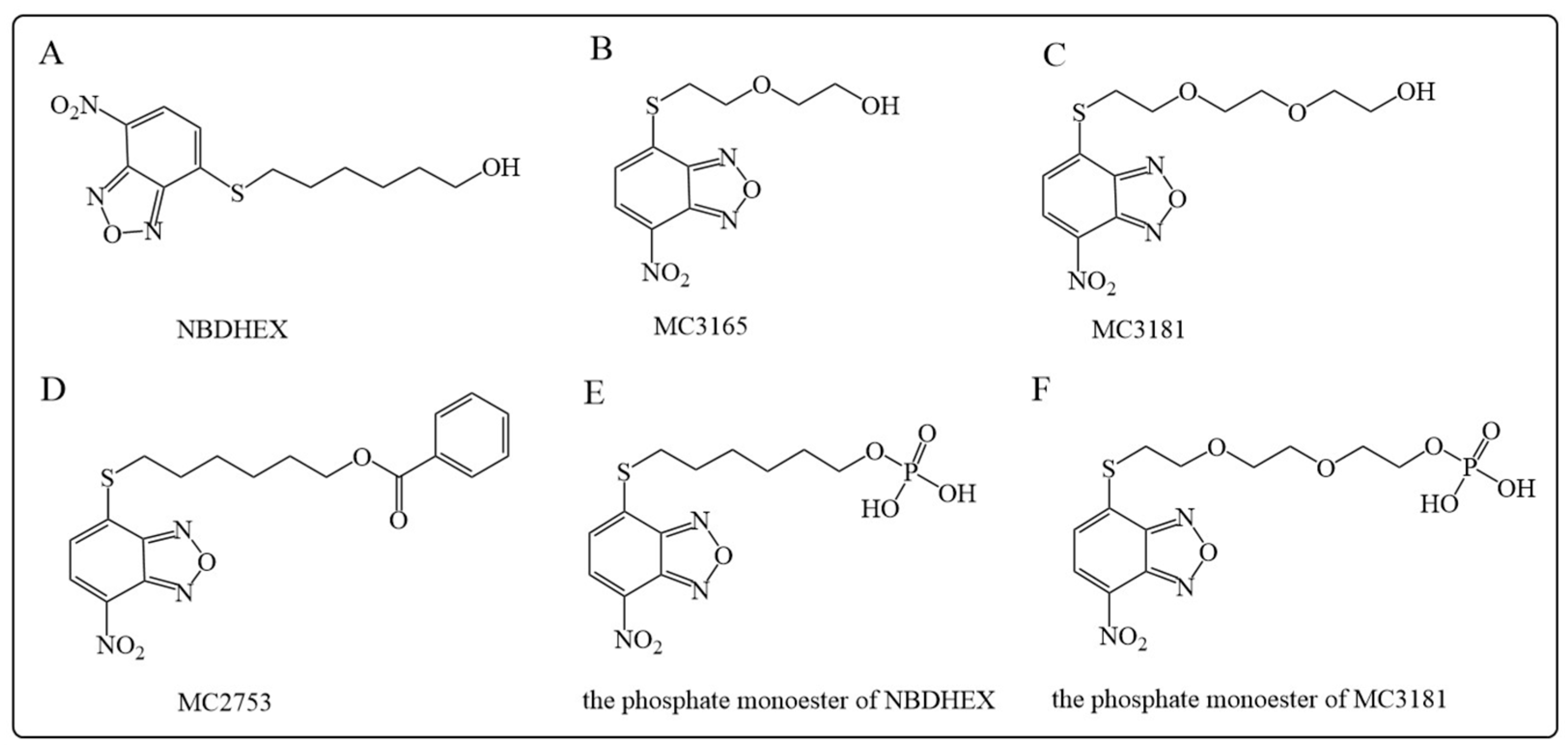
5.1.2. NBDHEX and Its Analogues
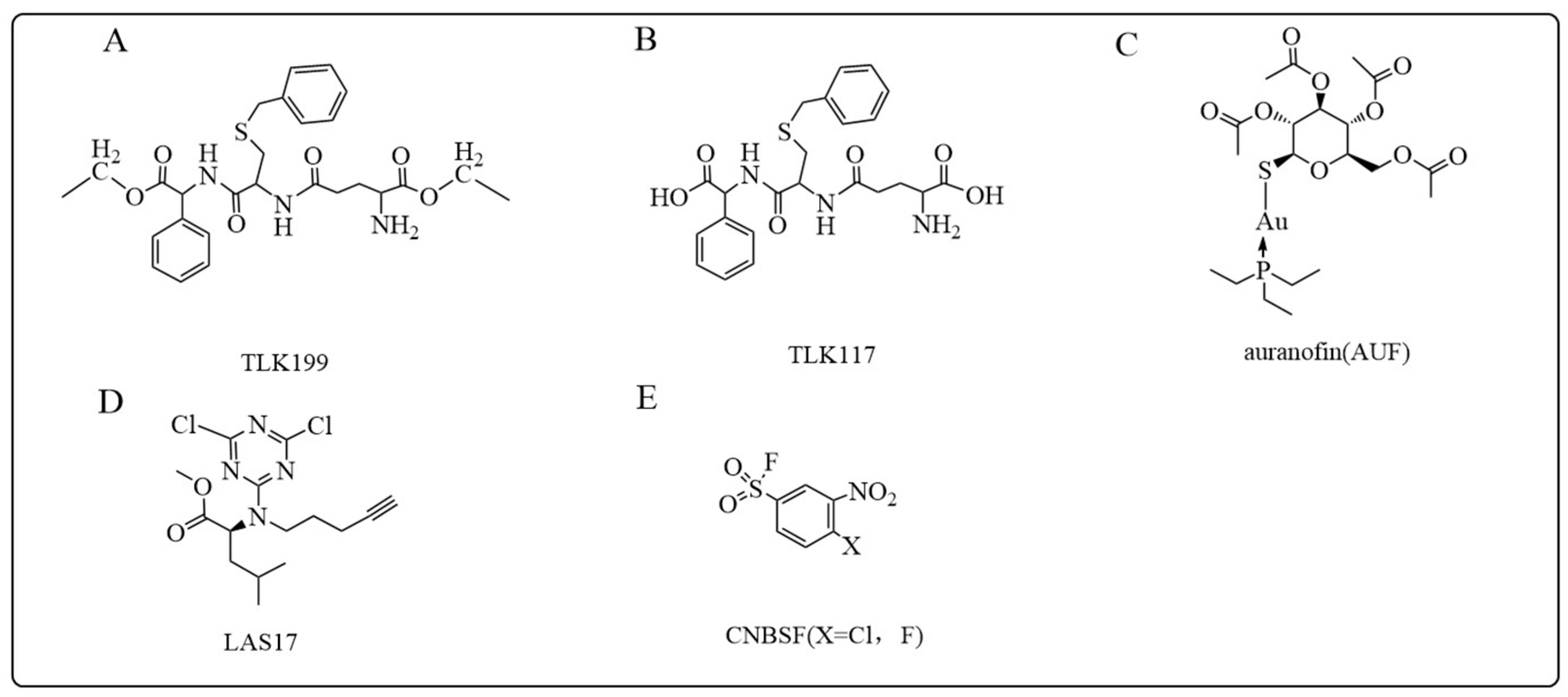
5.1.3. TLK199 and TLK117
5.1.4. Auranofin
5.1.5. Arsenic Compounds
5.1.6. LAS17
5.1.7. CNBSF
5.1.8. Other GSTP Inhibitors
5.2. GSTA Inhibitors
5.3. GSTM Inhibitors
5.4. GSTO Inhibitors
5.4.1. Alpha-Tocopherol (Vitamin E)
5.4.2. α-Chloroacetamide (CA)

6. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Booth, J.; Boyland, E.; Sims, P. An enzyme from rat liver catalysing conjugations with glutathione. Biochem. J. 1961, 79, 516–524. [Google Scholar] [CrossRef]
- Chatterjee, A.; Gupta, S. The multifaceted role of glutathione S-transferases in cancer. Cancer Lett. 2018, 433, 33–42. [Google Scholar] [CrossRef]
- Singh, R.R.; Reindl, K.M. Glutathione S-Transferases in Cancer. Antioxidants 2021, 10, 701. [Google Scholar] [CrossRef] [PubMed]
- Flanagan, J.U.; Smythe, M.L. Sigma-class glutathione transferases. Drug Metab. Rev. 2011, 43, 194–214. [Google Scholar] [CrossRef]
- Eaton, D.L.; Bammler, T.K. Concise review of the glutathione S-transferases and their significance to toxicology. Toxicol. Sci. 1999, 49, 156–164. [Google Scholar] [CrossRef]
- Hayes, J.D.; Flanagan, J.U.; Jowsey, I.R. Glutathione transferases. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 51–88. [Google Scholar] [CrossRef]
- Townsend, D.M.; Tew, K.D. The role of glutathione-S-transferase in anti-cancer drug resistance. Oncogene 2003, 22, 7369–7375. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, S.; Atkins, W.M. The chemistry and biology of inhibitors and pro-drugs targeted to glutathione S-transferases. Cell. Mol. Life Sci. 2005, 62, 1221–1233. [Google Scholar] [CrossRef] [PubMed]
- Bocedi, A.; Noce, A.; Marrone, G.; Noce, G.; Cattani, G.; Gambardella, G.; Di Lauro, M.; Di Daniele, N.; Ricci, G. Glutathione Transferase P1-1 an Enzyme Useful in Biomedicine and as Biomarker in Clinical Practice and in Environmental Pollution. Nutrients 2019, 11, 1741. [Google Scholar] [CrossRef]
- Wu, B.; Dong, D. Human cytosolic glutathione transferases: Structure, function, and drug discovery. Trends Pharmacol. Sci. 2012, 33, 656–668. [Google Scholar] [CrossRef]
- De Luca, A.; Mei, G.; Rosato, N.; Nicolai, E.; Federici, L.; Palumbo, C.; Pastore, A.; Serra, M.; Caccuri, A.M. The fine-tuning of TRAF2-GSTP1-1 interaction: Effect of ligand binding and in situ detection of the complex. Cell Death Dis. 2014, 5, e1015. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhou, T.; Duan, J.; Xiao, Z.; Li, G.; Xu, F. Inhibition of P-glycoprotein and glutathione S-transferase-pi mediated resistance by fluoxetine in MCF-7/ADM cells. Biomed. Pharmacother. 2013, 67, 757–762. [Google Scholar] [CrossRef] [PubMed]
- Townsend, D.M.; Findlay, V.L.; Tew, K.D. Glutathione S-transferases as regulators of kinase pathways and anticancer drug targets. Methods Enzymol. 2005, 401, 287–307. [Google Scholar] [CrossRef]
- Board, P.G.; Menon, D. Glutathione transferases, regulators of cellular metabolism and physiology. Biochim. Biophys. Acta 2013, 1830, 3267–3288. [Google Scholar] [CrossRef] [PubMed]
- Parsons, J.K.; Nelson, C.P.; Gage, W.R.; Nelson, W.G.; Kensler, T.W.; De Marzo, A.M. GSTA1 expression in normal, preneoplastic, and neoplastic human prostate tissue. Prostate 2001, 49, 30–37. [Google Scholar] [CrossRef]
- Mohana, K.; Achary, A. Human cytosolic glutathione-S-transferases: Quantitative analysis of expression, comparative analysis of structures and inhibition strategies of isozymes involved in drug resistance. Drug Metab. Rev. 2017, 49, 318–337. [Google Scholar] [CrossRef]
- Tetlow, N.; Board, P.G. Functional polymorphism of human glutathione transferase A2. Pharmacogenetics 2004, 14, 111–116. [Google Scholar] [CrossRef]
- Singh, S. Cytoprotective and regulatory functions of glutathione S-transferases in cancer cell proliferation and cell death. Cancer Chemother. Pharmacol. 2015, 75, 1–15. [Google Scholar] [CrossRef]
- Gonera, A.; Wawryka, J.; Sobkowicz, A.; Biezunska-Kusiak, K.; Dubinska-Magiera, M.; Krajewski, A.; Choromanska, A. SKOV-3 and Me45 cell response to cisplatin-based chemotherapy: An in vitro study. Folia Biol. 2014, 60, 213–219. [Google Scholar]
- Hayes, P.C.; Bouchier, I.A.; Beckett, G.J. Glutathione S-transferase in humans in health and disease. Gut 1991, 32, 813–818. [Google Scholar] [CrossRef]
- Piaggi, S.; Raggi, C.; Corti, A.; Pitzalis, E.; Mascherpa, M.C.; Saviozzi, M.; Pompella, A.; Casini, A.F. Glutathione transferase omega 1-1 (GSTO1-1) plays an anti-apoptotic role in cell resistance to cisplatin toxicity. Carcinogenesis 2010, 31, 804–811. [Google Scholar] [CrossRef]
- Zhang, J.; Ye, Z.W.; Janssen-Heininger, Y.; Townsend, D.M.; Tew, K.D. Development of Telintra as an Inhibitor of Glutathione S-Transferase P. Handb. Exp. Pharmacol. 2021, 264, 71–91. [Google Scholar] [CrossRef] [PubMed]
- Castro-Caldas, M.; Carvalho, A.N.; Rodrigues, E.; Henderson, C.; Wolf, C.R.; Gama, M.J. Glutathione S-transferase pi mediates MPTP-induced c-Jun N-terminal kinase activation in the nigrostriatal pathway. Mol. Neurobiol. 2012, 45, 466–477. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Dhull, D.K.; Gupta, V.; Channana, P.; Singh, A.; Bhardwaj, M.; Ruhal, P.; Mittal, R. Role of Glutathione-S-transferases in neurological problems. Expert Opin. Ther. Pat. 2017, 27, 299–309. [Google Scholar] [CrossRef]
- Wan Osman, W.H.; Mikami, B.; Saka, N.; Kondo, K.; Nagata, T.; Katahira, M. Structure of a serine-type glutathione S-transferase of Ceriporiopsis subvermispora and identification of the enzymatically important non-canonical residues by functional mutagenesis. Biochem. Biophys. Res. Commun. 2019, 510, 177–183. [Google Scholar] [CrossRef]
- Armstrong, R.N. Glutathione S-transferases: Structure and mechanism of an archetypical detoxication enzyme. Adv. Enzymol. Relat. Areas Mol. Biol. 1994, 69, 1–44. [Google Scholar] [CrossRef]
- Frova, C. Glutathione transferases in the genomics era: New insights and perspectives. Biomol. Eng. 2006, 23, 149–169. [Google Scholar] [CrossRef]
- Allocati, N.; Casalone, E.; Masulli, M.; Ceccarelli, I.; Carletti, E.; Parker, M.W.; Di Ilio, C. Functional analysis of the evolutionarily conserved proline 53 residue inProteus mirabilisglutathione transferase B1-1. FEBS Lett. 1999, 445, 347–350. [Google Scholar] [CrossRef] [PubMed]
- Oakley, A. Glutathione transferases: A structural perspective. Drug Metab. Rev. 2011, 43, 138–151. [Google Scholar] [CrossRef] [PubMed]
- Lawless, M.J.; Pettersson, J.R.; Rule, G.S.; Lanni, F.; Saxena, S. ESR Resolves the C Terminus Structure of the Ligand-free Human Glutathione S-Transferase A1-1. Biophys. J. 2018, 114, 592–601. [Google Scholar] [CrossRef]
- Rushmore, T.H.; Pickett, C.B. Glutathione S-transferases, structure, regulation, and therapeutic implications. J. Biol. Chem. 1993, 268, 11475–11478. [Google Scholar] [CrossRef] [PubMed]
