
The Roles of NFR2-Regulated Oxidative Stress and Mitochondrial Quality Control in Chronic Liver Diseases

Abstract
:1. Introduction
2. Oxidative Stress and Antioxidant Defense Mechanisms in CLDs
2.1. Definition of Oxidative Stress
2.2. The Formation of Reactive Oxygen Species
2.3. Antioxidant Defense Mechanism
2.3.1. Enzymatic Antioxidants
2.3.2. Non-Enzymatic Antioxidants
2.4. Oxidative Stress Biomarkers in CLD
3. Oxidative Stress and NRF2 Signaling Pathways in Chronic Liver Disease
3.1. The NRF2-KEAP1 Pathway as a Key Regulator of Oxidative Stress in Liver Health
3.2. The NRF2-Mediated Regulation of Lipid Metabolism in CLDs
3.3. NRF2-Mediated Protection against Lipid Peroxidation
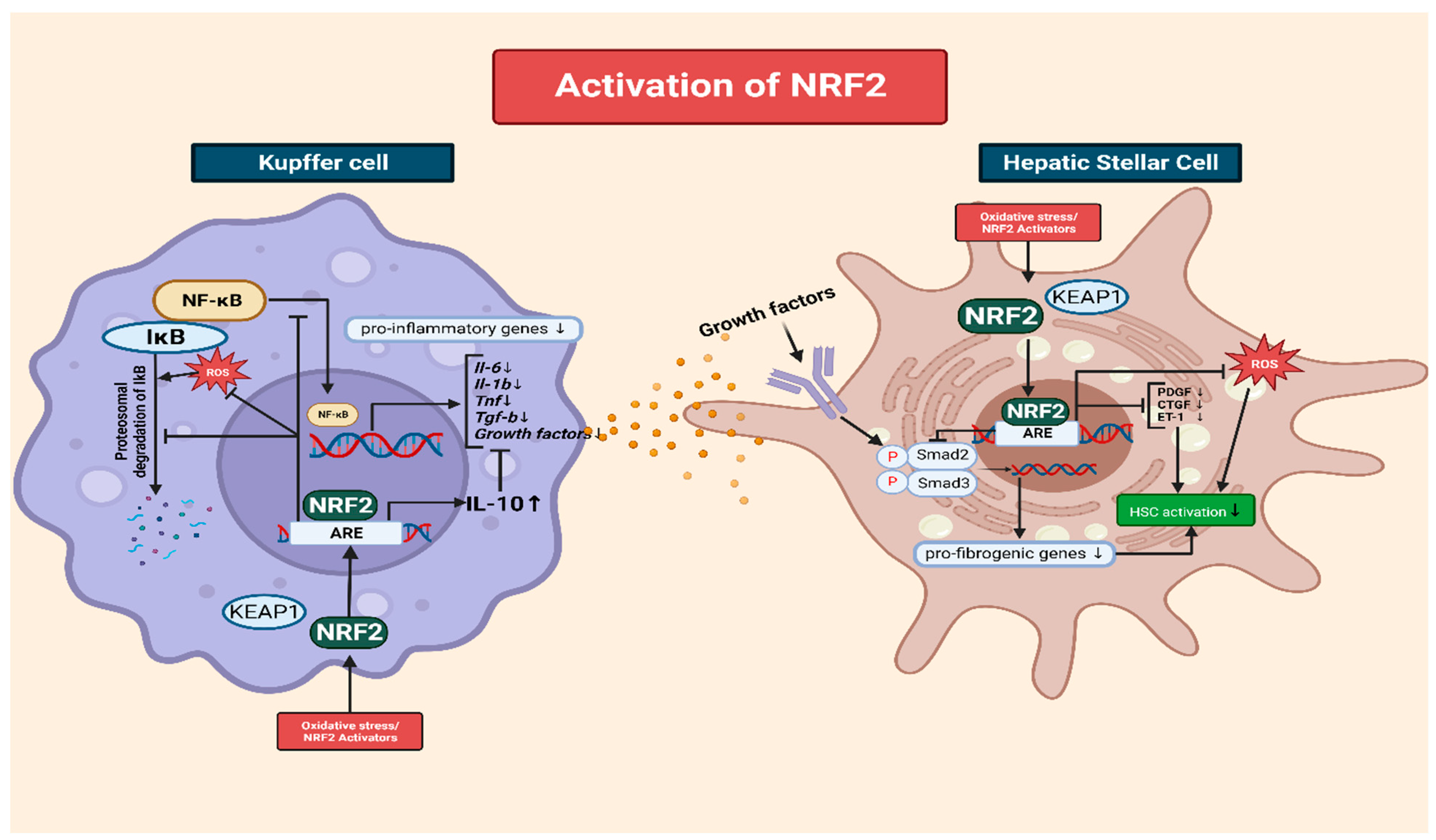
3.4. The Interplay of NRF2 and NF-κB in the Modulation of Inflammation via Kupffer Cells
3.5. The Role of NRF2 in Mitigating Oxidative-Stress-Induced HSC Activation
4. Crosstalk between NRF2 and Mitochondria Quality Control in Chronic Liver Disease
4.1. The Role of Mitochondria in the Formation of ROS
4.2. The Importance of Mitochondrial Metabolism in the Progression of Chronic Diseases
4.3. NRF2 Mediates Mitophagy and Mitochondrial Turnover
4.4. The Interplay of Mitochondria and NRF2 in CLD
5. Antioxidant Drugs (Clinical Trials) for the Treatment of CLD
5.1. Targeting the KEAP1-NRF2 Complex as a Therapeutic Strategy in Liver Diseases
5.2. Efficacy of Keap1-NRF2-Targeting Therapeutic Agents in Liver Disease Treatment
6. Challenges and Limitations: The Double-Edged Role of NRF2 in Chronic Liver Diseases
7. Concluding Remarks and Perspectives
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Asrani, S.K.; Devarbhavi, H.; Eaton, J.; Kamath, P.S. Burden of liver diseases in the world. J. Hepatol. 2019, 70, 151–171. [Google Scholar] [CrossRef]
- Mansouri, A.; Gattolliat, C.H.; Asselah, T. Mitochondrial Dysfunction and Signaling in Chronic Liver Diseases. Gastroenterology 2018, 155, 629–647. [Google Scholar] [CrossRef]
- Seen, S. Chronic liver disease and oxidative stress—A narrative review. Expert. Rev. Gastroenterol. Hepatol. 2021, 15, 1021–1035. [Google Scholar] [CrossRef] [PubMed]
- Moon, A.M.; Singal, A.G.; Tapper, E.B. Contemporary Epidemiology of Chronic Liver Disease and Cirrhosis. Clin. Gastroenterol. Hepatol. 2020, 18, 2650–2666. [Google Scholar] [CrossRef] [PubMed]
- Juanola, O.; Martinez-Lopez, S.; Frances, R.; Gomez-Hurtado, I. Non-Alcoholic Fatty Liver Disease: Metabolic, Genetic, Epigenetic and Environmental Risk Factors. Int. J. Environ. Res. Public Health 2021, 18, 5227. [Google Scholar] [CrossRef]
- Powell, E.E.; Wong, V.W.; Rinella, M. Non-alcoholic fatty liver disease. Lancet 2021, 397, 2212–2224. [Google Scholar] [CrossRef] [PubMed]
- Paik, J.M.; Kabbara, K.; Eberly, K.E.; Younossi, Y.; Henry, L.; Younossi, Z.M. Global burden of NAFLD and chronic liver disease among adolescents and young adults. Hepatology 2022, 75, 1204–1217. [Google Scholar] [CrossRef]