- Lian, L.Y. NMR structural studies of glutathione S-transferase. Cell. Mol. Life Sci. 1998, 54, 359–362. [Google Scholar] [CrossRef] [PubMed]
- Dourado, D.F.; Fernandes, P.A.; Ramos, M.J. Mammalian cytosolic glutathione transferases. Curr. Protein Pept. Sci. 2008, 9, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Dourado, D.F.; Fernandes, P.A.; Mannervik, B.; Ramos, M.J. Glutathione transferase: New model for glutathione activation. Chemistry 2008, 14, 9591–9598. [Google Scholar] [CrossRef]
- Ketterer, B.; Coles, B.; Meyer, D.J. The role of glutathione in detoxication. Environ. Health Perspect. 1983, 49, 59–69. [Google Scholar] [CrossRef]
- Pljesa-Ercegovac, M.; Savic-Radojevic, A.; Matic, M.; Coric, V.; Djukic, T.; Radic, T.; Simic, T. Glutathione Transferases: Potential Targets to Overcome Chemoresistance in Solid Tumors. Int. J. Mol. Sci. 2018, 19, 3785. [Google Scholar] [CrossRef] [PubMed]
- Ketterer, B. Detoxication reactions of glutathione and glutathione transferases. Xenobiotica 1986, 16, 957–973. [Google Scholar] [CrossRef]
- Dohnal, V.; Wu, Q.; Kuca, K. Metabolism of aflatoxins: Key enzymes and interindividual as well as interspecies differences. Arch. Toxicol. 2014, 88, 1635–1644. [Google Scholar] [CrossRef]
- Hayes, J.D.; Pulford, D.J. The glutathione S-transferase supergene family: Regulation of GST and the contribution of the isoenzymes to cancer chemoprotection and drug resistance. Crit. Rev. Biochem. Mol. Biol. 1995, 30, 445–600. [Google Scholar] [CrossRef]
- Josephy, P.D. Genetic variations in human glutathione transferase enzymes: Significance for pharmacology and toxicology. Hum. Genom. Proteom. 2010, 2010, 876940. [Google Scholar] [CrossRef]
- Chen, X.; Han, L.; Zhao, Y.; Huang, H.; Pan, H.; Zhang, C.; Chen, H.; Sun, S.; Yao, S.; Chen, X.; et al. Mechanistic Study of Icaritin-Induced Inactivation of Cytochrome P450 2C9. Drug Metab. Dispos. Biol. Fate Chem. 2023, 51, 771–781. [Google Scholar] [CrossRef]
- Zhao, Y.; Huang, H.; Lv, N.; Huang, C.; Chen, H.; Xing, H.; Guo, C.; Li, N.; Zhao, D.; Chen, X.; et al. Glutathione S-Transferases Mediate In Vitro and In Vivo Inactivation of Genipin: Implications for an Underlying Detoxification Mechanism. J. Agric. Food Chem. 2023, 71, 2399–2410. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Sun, C.; Zhang, Y.; Chen, X.; Huang, H.; Han, L.; Xing, H.; Zhao, D.; Chen, X.; Zhang, Y. Phase I Metabolism of Pterostilbene, a Dietary Resveratrol Derivative: Metabolite Identification, Species Differences, Isozyme Contribution, and Further Bioactivation. J. Agric. Food Chem. 2023, 71, 331–346. [Google Scholar] [CrossRef]
- Tsikas, D. Rat liver glutathione S-transferase-catalyzed conjugation of glutathione to the endogenous epoxides of oleic acid and cholesterol. Anal. Biochem. 2022, 644, 113994. [Google Scholar] [CrossRef] [PubMed]
- Tsikas, D.; Sawa, M.; Brunner, G.; Gutzki, F.; Meyer, H.; Frolich, J. Gas chromatography–mass spectrometry of cis-9,10-epoxyoctadecanoic acid (cis-EODA)I. Direct evidence for cis-EODA formation from oleic acid oxidation by liver microsomes and isolated hepatocytes. J. Chromatogr. B 2003, 784, 351–365. [Google Scholar] [CrossRef] [PubMed]
- Thum, T.; Batkai, S.; Malinski, P.G.; Becker, T.; Mevius, I.; Klempnauer, J.; Meyer, H.H.; Frolich, J.C.; Borlak, J.; Tsikas, D. Measurement and diagnostic use of hepatic cytochrome P450 metabolism of oleic acid in liver disease. Liver Int. 2010, 30, 1181–1188. [Google Scholar] [CrossRef] [PubMed]
- Balogh, L.M.; Atkins, W.M. Interactions of glutathione transferases with 4-hydroxynonenal. Drug Metab. Rev. 2011, 43, 165–178. [Google Scholar] [CrossRef]
- Nair, U.; Bartsch, H.; Nair, J. Lipid peroxidation-induced DNA damage in cancer-prone inflammatory diseases: A review of published adduct types and levels in humans. Free Radic. Biol. Med. 2007, 43, 1109–1120. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Bader Lange, M.L.; Sultana, R. Involvements of the lipid peroxidation product, HNE, in the pathogenesis and progression of Alzheimer’s disease. Biochim. Biophys. Acta 2010, 1801, 924–929. [Google Scholar] [CrossRef]
- Zhou, C.; Huang, Y.; Przedborski, S. Oxidative stress in Parkinson’s disease: A mechanism of pathogenic and therapeutic significance. Ann. N. Y. Acad. Sci. 2008, 1147, 93–104. [Google Scholar] [CrossRef]
- Ketterer, B. Glutathione S-transferases and prevention of cellular free radical damage. Free Radic. Res. 1998, 28, 647–658. [Google Scholar] [CrossRef]
- Grimsrud, P.A.; Picklo, M.J., Sr.; Griffin, T.J.; Bernlohr, D.A. Carbonylation of adipose proteins in obesity and insulin resistance: Identification of adipocyte fatty acid-binding protein as a cellular target of 4-hydroxynonenal. Mol. Cell. Proteom. 2007, 6, 624–637. [Google Scholar] [CrossRef]
- Leitinger, N. Cholesteryl ester oxidation products in atherosclerosis. Mol. Asp. Med. 2003, 24, 239–250. [Google Scholar] [CrossRef]
- Zimniak, P. Detoxification reactions: Relevance to aging. Ageing Res. Rev. 2008, 7, 281–300. [Google Scholar] [CrossRef]
- Laborde, E. Glutathione transferases as mediators of signaling pathways involved in cell proliferation and cell death. Cell Death Differ. 2010, 17, 1373–1380. [Google Scholar] [CrossRef] [PubMed]
- Gate, L.; Majumdar, R.S.; Lunk, A.; Tew, K.D. Increased myeloproliferation in glutathione S-transferase pi-deficient mice is associated with a deregulation of JNK and Janus kinase/STAT pathways. J. Biol. Chem. 2004, 279, 8608–8616. [Google Scholar] [CrossRef]
- Adler, V.; Yin, Z.; Tew, K.D.; Ronai, Z. Role of redox potential and reactive oxygen species in stress signaling. Oncogene 1999, 18, 6104–6111. [Google Scholar] [CrossRef]
- Adler, V.; Yin, Z.; Fuchs, S.Y.; Benezra, M.; Rosario, L.; Tew, K.D.; Pincus, M.R.; Sardana, M.; Henderson, C.J.; Wolf, C.R.; et al. Regulation of JNK signaling by GSTp. EMBO J. 1999, 18, 1321–1334. [Google Scholar] [CrossRef] [PubMed]
- Davis, R.J. Signal Transduction by the JNK Group of MAP Kinases. Cell 2000, 103, 239–252. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Li, B.; Wang, J.; Zhang, D.; Liu, Z.; Zhang, Z.; Zhang, W.; Wang, Y.; Bai, D.; Guan, J.; et al. Identification of Sensitivity Predictors of Neoadjuvant Chemotherapy for the Treatment of Adenocarcinoma of Gastroesophageal Junction. Oncol. Res. 2017, 25, 93–97. [Google Scholar] [CrossRef]
- Qiang, F.; Guangguo, R.; Yongtao, H.; Dandan, D.; Hong, Y. Multidrug resistance in primary tumors and metastases in patients with esophageal squamous cell carcinoma. Pathol. Oncol. Res. 2013, 19, 641–648. [Google Scholar] [CrossRef]
- Yang, L.; Du, C.; Wu, L.; Yu, J.; An, X.; Yu, W.; Cao, S.; Li, H.; Ren, X. Cytokine-Induced Killer Cells Modulates Resistance to Cisplatin in the A549/DDP Cell Line. J. Cancer 2017, 8, 3287–3295. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wei, J.; Tu, X.; Ye, X. Potential Role of GST-pi in Lung Cancer Stem Cell Cisplatin Resistance. BioMed Res. Int. 2021, 2021, 9142364. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Reichlin, A.; Santana, A.; Sokol, K.A.; Nussenzweig, M.C.; Choi, Y. TRAF2 Is Essential for JNK but Not NF-κB Activation and Regulates Lymphocyte Proliferation and Survival. Immunity 1997, 7, 703–713. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Fan, Y.; Xue, B.; Luo, L.; Shen, J.; Zhang, S.; Jiang, Y.; Yin, Z. Human glutathione S-transferase P1-1 interacts with TRAF2 and regulates TRAF2-ASK1 signals. Oncogene 2006, 25, 5787–5800. [Google Scholar] [CrossRef] [PubMed]
- Ichijo, H.; Nishida, E.; Irie, K.; ten Dijke, P.; Saitoh, M.; Moriguchi, T.; Takagi, M.; Matsumoto, K.; Miyazono, K.; Gotoh, Y. Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science 1997, 275, 90–94. [Google Scholar] [CrossRef]
- Cho, S.G.; Lee, Y.H.; Park, H.S.; Ryoo, K.; Kang, K.W.; Park, J.; Eom, S.J.; Kim, M.J.; Chang, T.S.; Choi, S.Y.; et al. Glutathione S-transferase mu modulates the stress-activated signals by suppressing apoptosis signal-regulating kinase 1. J. Biol. Chem. 2001, 276, 12749–12755. [Google Scholar] [CrossRef]
- Checa-Rojas, A.; Delgadillo-Silva, L.F.; Velasco-Herrera, M.D.C.; Andrade-Dominguez, A.; Gil, J.; Santillan, O.; Lozano, L.; Toledo-Leyva, A.; Ramirez-Torres, A.; Talamas-Rohana, P.; et al. GSTM3 and GSTP1: Novel players driving tumor progression in cervical cancer. Oncotarget 2018, 9, 21696–21714. [Google Scholar] [CrossRef]
- Dowling, R.J.; Zakikhani, M.; Fantus, I.G.; Pollak, M.; Sonenberg, N. Metformin inhibits mammalian target of rapamycin-dependent translation initiation in breast cancer cells. Cancer Res. 2007, 67, 10804–10812. [Google Scholar] [CrossRef]
- Louie, S.M.; Grossman, E.A.; Crawford, L.A.; Ding, L.; Camarda, R.; Huffman, T.R.; Miyamoto, D.K.; Goga, A.; Weerapana, E.; Nomura, D.K. GSTP1 Is a Driver of Triple-Negative Breast Cancer Cell Metabolism and Pathogenicity. Cell Chem. Biol. 2016, 23, 567–578. [Google Scholar] [CrossRef]
- Liu, X.; Sui, X.; Zhang, C.; Wei, K.; Bao, Y.; Xiong, J.; Zhou, Z.; Chen, Z.; Wang, C.; Zhu, H.; et al. Glutathione S-transferase A1 suppresses tumor progression and indicates better prognosis of human primary hepatocellular carcinoma. J. Cancer 2020, 11, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Saisawang, C.; Wongsantichon, J.; Robinson, R.C.; Ketterman, A.J. Glutathione transferase Omega 1-1 (GSTO1-1) modulates Akt and MEK1/2 signaling in human neuroblastoma cell SH-SY5Y. Proteins 2019, 87, 588–595. [Google Scholar] [CrossRef]
- Kou, X.; Chen, N.; Feng, Z.; Luo, L.; Yin, Z. GSTP1 negatively regulates Stat3 activation in epidermal growth factor signaling. Oncol. Lett. 2013, 5, 1053–1057. [Google Scholar] [CrossRef] [PubMed]
- Knorre, D.G.; Kudryashova, N.V.; Godovikova, T.S. Chemical and functional aspects of posttranslational modification of proteins. Acta Naturae 2009, 1, 29–51. [Google Scholar] [CrossRef] [PubMed]
- Dominko, K.; Dikic, D. Glutathionylation: A regulatory role of glutathione in physiological processes. Arh. Hig. Rada Toksikol. 2018, 69, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Dalle-Donne, I.; Rossi, R.; Colombo, G.; Giustarini, D.; Milzani, A. Protein S-glutathionylation: A regulatory device from bacteria to humans. Trends Biochem. Sci. 2009, 34, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Musaogullari, A.; Chai, Y.C. Redox Regulation by Protein S-Glutathionylation: From Molecular Mechanisms to Implications in Health and Disease. Int. J. Mol. Sci. 2020, 21, 8113. [Google Scholar] [CrossRef] [PubMed]
- Gallogly, M.M.; Mieyal, J.J. Mechanisms of reversible protein glutathionylation in redox signaling and oxidative stress. Curr. Opin. Pharmacol. 2007, 7, 381–391. [Google Scholar] [CrossRef]
- Ghezzi, P.; Bonetto, V.; Fratelli, M. Thiol-disulfide balance: From the concept of oxidative stress to that of redox regulation. Antioxid. Redox Signal. 2005, 7, 964–972. [Google Scholar] [CrossRef]
- Sciskalska, M.; Milnerowicz, H. The role of GSTpi isoform in the cells signalling and anticancer therapy. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 8537–8550. [Google Scholar] [CrossRef]
- Townsend, D.M.; Manevich, Y.; He, L.; Hutchens, S.; Pazoles, C.J.; Tew, K.D. Novel role for glutathione S-transferase pi. Regulator of protein S-Glutathionylation following oxidative and nitrosative stress. J. Biol. Chem. 2009, 284, 436–445. [Google Scholar] [CrossRef] [PubMed]
- Cha, S.J.; Lee, S.; Choi, H.J.; Han, Y.J.; Jeon, Y.M.; Jo, M.; Lee, S.; Nahm, M.; Lim, S.M.; Kim, S.H.; et al. Therapeutic modulation of GSTO activity rescues FUS-associated neurotoxicity via deglutathionylation in ALS disease models. Dev. Cell 2022, 57, 783–798.e8. [Google Scholar] [CrossRef] [PubMed]
- Board, P.G.; Menon, D. Structure, function and disease relevance of Omega-class glutathione transferases. Arch. Toxicol. 2016, 90, 1049–1067. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.W.; Zhang, J.; Ancrum, T.; Manevich, Y.; Townsend, D.M.; Tew, K.D. Glutathione S-Transferase P-Mediated Protein S-Glutathionylation of Resident Endoplasmic Reticulum Proteins Influences Sensitivity to Drug-Induced Unfolded Protein Response. Antioxid. Redox Signal. 2017, 26, 247–261. [Google Scholar] [CrossRef] [PubMed]
- Ghezzi, P. Protein glutathionylation in health and disease. Biochim. Biophys. Acta 2013, 1830, 3165–3172. [Google Scholar] [CrossRef]
- Manevich, Y.; Fisher, A.B. Peroxiredoxin 6, a 1-Cys peroxiredoxin, functions in antioxidant defense and lung phospholipid metabolism. Free Radic. Biol. Med. 2005, 38, 1422–1432. [Google Scholar] [CrossRef] [PubMed]
- Noguera-Mazon, V.; Lemoine, J.; Walker, O.; Rouhier, N.; Salvador, A.; Jacquot, J.P.; Lancelin, J.M.; Krimm, I. Glutathionylation induces the dissociation of 1-Cys D-peroxiredoxin non-covalent homodimer. J. Biol. Chem. 2006, 281, 31736–31742. [Google Scholar] [CrossRef]
- Yusuf, M.A.; Chuang, T.; Bhat, G.J.; Srivenugopal, K.S. Cys-141 glutathionylation of human p53: Studies using specific polyclonal antibodies in cancer samples and cell lines. Free Radic. Biol. Med. 2010, 49, 908–917. [Google Scholar] [CrossRef]
- Velu, C.S.; Niture, S.K.; Doneanu, C.E.; Pattabiraman, N.; Srivenugopal, K.S. Human p53 is inhibited by glutathionylation of cysteines present in the proximal DNA-binding domain during oxidative stress. Biochemistry 2007, 46, 7765–7780. [Google Scholar] [CrossRef]
- Cha, S.J.; Kim, H.; Choi, H.J.; Lee, S.; Kim, K. Protein Glutathionylation in the Pathogenesis of Neurodegenerative Diseases. Oxid. Med. Cell. Longev. 2017, 2017, 2818565. [Google Scholar] [CrossRef]
- Jeon, D.; Park, H.J.; Kim, H.S. Protein S-glutathionylation induced by hypoxia increases hypoxia-inducible factor-1alpha in human colon cancer cells. Biochem. Biophys. Res. Commun. 2018, 495, 212–216. [Google Scholar] [CrossRef]
- Zhang, J.; Ye, Z.W.; Chen, W.; Culpepper, J.; Jiang, H.; Ball, L.E.; Mehrotra, S.; Blumental-Perry, A.; Tew, K.D.; Townsend, D.M. Altered redox regulation and S-glutathionylation of BiP contribute to bortezomib resistance in multiple myeloma. Free Radic. Biol. Med. 2020, 160, 755–767. [Google Scholar] [CrossRef] [PubMed]