- Pierantonelli, I.; Svegliati-Baroni, G. Nonalcoholic Fatty Liver Disease: Basic Pathogenetic Mechanisms in the Progression from NAFLD to NASH. Transplantation 2019, 103, e1–e13. [Google Scholar] [CrossRef]
- Kawano, Y.; Cohen, D.E. Mechanisms of hepatic triglyceride accumulation in non-alcoholic fatty liver disease. J. Gastroenterol. 2013, 48, 434–441. [Google Scholar] [CrossRef]
- Sakurai, Y.; Kubota, N.; Yamauchi, T.; Kadowaki, T. Role of Insulin Resistance in MAFLD. Int. J. Mol. Sci. 2021, 22, 4156. [Google Scholar] [CrossRef]
- Wang, X.; Rao, H.; Liu, F.; Wei, L.; Li, H.; Wu, C. Recent Advances in Adipose Tissue Dysfunction and Its Role in the Pathogenesis of Non-Alcoholic Fatty Liver Disease. Cells 2021, 10, 3300. [Google Scholar] [CrossRef] [PubMed]
- Wree, A.; Broderick, L.; Canbay, A.; Hoffman, H.M.; Feldstein, A.E. From NAFLD to NASH to cirrhosis-new insights into disease mechanisms. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 627–636. [Google Scholar] [CrossRef]
- Buchanan, R.; Sinclair, J.M.A. Alcohol use disorder and the liver. Addiction 2021, 116, 1270–1278. [Google Scholar] [CrossRef] [PubMed]
- Hyun, J.; Han, J.; Lee, C.; Yoon, M.; Jung, Y. Pathophysiological Aspects of Alcohol Metabolism in the Liver. Int. J. Mol. Sci. 2021, 22, 5717. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.K.; Yates, E.; Lilly, K.; Dhanda, A.D. Oxidative stress in alcohol-related liver disease. World J. Hepatol. 2020, 12, 332–349. [Google Scholar] [CrossRef] [PubMed]
- Kruman, I.I.; Henderson, G.I.; Bergeson, S.E. DNA damage and neurotoxicity of chronic alcohol abuse. Exp. Biol. Med. 2012, 237, 740–747. [Google Scholar] [CrossRef]
- Smathers, R.L.; Galligan, J.J.; Stewart, B.J.; Petersen, D.R. Overview of lipid peroxidation products and hepatic protein modification in alcoholic liver disease. Chem. Biol. Interact. 2011, 192, 107–112. [Google Scholar] [CrossRef]
- Malnick, S.D.H.; Alin, P.; Somin, M.; Neuman, M.G. Fatty Liver Disease-Alcoholic and Non-Alcoholic: Similar but Different. Int. J. Mol. Sci. 2022, 23, 16226. [Google Scholar] [CrossRef]
- Mallet, M.; Silaghi, C.A.; Sultanik, P.; Conti, F.; Rudler, M.; Ratziu, V.; Thabut, D.; Pais, R. Current challenges and future perspectives in treating patients with NAFLD-related cirrhosis. Hepatology 2023. [Google Scholar] [CrossRef]
- Gines, P.; Krag, A.; Abraldes, J.G.; Sola, E.; Fabrellas, N.; Kamath, P.S. Liver cirrhosis. Lancet 2021, 398, 1359–1376. [Google Scholar] [CrossRef]