- Belcastro, E.; Gaucher, C.; Corti, A.; Leroy, P.; Lartaud, I.; Pompella, A. Regulation of protein function by S-nitrosation and S-glutathionylation: Processes and targets in cardiovascular pathophysiology. Biol. Chem. 2017, 398, 1267–1293. [Google Scholar] [CrossRef] [PubMed]
- Halloran, M.; Parakh, S.; Atkin, J.D. The role of s-nitrosylation and s-glutathionylation of protein disulphide isomerase in protein misfolding and neurodegeneration. Int. J. Cell Biol. 2013, 2013, 797914. [Google Scholar] [CrossRef] [PubMed]
- Sabens Liedhegner, E.A.; Gao, X.H.; Mieyal, J.J. Mechanisms of altered redox regulation in neurodegenerative diseases--focus on S--glutathionylation. Antioxid. Redox Signal. 2012, 16, 543–566. [Google Scholar] [CrossRef] [PubMed]
- Leonard, G.D.; Fojo, T.; Bates, S.E. The role of ABC transporters in clinical practice. Oncologist 2003, 8, 411–424. [Google Scholar] [CrossRef] [PubMed]
- Depeille, P.; Cuq, P.; Mary, S.; Passagne, I.; Evrard, A.; Cupissol, D.; Vian, L. Glutathione S-transferase M1 and multidrug resistance protein 1 act in synergy to protect melanoma cells from vincristine effects. Mol. Pharmacol. 2004, 65, 897–905. [Google Scholar] [CrossRef]
- Tew, K.D.; Monks, A.; Barone, L.; Rosser, D.; Akerman, G.; Montali, J.A.; Wheatley, J.B.; Schmidt, D.E., Jr. Glutathione-associated enzymes in the human cell lines of the National Cancer Institute Drug Screening Program. Mol. Pharmacol. 1996, 50, 149–159. [Google Scholar]
- Mousseau, M.; Chauvin, C.; Nissou, M.F.; Chaffanet, M.; Plantaz, D.; Pasquier, B.; Schaerer, R.; Benabid, A. A study of the expression of four chemoresistance-related genes in human primary and metastatic brain tumours. Eur. J. Cancer 1993, 29, 753–759. [Google Scholar] [CrossRef]
- Fruehauf, J.P.; Brem, H.; Brem, S.; Sloan, A.; Barger, G.; Huang, W.; Parker, R. In vitro drug response and molecular markers associated with drug resistance in malignant gliomas. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2006, 12, 4523–4532. [Google Scholar] [CrossRef]
- Geng, M.; Wang, L.; Chen, X.; Cao, R.; Li, P. The association between chemosensitivity and Pgp, GST-π and Topo II expression in gastric cancer. Diagn. Pathol. 2013, 8, 198. [Google Scholar] [CrossRef]
- Yu, D.S.; Hsieh, D.S.; Chang, S.Y. Increasing expression of GST-pi MIF, and ID1 genes in chemoresistant prostate cancer cells. Arch. Androl. 2006, 52, 275–281. [Google Scholar] [CrossRef]
- Wang, Z.; Liang, S.; Lian, X.; Liu, L.; Zhao, S.; Xuan, Q.; Guo, L.; Liu, H.; Yang, Y.; Dong, T.; et al. Identification of proteins responsible for adriamycin resistance in breast cancer cells using proteomics analysis. Sci. Rep. 2015, 5, 9301. [Google Scholar] [CrossRef] [PubMed]
- Smitherman, P.K.; Townsend, A.J.; Kute, T.E.; Morrow, C.S. Role of multidrug resistance protein 2 (MRP2, ABCC2) in alkylating agent detoxification: MRP2 potentiates glutathione S-transferase A1-1-mediated resistance to chlorambucil cytotoxicity. J. Pharmacol. Exp. Ther. 2004, 308, 260–267. [Google Scholar] [CrossRef]
- Manupati, K.; Debnath, S.; Goswami, K.; Bhoj, P.S.; Chandak, H.S.; Bahekar, S.P.; Das, A. Glutathione S-transferase omega 1 inhibition activates JNK-mediated apoptotic response in breast cancer stem cells. FEBS J. 2019, 286, 2167–2192. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Liu, F.; Wang, C.; Wang, C.; Tang, Y.; Jiang, Z. Glutathione S-transferase A1 mediates nicotine-induced lung cancer cell metastasis by promoting epithelial-mesenchymal transition. Exp. Ther. Med. 2017, 14, 1783–1788. [Google Scholar] [CrossRef]
- Ezgu, M.C.; Kural, C.; Simsek, G.G.; Kaygin, P.; Oguztuzun, S.; Kirik, A.; Yasar, S.; Kose, G.; Sarialtin, S.Y.; Coban, T.; et al. Chemoresistance in Malignant Intracranial Tumors: Longer Survival with Negative MDR1 Expression. Turk. Neurosurg. 2021, 31, 447–459. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.J.; Wang, D.D.; Li, J.; Xu, H.Z.; Shen, H.Y.; Chen, X.; Zhou, S.Y.; Zhong, S.L.; Zhao, J.H.; Tang, J.H. Predictive role of GSTP1-containing exosomes in chemotherapy-resistant breast cancer. Gene 2017, 623, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Rolland, D.; Raharijaona, M.; Barbarat, A.; Houlgatte, R.; Thieblemont, C. Inhibition of GST-pi nuclear transfer increases mantle cell lymphoma sensitivity to cisplatin, cytarabine, gemcitabine, bortezomib and doxorubicin. Anticancer. Res. 2010, 30, 3951–3957. [Google Scholar]
- Tang, Y.; Xuan, X.Y.; Li, M.; Dong, Z.M. Roles of GST-π and polβ genes in chemoresistance of esophageal carcinoma cells. Asian Pac. J. Cancer Prev. APJCP 2013, 14, 7375–7379. [Google Scholar] [CrossRef]
- Wang, X.; Li, Y.; Chen, W.; Wang, Y.; Hui, L.; Liu, J.; Li, N.; Zhang, L.; Zou, Y.; Wang, F. Nrf-2/Gst-α mediated imatinib resistance through rapid 4-HNE clearance. Exp. Cell Res. 2017, 353, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Zou, M.; Hu, X.; Xu, B.; Tong, T.; Jing, Y.; Xi, L.; Zhou, W.; Lu, J.; Wang, X.; Yang, X.; et al. Glutathione Stransferase isozyme alpha 1 is predominantly involved in the cisplatin resistance of common types of solid cancer. Oncol. Rep. 2019, 41, 989–998. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.T.; Song, K.; Zhou, J.; Shi, Y.H.; Liu, W.R.; Tian, M.X.; Jin, L.; Shi, G.M.; Gao, Q.; Ding, Z.B.; et al. Autophagy activation contributes to glutathione transferase Mu 1-mediated chemoresistance in hepatocellular carcinoma. Oncol. Lett. 2018, 16, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Attaoua, C.; Vincent, L.A.; Abdel Jaoued, A.; Hadj-Kaddour, K.; Bai, Q.; De Vos, J.; Vian, L.; Cuq, P. Differential involvement of glutathione S-transferase mu 1 and multidrug resistance protein 1 in melanoma acquired resistance to vinca alkaloids. Fundam. Clin. Pharmacol. 2015, 29, 62–71. [Google Scholar] [CrossRef]
- Cheng, S.Y.; Chen, N.F.; Wen, Z.H.; Yao, Z.K.; Tsui, K.H.; Kuo, H.M.; Chen, W.F. Glutathione S-Transferase M3 Is Associated with Glycolysis in Intrinsic Temozolomide-Resistant Glioblastoma Multiforme Cells. Int. J. Mol. Sci. 2021, 22, 7080. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Xie, S.; Zhou, L.; Tang, X.; Guan, X.; Deng, M.; Zheng, H.; Wang, Y.; Lu, R.; Guo, L. Up-regulation of GSTT1 in serous ovarian cancer associated with resistance to TAXOL/carboplatin. J. Ovarian Res. 2021, 14, 122. [Google Scholar] [CrossRef]
- Diedrich, A.; Bock, H.C.; Konig, F.; Schulz, T.G.; Ludwig, H.C.; Herken, R.; Quondamatteo, F. Expression of glutathione S-transferase T1 (GSTT1) in human brain tumours. Histol. Histopathol. 2006, 21, 1199–1207. [Google Scholar] [CrossRef]
- Kobayashi, Y. A study on diagnosis of oral squamous cell carcinoma (oral SCC) by glutathione S-transferase-pi (GST-pi). Kokubyo Gakkai Zasshi J. Stomatol. Soc. Jpn. 1999, 66, 46–56. [Google Scholar] [CrossRef]
- Goto, S.; Ihara, Y.; Urata, Y.; Izumi, S.; Abe, K.; Koji, T.; Kondo, T. Doxorubicin-induced DNA intercalation and scavenging by nuclear glutathione S-transferase pi. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2001, 15, 2702–2714. [Google Scholar] [CrossRef]
- Ali-Osman, F.; Brunner, J.M.; Kutluk, T.M.; Hess, K. Prognostic significance of glutathione S-transferase pi expression and subcellular localization in human gliomas. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 1997, 3, 2253–2261. [Google Scholar]
- Shiratori, Y.; Soma, Y.; Maruyama, H.; Sato, S.; Takano, A.; Sato, K. Immunohistochemical detection of the placental form of glutathione S-transferase in dysplastic and neoplastic human uterine cervix lesions. Cancer Res. 1987, 47, 6806–6809. [Google Scholar]
- Chen, Z.; Hao, W.; Tang, J.; Gao, W.-Q.; Xu, H. CSTF2 Promotes Hepatocarcinogenesis and Hepatocellular Carcinoma Progression via Aerobic Glycolysis. Front. Oncol. 2022, 12, 897804. [Google Scholar] [CrossRef]
- Vaupel, P.; Schmidberger, H.; Mayer, A. The Warburg effect: Essential part of metabolic reprogramming and central contributor to cancer progression. Int. J. Radiat. Biol. 2019, 95, 912–919. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Xiong, Y.; Qiao, T.; Li, X.; Jia, L.; Han, Y. Lactate dehydrogenase A: A key player in carcinogenesis and potential target in cancer therapy. Cancer Med. 2018, 7, 6124–6136. [Google Scholar] [CrossRef] [PubMed]
- Ganapathy-Kanniappan, S.; Geschwind, J.F. Tumor glycolysis as a target for cancer therapy: Progress and prospects. Mol. Cancer 2013, 12, 152. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Yang, J.; Ding, C.; Li, J.; You, L.; Dai, M.; Zhao, Y. Glutathione S-Transferase Mu-3 Predicts a Better Prognosis and Inhibits Malignant Behavior and Glycolysis in Pancreatic Cancer. Front. Oncol. 2020, 10, 1539. [Google Scholar] [CrossRef]
- Wang, Z.; Jia, R.; Wang, L.; Yang, Q.; Hu, X.; Fu, Q.; Zhang, X.; Li, W.; Ren, Y. The Emerging Roles of Rad51 in Cancer and Its Potential as a Therapeutic Target. Front. Oncol. 2022, 12, 935593. [Google Scholar] [CrossRef]
- Lock, R.B.; Ross, W.E. DNA topoisomerases in cancer therapy. Anti-Cancer Drug Des. 1987, 2, 151–164. [Google Scholar]
- Kundu, M.; Thompson, C.B. Autophagy: Basic principles and relevance to disease. Annu. Rev. Pathol. 2008, 3, 427–455. [Google Scholar] [CrossRef]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef]
- Yang, S.; Wang, X.; Contino, G.; Liesa, M.; Sahin, E.; Ying, H.; Bause, A.; Li, Y.; Stommel, J.M.; Dell’antonio, G.; et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 2011, 25, 717–729. [Google Scholar] [CrossRef] [PubMed]
- Martin, A.P.; Mitchell, C.; Rahmani, M.; Nephew, K.P.; Grant, S.; Dent, P. Inhibition of MCL-1 enhances lapatinib toxicity and overcomes lapatinib resistance via BAK-dependent autophagy. Cancer Biol. Ther. 2009, 8, 2084–2096. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J. Ferroptosis: Bug or feature? Immunol. Rev. 2017, 277, 150–157. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.W.; Amante, J.J.; Goel, H.L.; Mercurio, A.M. The α6β4 integrin promotes resistance to ferroptosis. J. Cell Biol. 2017, 216, 4287–4297. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, X.; Jin, S.; Chen, Y.; Guo, R. Ferroptosis in cancer therapy: A novel approach to reversing drug resistance. Mol. Cancer 2022, 21, 47. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Lim, S.O.; Yan, M.; Hsu, J.L.; Yao, J.; Wei, Y.; Chang, S.S.; Yamaguchi, H.; Lee, H.H.; Ke, B.; et al. TYRO3 induces anti-PD-1/PD-L1 therapy resistance by limiting innate immunity and tumoral ferroptosis. J. Clin. Investig. 2021, 131, e139434. [Google Scholar] [CrossRef]
- Wang, Q.; Bin, C.; Xue, Q.; Gao, Q.; Huang, A.; Wang, K.; Tang, N. GSTZ1 sensitizes hepatocellular carcinoma cells to sorafenib-induced ferroptosis via inhibition of NRF2/GPX4 axis. Cell Death Dis. 2021, 12, 426. [Google Scholar] [CrossRef]
- Kuang, F.; Liu, J.; Xie, Y.; Tang, D.; Kang, R. MGST1 is a redox-sensitive repressor of ferroptosis in pancreatic cancer cells. Cell Chem. Biol. 2021, 28, 765–775.e765. [Google Scholar] [CrossRef]
- Pearce, R.K.; Owen, A.; Daniel, S.; Jenner, P.; Marsden, C.D. Alterations in the distribution of glutathione in the substantia nigra in Parkinson’s disease. J. Neural Transm. 1997, 104, 661–677. [Google Scholar] [CrossRef]
- Sian, J.; Dexter, D.T.; Lees, A.J.; Daniel, S.; Agid, Y.; Javoy-Agid, F.; Jenner, P.; Marsden, C.D. Alterations in glutathione levels in Parkinson’s disease and other neurodegenerative disorders affecting basal ganglia. Ann. Neurol. 1994, 36, 348–355. [Google Scholar] [CrossRef]
- Shi, M.; Bradner, J.; Bammler, T.K.; Eaton, D.L.; Zhang, J.; Ye, Z.; Wilson, A.M.; Montine, T.J.; Pan, C.; Zhang, J. Identification of glutathione S-transferase pi as a protein involved in Parkinson disease progression. Am. J. Pathol. 2009, 175, 54–65. [Google Scholar] [CrossRef]
- Whitworth, A.J.; Theodore, D.A.; Greene, J.C.; Benes, H.; Wes, P.D.; Pallanck, L.J. Increased glutathione S-transferase activity rescues dopaminergic neuron loss in a Drosophila model of Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2005, 102, 8024–8029. [Google Scholar] [CrossRef]
- Laliberte, R.E.; Perregaux, D.G.; Hoth, L.R.; Rosner, P.J.; Jordan, C.K.; Peese, K.M.; Eggler, J.F.; Dombroski, M.A.; Geoghegan, K.F.; Gabel, C.A. Glutathione s-transferase omega 1-1 is a target of cytokine release inhibitory drugs and may be responsible for their effect on interleukin-1beta posttranslational processing. J. Biol. Chem. 2003, 278, 16567–16578. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.J.; Oliveira, S.A.; Xu, P.; Martin, E.R.; Stenger, J.E.; Scherzer, C.R.; Hauser, M.A.; Scott, W.K.; Small, G.W.; Nance, M.A.; et al. Glutathione S-transferase omega-1 modifies age-at-onset of Alzheimer disease and Parkinson disease. Hum. Mol. Genet. 2003, 12, 3259–3267. [Google Scholar] [CrossRef]
- Baez, S.; Segura-Aguilar, J.; Widersten, M.; Johansson, A.S.; Mannervik, B. Glutathione transferases catalyse the detoxication of oxidized metabolites (o-quinones) of catecholamines and may serve as an antioxidant system preventing degenerative cellular processes. Biochem. J. 1997, 324 Pt 1, 25–28. [Google Scholar] [CrossRef] [PubMed]
- Ercegovac, M.; Jovic, N.; Sokic, D.; Savic-Radojevic, A.; Coric, V.; Radic, T.; Nikolic, D.; Kecmanovic, M.; Matic, M.; Simic, T.; et al. GSTA1, GSTM1, GSTP1 and GSTT1 polymorphisms in progressive myoclonus epilepsy: A Serbian case-control study. Seizure 2015, 32, 30–36. [Google Scholar] [CrossRef]
- Shang, W.; Liu, W.H.; Zhao, X.H.; Sun, Q.J.; Bi, J.Z.; Chi, Z.F. Expressions of glutathione S-transferase alpha, mu, and pi in brains of medically intractable epileptic patients. BMC Neurosci. 2008, 9, 67. [Google Scholar] [CrossRef]