- Louvet, A.; Mathurin, P. Alcoholic liver disease: Mechanisms of injury and targeted treatment. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 231–242. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishna, G.; Rastogi, A.; Trehanpati, N.; Sen, B.; Khosla, R.; Sarin, S.K. From cirrhosis to hepatocellular carcinoma: New molecular insights on inflammation and cellular senescence. Liver Cancer 2013, 2, 367–383. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Tian, R.; She, Z.; Cai, J.; Li, H. Role of oxidative stress in the pathogenesis of nonalcoholic fatty liver disease. Free Radic. Biol. Med. 2020, 152, 116–141. [Google Scholar] [CrossRef]
- Cichoz-Lach, H.; Michalak, A. Oxidative stress as a crucial factor in liver diseases. World J. Gastroenterol. 2014, 20, 8082–8091. [Google Scholar] [CrossRef]
- Ha, H.L.; Shin, H.J.; Feitelson, M.A.; Yu, D.Y. Oxidative stress and antioxidants in hepatic pathogenesis. World J. Gastroenterol. 2010, 16, 6035–6043. [Google Scholar] [CrossRef]
- Uchida, D.; Takaki, A.; Oyama, A.; Adachi, T.; Wada, N.; Onishi, H.; Okada, H. Oxidative Stress Management in Chronic Liver Diseases and Hepatocellular Carcinoma. Nutrients 2020, 12, 1576. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, P.; Terracciano, L.; D’Angelo, S.; Ferbo, U.; Bracigliano, A.; Tarantino, L.; Perrella, A.; Perrella, O.; De Chiara, G.; Panico, L.; et al. Oxidative stress and steatosis are cofactors of liver injury in primary biliary cirrhosis. J. Gastroenterol. 2010, 45, 1053–1062. [Google Scholar] [CrossRef]
- Valko, M.; Rhodes, C.J.; Moncol, J.; Izakovic, M.; Mazur, M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem. Biol. Interact. 2006, 160, 1–40. [Google Scholar] [CrossRef]
- Willcox, J.K.; Ash, S.L.; Catignani, G.L. Antioxidants and prevention of chronic disease. Crit. Rev. Food Sci. Nutr. 2004, 44, 275–295. [Google Scholar] [CrossRef]
- Betteridge, D.J. What is oxidative stress? Metabolism 2000, 49, 3–8. [Google Scholar] [CrossRef]
- Collin, F. Chemical Basis of Reactive Oxygen Species Reactivity and Involvement in Neurodegenerative Diseases. Int. J. Mol. Sci. 2019, 20, 2407. [Google Scholar] [CrossRef] [PubMed]
- Lobo, V.; Patil, A.; Phatak, A.; Chandra, N. Free radicals, antioxidants and functional foods: Impact on human health. Pharmacogn. Rev. 2010, 4, 118–126. [Google Scholar] [CrossRef]
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003, 552, 335–344. [Google Scholar] [CrossRef]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef]
- Veith, A.; Moorthy, B. Role of Cytochrome P450s in the Generation and Metabolism of Reactive Oxygen Species. Curr. Opin. Toxicol. 2018, 7, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Canton, M.; Sanchez-Rodriguez, R.; Spera, I.; Venegas, F.C.; Favia, M.; Viola, A.; Castegna, A. Reactive Oxygen Species in Macrophages: Sources and Targets. Front. Immunol. 2021, 12, 734229. [Google Scholar] [CrossRef] [PubMed]
- Forman, H.J.; Zhang, H. Targeting oxidative stress in disease: Promise and limitations of antioxidant therapy. Nat. Rev. Drug Discov. 2021, 20, 689–709. [Google Scholar] [CrossRef] [PubMed]
- Juan, C.A.; Perez de la Lastra, J.M.; Plou, F.J.; Perez-Lebena, E. The Chemistry of Reactive Oxygen Species (ROS) Revisited: Outlining Their Role in Biological Macromolecules (DNA, Lipids and Proteins) and Induced Pathologies. Int. J. Mol. Sci. 2021, 22, 4642. [Google Scholar] [CrossRef]
- Liguori, I.; Russo, G.; Curcio, F.; Bulli, G.; Aran, L.; Della-Morte, D.; Gargiulo, G.; Testa, G.; Cacciatore, F.; Bonaduce, D.; et al. Oxidative stress, aging, and diseases. Clin. Interv. Aging 2018, 13, 757–772. [Google Scholar] [CrossRef] [PubMed]
- Irato, P.; Santovito, G. Enzymatic and Non-Enzymatic Molecules with Antioxidant Function. Antioxidants 2021, 10, 579. [Google Scholar] [CrossRef] [PubMed]
- Rosa, A.C.; Corsi, D.; Cavi, N.; Bruni, N.; Dosio, F. Superoxide Dismutase Administration: A Review of Proposed Human Uses. Molecules 2021, 26, 1844. [Google Scholar] [CrossRef]
- Trist, B.G.; Hilton, J.B.; Hare, D.J.; Crouch, P.J.; Double, K.L. Superoxide Dismutase 1 in Health and Disease: How a Frontline Antioxidant Becomes Neurotoxic. Angew. Chem. Int. Ed. Engl. 2021, 60, 9215–9246. [Google Scholar] [CrossRef] [PubMed]
- Alfonso-Prieto, M.; Biarnes, X.; Vidossich, P.; Rovira, C. The molecular mechanism of the catalase reaction. J. Am. Chem. Soc. 2009, 131, 11751–11761. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.K.; Cho, H.W.; Song, S.E.; Song, D.K. Catalase and nonalcoholic fatty liver disease. Pflugers Arch. 2018, 470, 1721–1737. [Google Scholar] [CrossRef] [PubMed]
- Szuster-Ciesielska, A.; Daniluk, J.; Kandefer-Szerszen, M. Oxidative stress in the blood of patients with alcohol-related liver cirrhosis. Med. Sci. Monit. 2002, 8, CR419–CR424. [Google Scholar]
- Handy, D.E.; Loscalzo, J. The role of glutathione peroxidase-1 in health and disease. Free Radic. Biol. Med. 2022, 188, 146–161. [Google Scholar] [CrossRef]
- Jiao, Y.; Wang, Y.; Guo, S.; Wang, G. Glutathione peroxidases as oncotargets. Oncotarget 2017, 8, 80093–80102. [Google Scholar] [CrossRef]
- Couto, N.; Wood, J.; Barber, J. The role of glutathione reductase and related enzymes on cellular redox homoeostasis network. Free Radic. Biol. Med. 2016, 95, 27–42. [Google Scholar] [CrossRef]
- Arner, E.S.; Holmgren, A. Physiological functions of thioredoxin and thioredoxin reductase. Eur. J. Biochem. 2000, 267, 6102–6109. [Google Scholar] [CrossRef]
- Prieto-Alamo, M.J.; Jurado, J.; Gallardo-Madueno, R.; Monje-Casas, F.; Holmgren, A.; Pueyo, C. Transcriptional regulation of glutaredoxin and thioredoxin pathways and related enzymes in response to oxidative stress. J. Biol. Chem. 2000, 275, 13398–13405. [Google Scholar] [CrossRef]
- Gaucher, C.; Boudier, A.; Bonetti, J.; Clarot, I.; Leroy, P.; Parent, M. Glutathione: Antioxidant Properties Dedicated to Nanotechnologies. Antioxidants 2018, 7, 62. [Google Scholar] [CrossRef] [PubMed]