- He, N.; Bai, S.; Huang, Y.; Xing, Y.; Chen, L.; Yu, F.; Lv, C. Evaluation of Glutathione S-Transferase Inhibition Effects on Idiopathic Pulmonary Fibrosis Therapy with a Near-Infrared Fluorescent Probe in Cell and Mice Models. Anal. Chem. 2019, 91, 5424–5432. [Google Scholar] [CrossRef] [PubMed]
- McMillan, D.H.; van der Velden, J.L.; Lahue, K.G.; Qian, X.; Schneider, R.W.; Iberg, M.S.; Nolin, J.D.; Abdalla, S.; Casey, D.T.; Tew, K.D.; et al. Attenuation of lung fibrosis in mice with a clinically relevant inhibitor of glutathione-S-transferase π. JCI Insight 2016, 1, e85717. [Google Scholar] [CrossRef]
- Robin, S.K.D.; Ansari, M.; Uppugunduri, C.R.S. Spectrophotometric Screening for Potential Inhibitors of Cytosolic Glutathione S-Transferases. J. Vis. Exp. JoVE 2020, 164, e61347. [Google Scholar] [CrossRef]
- Melvin, K.E.; Farrelly, R.O.; North, J.D. Ethacrynic acid: A new oral diuretic. Br. Med. J. 1963, 1, 1521–1524. [Google Scholar] [CrossRef]
- Dollery, C.T.; Parry, E.H.; Young, D.S. Diuretic and Hypotensive Properties of Ethacrynic Acid: A Comparison with Hydrochlorothiazide. Lancet 1964, 1, 947–952. [Google Scholar] [CrossRef] [PubMed]
- Sau, A.; Pellizzari Tregno, F.; Valentino, F.; Federici, G.; Caccuri, A.M. Glutathione transferases and development of new principles to overcome drug resistance. Arch. Biochem. Biophys. 2010, 500, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Awasthi, S.; Srivastava, S.K.; Ahmad, F.; Ahmad, H.; Ansari, G.A. Interactions of glutathione S-transferase-pi with ethacrynic acid and its glutathione conjugate. Biochim. Biophys. Acta 1993, 1164, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Punganuru, S.R.; Mostofa, A.G.M.; Madala, H.R.; Basak, D.; Srivenugopal, K.S. Potent anti-proliferative actions of a non-diuretic glucosamine derivative of ethacrynic acid. Bioorganic Med. Chem. Lett. 2016, 26, 2829–2833. [Google Scholar] [CrossRef] [PubMed]
- Parker, L.J.; Italiano, L.C.; Morton, C.J.; Hancock, N.C.; Ascher, D.B.; Aitken, J.B.; Harris, H.H.; Campomanes, P.; Rothlisberger, U.; De Luca, A.; et al. Studies of glutathione transferase P1-1 bound to a platinum(IV)-based anticancer compound reveal the molecular basis of its activation. Chemistry 2011, 17, 7806–7816. [Google Scholar] [CrossRef] [PubMed]
- Ang, W.H.; Khalaila, I.; Allardyce, C.S.; Juillerat-Jeanneret, L.; Dyson, P.J. Rational design of platinum(IV) compounds to overcome glutathione-S-transferase mediated drug resistance. J. Am. Chem. Soc. 2005, 127, 1382–1383. [Google Scholar] [CrossRef]
- Biancalana, L.; Batchelor, L.K.; Pereira, S.A.P.; Tseng, P.J.; Zacchini, S.; Pampaloni, G.; Saraiva, L.; Dyson, P.J.; Marchetti, F. Bis-conjugation of Bioactive Molecules to Cisplatin-like Complexes through (2,2′-Bipyridine)-4,4′-Dicarboxylic Acid with Optimal Cytotoxicity Profile Provided by the Combination Ethacrynic Acid/Flurbiprofen. Chemistry 2020, 26, 17525–17535. [Google Scholar] [CrossRef]
- Sha, H.H.; Wang, Z.; Dong, S.C.; Hu, T.M.; Liu, S.W.; Zhang, J.Y.; Wu, Y.; Ma, R.; Wu, J.Z.; Chen, D.; et al. 6-(7-nitro-2,1,3-benzoxadiazol-4-ylthio) hexanol: A promising new anticancer compound. Biosci. Rep. 2018, 38, BSR20171440. [Google Scholar] [CrossRef]
- Turella, P.; Cerella, C.; Filomeni, G.; Bullo, A.; De Maria, F.; Ghibelli, L.; Ciriolo, M.R.; Cianfriglia, M.; Mattei, M.; Federici, G.; et al. Proapoptotic activity of new glutathione S-transferase inhibitors. Cancer Res. 2005, 65, 3751–3761. [Google Scholar] [CrossRef]
- Zhang, J.; Grek, C.; Ye, Z.W.; Manevich, Y.; Tew, K.D.; Townsend, D.M. Pleiotropic functions of glutathione S-transferase P. Adv. Cancer Res. 2014, 122, 143–175. [Google Scholar] [CrossRef]
- Pasello, M.; Michelacci, F.; Scionti, I.; Hattinger, C.M.; Zuntini, M.; Caccuri, A.M.; Scotlandi, K.; Picci, P.; Serra, M. Overcoming glutathione S-transferase P1-related cisplatin resistance in osteosarcoma. Cancer Res. 2008, 68, 6661–6668. [Google Scholar] [CrossRef] [PubMed]
- Sciarretta, F.; Fulci, C.; Palumbo, C.; Rotili, D.; Tentori, L.; Graziani, G.; Caccuri, A.M. Effects of Glutathione Transferase-Targeting Nitrobenzoxadiazole Compounds in Relation to PD-L1 Status in Human Melanoma Cells. Chemotherapy 2019, 64, 138–145. [Google Scholar] [CrossRef]
- Turella, P.; Filomeni, G.; Dupuis, M.L.; Ciriolo, M.R.; Molinari, A.; De Maria, F.; Tombesi, M.; Cianfriglia, M.; Federici, G.; Ricci, G.; et al. A strong glutathione S-transferase inhibitor overcomes the P-glycoprotein-mediated resistance in tumor cells. 6-(7-Nitro-2,1,3-benzoxadiazol-4-ylthio)hexanol (NBDHEX) triggers a caspase-dependent apoptosis in MDR1-expressing leukemia cells. J. Biol. Chem. 2006, 281, 23725–23732. [Google Scholar] [CrossRef] [PubMed]
- Ricci, G.; De Maria, F.; Antonini, G.; Turella, P.; Bullo, A.; Stella, L.; Filomeni, G.; Federici, G.; Caccuri, A.M. 7-Nitro-2,1,3-benzoxadiazole derivatives, a new class of suicide inhibitors for glutathione S-transferases. Mechanism of action of potential anticancer drugs. J. Biol. Chem. 2005, 280, 26397–26405. [Google Scholar] [CrossRef]
- De Luca, A.; Rotili, D.; Carpanese, D.; Lenoci, A.; Calderan, L.; Scimeca, M.; Mai, A.; Bonanno, E.; Rosato, A.; Geroni, C.; et al. A novel orally active water-soluble inhibitor of human glutathione transferase exerts a potent and selective antitumor activity against human melanoma xenografts. Oncotarget 2015, 6, 4126–4143. [Google Scholar] [CrossRef] [PubMed]
- Fulci, C.; Rotili, D.; De Luca, A.; Stella, L.; Morozzo Della Rocca, B.; Forgione, M.; Di Paolo, V.; Mai, A.; Falconi, M.; Quintieri, L.; et al. A new nitrobenzoxadiazole-based GSTP1-1 inhibitor with a previously unheard of mechanism of action and high stability. J. Enzym. Inhib. Med. Chem. 2017, 32, 240–247. [Google Scholar] [CrossRef]
- Graziani, G.; Artuso, S.; De Luca, A.; Muzi, A.; Rotili, D.; Scimeca, M.; Atzori, M.G.; Ceci, C.; Mai, A.; Leonetti, C.; et al. A new water soluble MAPK activator exerts antitumor activity in melanoma cells resistant to the BRAF inhibitor vemurafenib. Biochem. Pharmacol. 2015, 95, 16–27. [Google Scholar] [CrossRef]
- Palumbo, C.; De Luca, A.; Rosato, N.; Forgione, M.; Rotili, D.; Caccuri, A.M. c-Jun N-terminal kinase activation by nitrobenzoxadiazoles leads to late-stage autophagy inhibition. J. Transl. Med. 2016, 14, 37. [Google Scholar] [CrossRef]
- Di Paolo, V.; Fulci, C.; Rotili, D.; De Luca, A.; Tomassi, S.; Serra, M.; Scimeca, M.; Geroni, C.; Quintieri, L.; Caccuri, A.M. Characterization of water-soluble esters of nitrobenzoxadiazole-based GSTP1-1 inhibitors for cancer treatment. Biochem. Pharmacol. 2020, 178, 114060. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, D.; Batist, G. TLK-199 (Telik). IDrugs Investig. Drugs J. 2005, 8, 662–669. [Google Scholar]
- Raza, A.; Galili, N.; Callander, N.; Ochoa, L.; Piro, L.; Emanuel, P.; Williams, S.; Burris, H., 3rd; Faderl, S.; Estrov, Z.; et al. Phase 1-2a multicenter dose-escalation study of ezatiostat hydrochloride liposomes for injection (Telintra, TLK199), a novel glutathione analog prodrug in patients with myelodysplastic syndrome. J. Hematol. Oncol. 2009, 2, 20. [Google Scholar] [CrossRef] [PubMed]