- Padayatty, S.J.; Katz, A.; Wang, Y.; Eck, P.; Kwon, O.; Lee, J.H.; Chen, S.; Corpe, C.; Dutta, A.; Dutta, S.K.; et al. Vitamin C as an antioxidant: Evaluation of its role in disease prevention. J. Am. Coll. Nutr. 2003, 22, 18–35. [Google Scholar] [CrossRef]
- Chow, C.K. Vitamin E and oxidative stress. Free Radic. Biol. Med. 1991, 11, 215–232. [Google Scholar] [CrossRef]
- Perumpail, B.J.; Li, A.A.; John, N.; Sallam, S.; Shah, N.D.; Kwong, W.; Cholankeril, G.; Kim, D.; Ahmed, A. The Role of Vitamin E in the Treatment of NAFLD. Diseases 2018, 6, 86. [Google Scholar] [CrossRef]
- Johra, F.T.; Bepari, A.K.; Bristy, A.T.; Reza, H.M. A Mechanistic Review of beta-Carotene, Lutein, and Zeaxanthin in Eye Health and Disease. Antioxidants 2020, 9, 1046. [Google Scholar] [CrossRef]
- Elvira-Torales, L.I.; Garcia-Alonso, J.; Periago-Caston, M.J. Nutritional Importance of Carotenoids and Their Effect on Liver Health: A Review. Antioxidants 2019, 8, 229. [Google Scholar] [CrossRef] [PubMed]
- Stice, C.P.; Wang, X.D. Carotenoids and alcoholic liver disease. Hepatobiliary Surg. Nutr. 2013, 2, 244–247. [Google Scholar] [CrossRef]
- Panche, A.N.; Diwan, A.D.; Chandra, S.R. Flavonoids: An overview. J. Nutr. Sci. 2016, 5, e47. [Google Scholar] [CrossRef] [PubMed]
- Tan, P.; Jin, L.; Qin, X.; He, B. Natural flavonoids: Potential therapeutic strategies for non-alcoholic fatty liver disease. Front. Pharmacol. 2022, 13, 1005312. [Google Scholar] [CrossRef]
- Tripathi, A.K.; Ray, A.K.; Mishra, S.K.; Bishen, S.M.; Mishra, H.; Khurana, A. Molecular and Therapeutic Insights of Alpha-Lipoic Acid as a Potential Molecule for Disease Prevention. Rev. Bras. Farmacogn. 2023, 33, 272–287. [Google Scholar] [CrossRef]
- Stankovic, M.N.; Mladenovic, D.; Ninkovic, M.; Ethuricic, I.; Sobajic, S.; Jorgacevic, B.; de Luka, S.; Vukicevic, R.J.; Radosavljevic, T.S. The effects of alpha-lipoic acid on liver oxidative stress and free fatty acid composition in methionine-choline deficient diet-induced NAFLD. J. Med. Food 2014, 17, 254–261. [Google Scholar] [CrossRef] [PubMed]
- Foo, N.P.; Lin, S.H.; Lee, Y.H.; Wu, M.J.; Wang, Y.J. alpha-Lipoic acid inhibits liver fibrosis through the attenuation of ROS-triggered signaling in hepatic stellate cells activated by PDGF and TGF-beta. Toxicology 2011, 282, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Ayala, A.; Munoz, M.F.; Arguelles, S. Lipid peroxidation: Production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid. Med. Cell. Longev. 2014, 2014, 360438. [Google Scholar] [CrossRef] [PubMed]
- Omari Shekaftik, S.; Nasirzadeh, N. 8-Hydroxy-2′-deoxyguanosine (8-OHdG) as a biomarker of oxidative DNA damage induced by occupational exposure to nanomaterials: A systematic review. Nanotoxicology 2021, 15, 850–864. [Google Scholar] [CrossRef] [PubMed]
- Katerji, M.; Filippova, M.; Duerksen-Hughes, P. Approaches and Methods to Measure Oxidative Stress in Clinical Samples: Research Applications in the Cancer Field. Oxid. Med. Cell. Longev. 2019, 2019, 1279250. [Google Scholar] [CrossRef]
- Shearn, C.T.; Saba, L.M.; Roede, J.R.; Orlicky, D.J.; Shearn, A.H.; Petersen, D.R. Differential carbonylation of proteins in end-stage human fatty and nonfatty NASH. Free Radic. Biol. Med. 2017, 113, 280–290. [Google Scholar] [CrossRef]
- Chienwichai, P.; Reamtong, O.; Boonyuen, U.; Pisitkun, T.; Somparn, P.; Tharnpoophasiam, P.; Worakhunpiset, S.; Topanurak, S. Hepatic protein Carbonylation profiles induced by lipid accumulation and oxidative stress for investigating cellular response to non-alcoholic fatty liver disease in vitro. Proteome Sci. 2019, 17, 1. [Google Scholar] [CrossRef]
- Vairetti, M.; Di Pasqua, L.G.; Cagna, M.; Richelmi, P.; Ferrigno, A.; Berardo, C. Changes in Glutathione Content in Liver Diseases: An Update. Antioxidants 2021, 10, 364. [Google Scholar] [CrossRef] [PubMed]
- Galicia-Moreno, M.; Rosique-Oramas, D.; Medina-Avila, Z.; Alvarez-Torres, T.; Falcon, D.; Higuera-de la Tijera, F.; Bejar, Y.L.; Cordero-Perez, P.; Munoz-Espinosa, L.; Perez-Hernandez, J.L.; et al. Behavior of Oxidative Stress Markers in Alcoholic Liver Cirrhosis Patients. Oxid. Med. Cell. Longev. 2016, 2016, 9370565. [Google Scholar] [CrossRef]
- Sfarti, C.; Ciobica, A.; Balmus, I.M.; Ilie, O.D.; Trifan, A.; Petrea, O.; Cojocariu, C.; Girleanu, I.; Singeap, A.M.; Stanciu, C. Systemic Oxidative Stress Markers in Cirrhotic Patients with Hepatic Encephalopathy: Possible Connections with Systemic Ammoniemia. Medicina 2020, 56, 196. [Google Scholar] [CrossRef] [PubMed]
- Horoz, M.; Bolukbas, C.; Bolukbas, F.F.; Sabuncu, T.; Aslan, M.; Sarifakiogullari, S.; Gunaydin, N.; Erel, O. Measurement of the total antioxidant response using a novel automated method in subjects with nonalcoholic steatohepatitis. BMC Gastroenterol. 2005, 5, 35. [Google Scholar] [CrossRef]
- Perrone, A.; Giovino, A.; Benny, J.; Martinelli, F. Advanced Glycation End Products (AGEs): Biochemistry, Signaling, Analytical Methods, and Epigenetic Effects. Oxid. Med. Cell. Longev. 2020, 2020, 3818196. [Google Scholar] [CrossRef]
- Liu, H.; Han, T.; Tian, J.; Zhu, Z.Y.; Liu, Y.; Li, Y.; Xiao, S.X.; Li, Y.; Feng, Y.Y. Monitoring oxidative stress in acute-on-chronic liver failure by advanced oxidation protein products. Hepatol. Res. 2012, 42, 171–180. [Google Scholar] [CrossRef]
- Ngo, V.; Duennwald, M.L. Nrf2 and Oxidative Stress: A General Overview of Mechanisms and Implications in Human Disease. Antioxidants 2022, 11, 2345. [Google Scholar] [CrossRef]
- He, F.; Ru, X.; Wen, T. NRF2, a Transcription Factor for Stress Response and Beyond. Int. J. Mol. Sci. 2020, 21, 4777. [Google Scholar] [CrossRef] [PubMed]
- Galicia-Moreno, M.; Lucano-Landeros, S.; Monroy-Ramirez, H.C.; Silva-Gomez, J.; Gutierrez-Cuevas, J.; Santos, A.; Armendariz-Borunda, J. Roles of Nrf2 in Liver Diseases: Molecular, Pharmacological, and Epigenetic Aspects. Antioxidants 2020, 9, 980. [Google Scholar] [CrossRef] [PubMed]