- Ruscoe, J.E.; Rosario, L.A.; Wang, T.; Gaté, L.; Arifoglu, P.; Wolf, C.R.; Henderson, C.J.; Ronai, Z.; Tew, K.D. Pharmacologic or genetic manipulation of glutathione S-transferase P1-1 (GSTpi) influences cell proliferation pathways. J. Pharmacol. Exp. Ther. 2001, 298, 339–345. [Google Scholar] [PubMed]
- Mahadevan, D.; Sutton, G.R. Ezatiostat hydrochloride for the treatment of myelodysplastic syndromes. Expert Opin. Investig. Drugs 2015, 24, 725–733. [Google Scholar] [CrossRef] [PubMed]
- Kean, W.F.; Hart, L.; Buchanan, W.W. Auranofin. Br. J. Rheumatol. 1997, 36, 560–572. [Google Scholar] [CrossRef]
- Abdalbari, F.H.; Telleria, C.M. The gold complex auranofin: New perspectives for cancer therapy. Discov. Oncol. 2021, 12, 42. [Google Scholar] [CrossRef]
- Massai, L.; Cirri, D.; Marzo, T.; Messori, L. Auranofin and its analogs as prospective agents for the treatment of colorectal cancer. Cancer Drug Resist. 2022, 5, 1–14. [Google Scholar] [CrossRef]
- Ito, K.; Nishida, Y.; Hamada, S.; Shimizu, K.; Sakai, T.; Ohkawara, B.; Alman, B.A.; Enomoto, A.; Ikuta, K.; Koike, H.; et al. Efficacy of auranofin as an inhibitor of desmoid progression. Sci. Rep. 2022, 12, 11918. [Google Scholar] [CrossRef]
- De Luca, A.; Hartinger, C.G.; Dyson, P.J.; Lo Bello, M.; Casini, A. A new target for gold(I) compounds: Glutathione-S-transferase inhibition by auranofin. J. Inorg. Biochem. 2013, 119, 38–42. [Google Scholar] [CrossRef]
- Bradley, D. Therapeutic needs revive arsenic compound. Pharm. Sci. Technol. Today 2000, 3, 401. [Google Scholar] [CrossRef]
- Murgo, A.J. Clinical trials of arsenic trioxide in hematologic and solid tumors: Overview of the National Cancer Institute Cooperative Research and Development Studies. Oncologist 2001, 6 (Suppl. S2), 22–28. [Google Scholar] [CrossRef]
- Bahlis, N.J.; McCafferty-Grad, J.; Jordan-McMurry, I.; Neil, J.; Reis, I.; Kharfan-Dabaja, M.; Eckman, J.; Goodman, M.; Fernandez, H.F.; Boise, L.H.; et al. Feasibility and correlates of arsenic trioxide combined with ascorbic acid-mediated depletion of intracellular glutathione for the treatment of relapsed/refractory multiple myeloma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2002, 8, 3658–3668. [Google Scholar]
- Akao, Y.; Yamada, H.; Nakagawa, Y. Arsenic-induced apoptosis in malignant cells in vitro. Leuk. Lymphoma 2000, 37, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.C.; Cao, E.H.; Li, J.F.; Ma, W.; Qin, J.F. Induction of apoptosis and inhibition of human gastric cancer MGC-803 cell growth by arsenic trioxide. Eur. J. Cancer 1999, 35, 1258–1263. [Google Scholar] [CrossRef]
- Xie, Y.; Liu, J.; Liu, Y.; Klaassen, C.D.; Waalkes, M.P. Toxicokinetic and genomic analysis of chronic arsenic exposure in multidrug-resistance mdr1a/1b(-/-) double knockout mice. Mol. Cell. Biochem. 2004, 255, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Parker, L.J.; Bocedi, A.; Ascher, D.B.; Aitken, J.B.; Harris, H.H.; Lo Bello, M.; Ricci, G.; Morton, C.J.; Parker, M.W. Glutathione transferase P1-1 as an arsenic drug-sequestering enzyme. Protein Sci. 2017, 26, 317–326. [Google Scholar] [CrossRef]
- Crawford, L.A.; Weerapana, E. A tyrosine-reactive irreversible inhibitor for glutathione S-transferase Pi (GSTP1). Mol. BioSyst. 2016, 12, 1768–1771. [Google Scholar] [CrossRef] [PubMed]
- Shishido, Y.; Tomoike, F.; Kuwata, K.; Fujikawa, H.; Sekido, Y.; Murakami-Tonami, Y.; Kameda, T.; Abe, N.; Kimura, Y.; Shuto, S.; et al. A Covalent Inhibitor for Glutathione S-Transferase Pi (GSTP(1-1)) in Human Cells. Chembiochem 2019, 20, 900–905. [Google Scholar] [CrossRef] [PubMed]
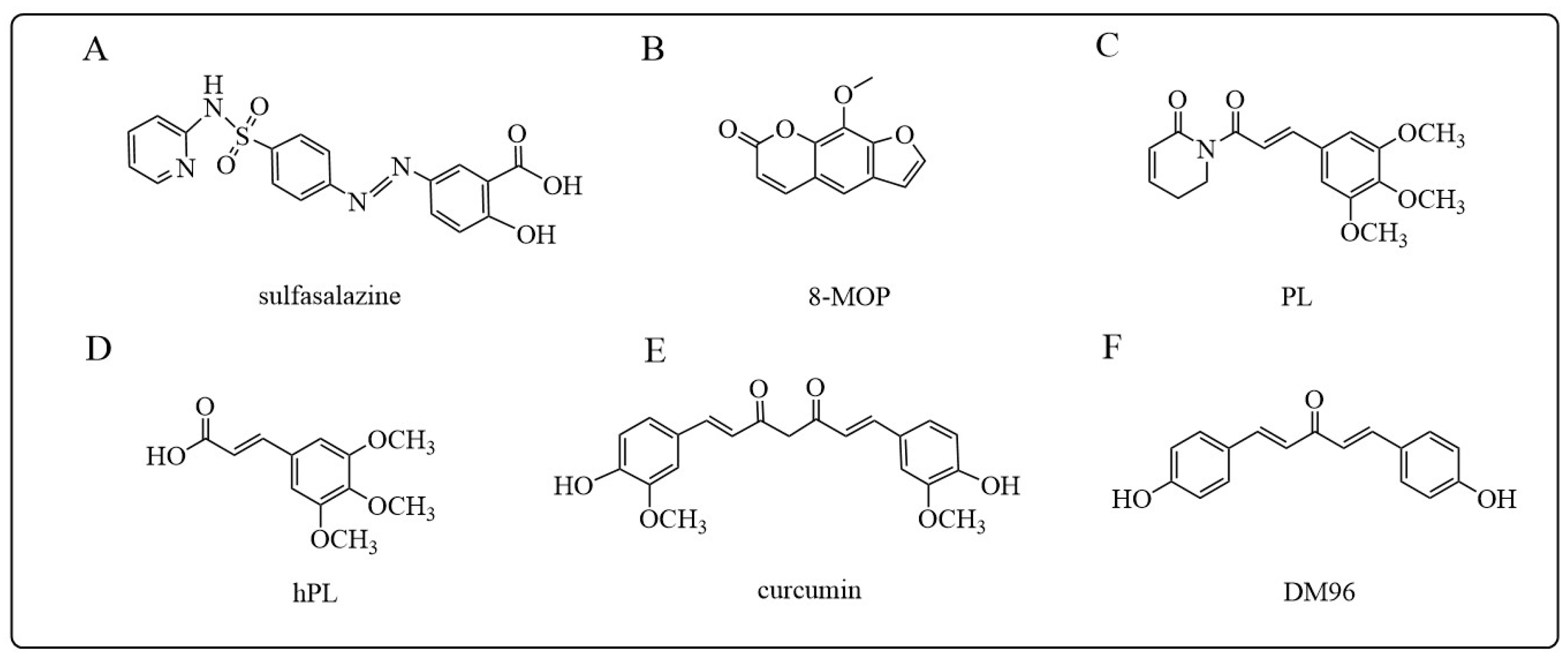
- Gupta, V.; Jani, J.P.; Jacobs, S.; Levitt, M.; Fields, L.; Awasthi, S.; Xu, B.H.; Sreevardhan, M.; Awasthi, Y.C.; Singh, S.V. Activity of melphalan in combination with the glutathione transferase inhibitor sulfasalazine. Cancer Chemother. Pharmacol. 1995, 36, 13–19. [Google Scholar] [CrossRef]
- Pathania, S.; Bhatia, R.; Baldi, A.; Singh, R.; Rawal, R.K. Drug metabolizing enzymes and their inhibitors’ role in cancer resistance. Biomed. Pharmacother. 2018, 105, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Tzaneva, S.; Kittler, H.; Thallinger, C.; Hönigsmann, H.; Tanew, A. Oral vs. bath PUVA using 8-methoxypsoralen for chronic palmoplantar eczema. Photodermatol. Photoimmunol. Photomed. 2009, 25, 101–105. [Google Scholar] [CrossRef]
- de Oliveira, D.M.; de Farias, M.T.; Teles, A.L.; Dos Santos Junior, M.C.; de Cerqueira, M.D.; Lima, R.M.; El-Bachá, R.S. 8-Methoxypsoralen is a competitive inhibitor of glutathione S-transferase P1-1. Front. Cell. Neurosci. 2014, 8, 308. [Google Scholar] [CrossRef]
- Bezerra, D.P.; Pessoa, C.; de Moraes, M.O.; Saker-Neto, N.; Silveira, E.R.; Costa-Lotufo, L.V. Overview of the therapeutic potential of piplartine (piperlongumine). Eur. J. Pharm. Sci. Off. J. Eur. Fed. Pharm. Sci. 2013, 48, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Harshbarger, W.; Gondi, S.; Ficarro, S.B.; Hunter, J.; Udayakumar, D.; Gurbani, D.; Singer, W.D.; Liu, Y.; Li, L.; Marto, J.A.; et al. Structural and Biochemical Analyses Reveal the Mechanism of Glutathione S-Transferase Pi 1 Inhibition by the Anti-cancer Compound Piperlongumine. J. Biol. Chem. 2017, 292, 112–120. [Google Scholar] [CrossRef]
- Roh, J.L.; Kim, E.H.; Park, J.Y.; Kim, J.W.; Kwon, M.; Lee, B.H. Piperlongumine selectively kills cancer cells and increases cisplatin antitumor activity in head and neck cancer. Oncotarget 2014, 5, 9227–9238. [Google Scholar] [CrossRef]
- Duvoix, A.; Morceau, F.; Delhalle, S.; Schmitz, M.; Schnekenburger, M.; Galteau, M.M.; Dicato, M.; Diederich, M. Induction of apoptosis by curcumin: Mediation by glutathione S-transferase P1-1 inhibition. Biochem. Pharmacol. 2003, 66, 1475–1483. [Google Scholar] [CrossRef]
- Pantiora, P.; Furlan, V.; Matiadis, D.; Mavroidi, B.; Perperopoulou, F.; Papageorgiou, A.C.; Sagnou, M.; Bren, U.; Pelecanou, M.; Labrou, N.E. Monocarbonyl Curcumin Analogues as Potent Inhibitors against Human Glutathione Transferase P1-1. Antioxidants 2022, 12, 63. [Google Scholar] [CrossRef]
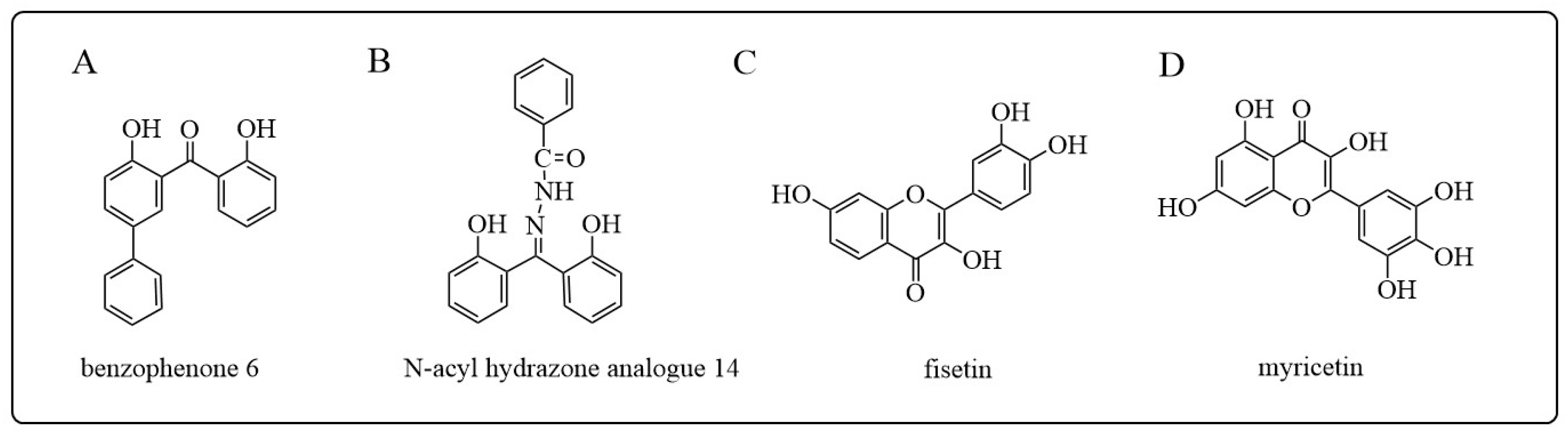
- Perperopoulou, F.D.; Tsoungas, P.G.; Thireou, T.N.; Rinotas, V.E.; Douni, E.K.; Eliopoulos, E.E.; Labrou, N.E.; Clonis, Y.D. 2,2′-Dihydroxybenzophenones and their carbonyl N-analogues as inhibitor scaffolds for MDR-involved human glutathione transferase isoenzyme A1-1. Bioorganic Med. Chem. 2014, 22, 3957–3970. [Google Scholar] [CrossRef]
- Pouliou, F.M.; Thireou, T.N.; Eliopoulos, E.E.; Tsoungas, P.G.; Labrou, N.E.; Clonis, Y.D. Isoenzyme- and allozyme-specific inhibitors: 2,2′-dihydroxybenzophenones and their carbonyl N-analogues that discriminate between human glutathione transferase A1-1 and P1-1 allozymes. Chem. Biol. Drug Des. 2015, 86, 1055–1063. [Google Scholar] [CrossRef] [PubMed]