- Baird, L.; Yamamoto, M. The Molecular Mechanisms Regulating the KEAP1-NRF2 Pathway. Mol. Cell. Biol. 2020, 40, e00099-20. [Google Scholar] [CrossRef]
- Bellezza, I.; Giambanco, I.; Minelli, A.; Donato, R. Nrf2-Keap1 signaling in oxidative and reductive stress. Biochim. Biophys. Acta Mol. Cell. Res. 2018, 1865, 721–733. [Google Scholar] [CrossRef] [PubMed]
- Robledinos-Anton, N.; Fernandez-Gines, R.; Manda, G.; Cuadrado, A. Activators and Inhibitors of NRF2: A Review of Their Potential for Clinical Development. Oxid. Med. Cell. Longev. 2019, 2019, 9372182. [Google Scholar] [CrossRef]
- Shaw, P.; Chattopadhyay, A. Nrf2-ARE signaling in cellular protection: Mechanism of action and the regulatory mechanisms. J. Cell. Physiol. 2020, 235, 3119–3130. [Google Scholar] [CrossRef]
- Zhou, J.; Zheng, Q.; Chen, Z. The Nrf2 Pathway in Liver Diseases. Front. Cell. Dev. Biol. 2022, 10, 826204. [Google Scholar] [CrossRef]
- Shin, S.; Wakabayashi, J.; Yates, M.S.; Wakabayashi, N.; Dolan, P.M.; Aja, S.; Liby, K.T.; Sporn, M.B.; Yamamoto, M.; Kensler, T.W. Role of Nrf2 in prevention of high-fat diet-induced obesity by synthetic triterpenoid CDDO-imidazolide. Eur. J. Pharmacol. 2009, 620, 138–144. [Google Scholar] [CrossRef]
- Qiu, S.; Liang, Z.; Wu, Q.; Wang, M.; Yang, M.; Chen, C.; Zheng, H.; Zhu, Z.; Li, L.; Yang, G. Hepatic lipid accumulation induced by a high-fat diet is regulated by Nrf2 through multiple pathways. FASEB J. 2022, 36, e22280. [Google Scholar] [CrossRef] [PubMed]
- Akl, M.G.; Li, L.; Baccetto, R.; Phanse, S.; Zhang, Q.; Trites, M.J.; McDonald, S.; Aoki, H.; Babu, M.; Widenmaier, S.B. Complementary gene regulation by NRF1 and NRF2 protects against hepatic cholesterol overload. Cell Rep. 2023, 42, 112399. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.K.; Yeager, R.L.; Tanaka, Y.; Klaassen, C.D. Enhanced expression of Nrf2 in mice attenuates the fatty liver produced by a methionine- and choline-deficient diet. Toxicol. Appl. Pharmacol. 2010, 245, 326–334. [Google Scholar] [CrossRef]
- Tanaka, Y.; Ikeda, T.; Yamamoto, K.; Ogawa, H.; Kamisako, T. Dysregulated expression of fatty acid oxidation enzymes and iron-regulatory genes in livers of Nrf2-null mice. J. Gastroenterol. Hepatol. 2012, 27, 1711–1717. [Google Scholar] [CrossRef] [PubMed]
- Dinkova-Kostova, A.T.; Abramov, A.Y. The emerging role of Nrf2 in mitochondrial function. Free Radic. Biol. Med. 2015, 88, 179–188. [Google Scholar] [CrossRef]
- Chambel, S.S.; Santos-Goncalves, A.; Duarte, T.L. The Dual Role of Nrf2 in Nonalcoholic Fatty Liver Disease: Regulation of Antioxidant Defenses and Hepatic Lipid Metabolism. Biomed. Res. Int. 2015, 2015, 597134. [Google Scholar] [CrossRef]