- Alqarni, M.H.; Foudah, A.I.; Muharram, M.M.; Labrou, N.E. The Interaction of the Flavonoid Fisetin with Human Glutathione Transferase A1-1. Metabolites 2021, 11, 190. [Google Scholar] [CrossRef] [PubMed]
- Alqarni, M.H.; Foudah, A.I.; Muharram, M.M.; Alam, A.; Labrou, N.E. Myricetin as a Potential Adjuvant in Chemotherapy: Studies on the Inhibition of Human Glutathione Transferase A1-1. Biomolecules 2022, 12, 1364. [Google Scholar] [CrossRef]
- Clipson, A.J.; Bhat, V.T.; McNae, I.; Caniard, A.M.; Campopiano, D.J.; Greaney, M.F. Bivalent enzyme inhibitors discovered using dynamic covalent chemistry. Chemistry 2012, 18, 10562–10570. [Google Scholar] [CrossRef] [PubMed]
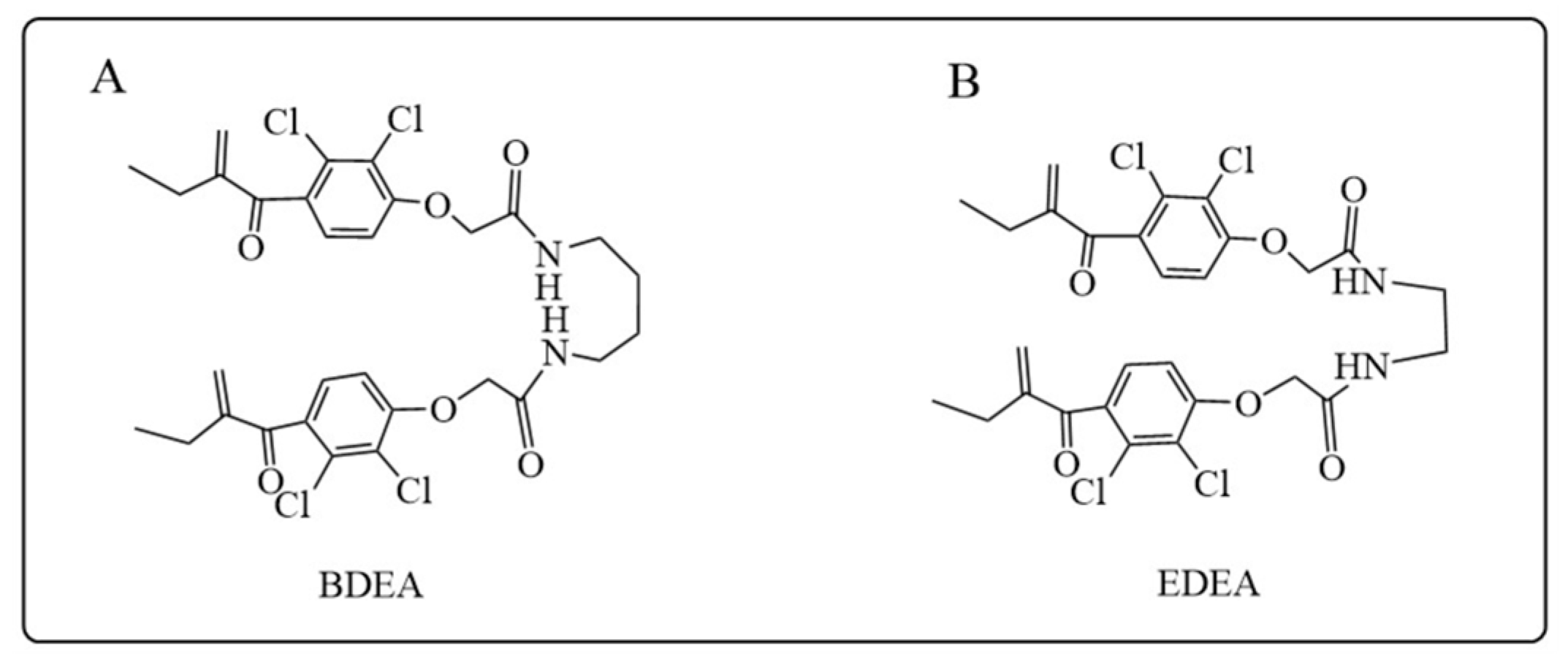
- Xu, B.; Tong, T.; Wang, X.; Liu, F.; Zhang, X.; Hu, X.; Li, X.; Yang, X.; Liao, F. Short divalent ethacrynic amides as pro-inhibitors of glutathione S-transferase isozyme Mu and potent sensitisers of cisplatin-resistant ovarian cancers. J. Enzym. Inhib. Med. Chem. 2022, 37, 728–742. [Google Scholar] [CrossRef]
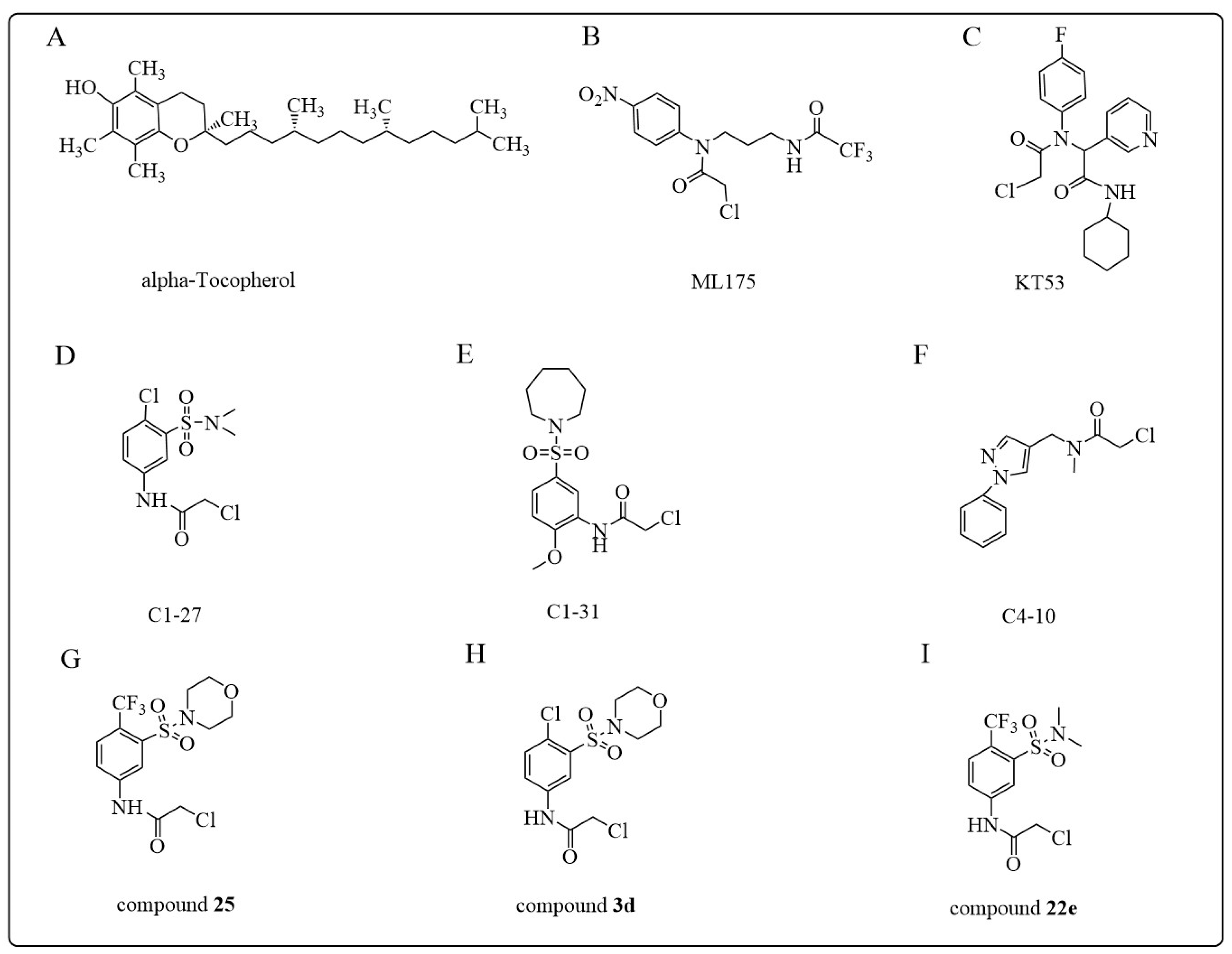
- Rota, C.; Rimbach, G.; Minihane, A.M.; Stoecklin, E.; Barella, L. Dietary vitamin E modulates differential gene expression in the rat hippocampus: Potential implications for its neuroprotective properties. Nutr. Neurosci. 2005, 8, 21–29. [Google Scholar] [CrossRef]
- Sampayo-Reyes, A.; Zakharyan, R.A. Inhibition of human glutathione S-transferase omega by tocopherol succinate. Biomed. Pharmacother. 2006, 60, 238–244. [Google Scholar] [CrossRef]
- Morris, M.C.; Evans, D.A.; Tangney, C.C.; Bienias, J.L.; Wilson, R.S.; Aggarwal, N.T.; Scherr, P.A. Relation of the tocopherol forms to incident Alzheimer disease and to cognitive change. Am. J. Clin. Nutr. 2005, 81, 508–514. [Google Scholar] [CrossRef] [PubMed]
- Sampayo-Reyes, A.; Zakharyan, R.A. Tocopherol esters inhibit human glutathione S-transferase omega. Acta Biochim. Pol. 2006, 53, 547–552. [Google Scholar] [CrossRef]
- Bachovchin, D.A.; Brown, S.J.; Rosen, H.; Cravatt, B.F. Identification of selective inhibitors of uncharacterized enzymes by high-throughput screening with fluorescent activity-based probes. Nat. Biotechnol. 2009, 27, 387–394. [Google Scholar] [CrossRef]
- Tsuboi, K.; Bachovchin, D.A.; Speers, A.E.; Brown, S.J.; Spicer, T.; Fernandez-Vega, V.; Ferguson, J.; Cravatt, B.F.; Hodder, P.; Rosen, H. Optimization and Characterization of an Inhibitor for Glutathione S-Tranferase Omega 1 (GSTO1). In Probe Reports from the NIH Molecular Libraries Program; National Center for Biotechnology Information (US): Bethesda, MD, USA, 2010. [Google Scholar]
- Dai, W.; Samanta, S.; Xue, D.; Petrunak, E.M.; Stuckey, J.A.; Han, Y.; Sun, D.; Wu, Y.; Neamati, N. Structure-Based Design of N-(5-Phenylthiazol-2-yl)acrylamides as Novel and Potent Glutathione S-Transferase Omega 1 Inhibitors. J. Med. Chem. 2019, 62, 3068–3087. [Google Scholar] [CrossRef] [PubMed]
- Ramkumar, K.; Samanta, S.; Kyani, A.; Yang, S.; Tamura, S.; Ziemke, E.; Stuckey, J.A.; Li, S.; Chinnaswamy, K.; Otake, H.; et al. Mechanistic evaluation and transcriptional signature of a glutathione S-transferase omega 1 inhibitor. Nat. Commun. 2016, 7, 13084. [Google Scholar] [CrossRef] [PubMed]
- Tsuboi, K.; Bachovchin, D.A.; Speers, A.E.; Spicer, T.P.; Fernandez-Vega, V.; Hodder, P.; Rosen, H.; Cravatt, B.F. Potent and selective inhibitors of glutathione S-transferase omega 1 that impair cancer drug resistance. J. Am. Chem. Soc. 2011, 133, 16605–16616. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Tummala, P.; Oakley, A.J.; Deora, G.S.; Nakano, Y.; Rooke, M.; Cuellar, M.E.; Strasser, J.M.; Dahlin, J.L.; Walters, M.A.; et al. Development of Benzenesulfonamide Derivatives as Potent Glutathione Transferase Omega-1 Inhibitors. J. Med. Chem. 2020, 63, 2894–2914. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Class | Gene | Chromosome | Human Isoform | Tissue and Organ Distribution | References |
|---|---|---|---|---|---|
| α | GSTA | 6p12.2 | GSTA1-1 | liver, kidney, adrenal gland, pancreas, testes, prostate | [14,15,16] |
| GSTA2-2 | liver, pancreas, kidney | [17,18] | |||
| GSTA3-3 | ovaries, testes, adrenal glands, placenta | [18] | |||
| GSTA4-4 | brain, placenta, skeletal muscle | [16] | |||
| GSTA5-5 | liver, kidney | [2] | |||
| μ | GSTM | 1p13.3 | GSTM1-1 | liver, testes, brain | [16,18] |
| GSTM2-2 | brain, testes, heart | [16,18] | |||
| GSTM3-3 | testes, brain | [18] | |||
| GSTM4-4 | duodenum, intestine | [2] | |||
| GSTM5-5 | brain | [16] | |||
| π | GSTP | 11q13 | GSTP1-1 | brain, heart, lungs, testes, pancreas, skin, kidney, bladder, prostate, colon | [16,19,20] |
| θ | GSTT | 22q11.23 | GSTT1-1 | kidney, liver, small intestine, brain, lung | [2,16,18] |
| GSTT2-2 | liver | [18] | |||
| σ | GSTS | 4q23.3 | GSTS1-1 | brain, heart, testicles | [16] |
| ω | GSTO | 10q25.1 | GSTO1-1 | liver, heart, | [18] |
| GSTO2-2 | testicles | [21] | |||
| ζ | GSTZ | 14q24.3 | GSTZ1-1 | liver, testicles | [16] |
| Isozyme Types | Types of Cancer | Anti-Tumor Drugs | References |
|---|---|---|---|
| GSTP1 | Breast cancer, ovarian cancer, colorectal cancer, lung cancer, gastric cancer, glioma, human squamous cell carcinoma, glioblastoma multiforme (GBM), bladder cancer, osteosarcoma, mantle cell lymphoma (MCL), acute lymphoblastic leukemia (ALL), prostate cancer, esophageal cancer | Cisplatin, carboplatin, doxorubicin, cyclophosphamide, paclitaxel, docetaxel, melphalan, etoposide, oxaliplatin, fluorouracil, irinotecan, cytarabine, gemcitabine, bortezomib | [18,103,107,108,109,110] |
| GSTA1 | Colorectal cancer, leukemia, lung cancer | Bacitracin, melphalan, chlorambucil, thiotepa, cyclophosphamide, imatinib, cisplatin | [18,104,106,111,112] |
| GSTM1 | Intracranial tumors (ICT), liver cancer, melanoma | Thiotepa, oxaliplatin, vincristine | [18,97,107,113,114] |
| GSTM3 | Breast cancer, glioblastoma multiforme (GBM) | BCNU, temozolomide (TMZ) | [18,103,105,115] |
| GSTO1 | Breast cancer, pancreatic cancer, ovarian cancer | Cisplatin | [21,103] |
| GSTT1 | Ovarian cancer, glioblastoma multiforme | Paclitaxel, carboplatin, BCNU | [18,116,117] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lv, N.; Huang, C.; Huang, H.; Dong, Z.; Chen, X.; Lu, C.; Zhang, Y. Overexpression of Glutathione S-Transferases in Human Diseases: Drug Targets and Therapeutic Implications. Antioxidants 2023, 12, 1970. https://doi.org/10.3390/antiox12111970
Lv N, Huang C, Huang H, Dong Z, Chen X, Lu C, Zhang Y. Overexpression of Glutathione S-Transferases in Human Diseases: Drug Targets and Therapeutic Implications. Antioxidants. 2023; 12(11):1970. https://doi.org/10.3390/antiox12111970
Chicago/Turabian StyleLv, Ning, Chunyan Huang, Haoyan Huang, Zhiqiang Dong, Xijing Chen, Chengcan Lu, and Yongjie Zhang. 2023. "Overexpression of Glutathione S-Transferases in Human Diseases: Drug Targets and Therapeutic Implications" Antioxidants 12, no. 11: 1970. https://doi.org/10.3390/antiox12111970