- Morita, M.; Ishida, N.; Uchiyama, K.; Yamaguchi, K.; Itoh, Y.; Shichiri, M.; Yoshida, Y.; Hagihara, Y.; Naito, Y.; Yoshikawa, T.; et al. Fatty liver induced by free radicals and lipid peroxidation. Free Radic. Res. 2012, 46, 758–765. [Google Scholar] [CrossRef]
- Martin-Fernandez, M.; Arroyo, V.; Carnicero, C.; Siguenza, R.; Busta, R.; Mora, N.; Antolin, B.; Tamayo, E.; Aspichueta, P.; Carnicero-Frutos, I.; et al. Role of Oxidative Stress and Lipid Peroxidation in the Pathophysiology of NAFLD. Antioxidants 2022, 11, 2217. [Google Scholar] [CrossRef] [PubMed]
- Su, L.J.; Zhang, J.H.; Gomez, H.; Murugan, R.; Hong, X.; Xu, D.; Jiang, F.; Peng, Z.Y. Reactive Oxygen Species-Induced Lipid Peroxidation in Apoptosis, Autophagy, and Ferroptosis. Oxid. Med. Cell. Longev. 2019, 2019, 5080843. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Xu, L.; Porter, N.A. Free radical lipid peroxidation: Mechanisms and analysis. Chem. Rev. 2011, 111, 5944–5972. [Google Scholar] [CrossRef]
- Winczura, A.; Zdzalik, D.; Tudek, B. Damage of DNA and proteins by major lipid peroxidation products in genome stability. Free Radic. Res. 2012, 46, 442–459. [Google Scholar] [CrossRef] [PubMed]
- Yesilova, Z.; Yaman, H.; Oktenli, C.; Ozcan, A.; Uygun, A.; Cakir, E.; Sanisoglu, S.Y.; Erdil, A.; Ates, Y.; Aslan, M.; et al. Systemic markers of lipid peroxidation and antioxidants in patients with nonalcoholic Fatty liver disease. Am. J. Gastroenterol. 2005, 100, 850–855. [Google Scholar] [CrossRef]
- Spiteller, G. Is lipid peroxidation of polyunsaturated acids the only source of free radicals that induce aging and age-related diseases? Rejuvenation Res. 2010, 13, 91–103. [Google Scholar] [CrossRef]
- Negre-Salvayre, A.; Auge, N.; Ayala, V.; Basaga, H.; Boada, J.; Brenke, R.; Chapple, S.; Cohen, G.; Feher, J.; Grune, T.; et al. Pathological aspects of lipid peroxidation. Free Radic. Res. 2010, 44, 1125–1171. [Google Scholar] [CrossRef] [PubMed]
- Dodson, M.; Castro-Portuguez, R.; Zhang, D.D. NRF2 plays a critical role in mitigating lipid peroxidation and ferroptosis. Redox Biol. 2019, 23, 101107. [Google Scholar] [CrossRef]
- Busch, C.J.; Hendrikx, T.; Weismann, D.; Jackel, S.; Walenbergh, S.M.; Rendeiro, A.F.; Weisser, J.; Puhm, F.; Hladik, A.; Goderle, L.; et al. Malondialdehyde epitopes are sterile mediators of hepatic inflammation in hypercholesterolemic mice. Hepatology 2017, 65, 1181–1195. [Google Scholar] [CrossRef]
- Yadav, U.C.; Ramana, K.V. Regulation of NF-κB-induced inflammatory signaling by lipid peroxidation-derived aldehydes. Oxid. Med. Cell. Longev. 2013, 2013, 690545. [Google Scholar] [CrossRef]
- Ma, F.; Luo, S.; Lu, C.; Jiang, X.; Chen, K.; Deng, J.; Ma, S.; Li, Z. The role of Nrf2 in periodontal disease by regulating lipid peroxidation, inflammation and apoptosis. Front. Endocrinol. 2022, 13, 963451. [Google Scholar] [CrossRef] [PubMed]
- Gruber, F.; Ornelas, C.M.; Karner, S.; Narzt, M.S.; Nagelreiter, I.M.; Gschwandtner, M.; Bochkov, V.; Tschachler, E. Nrf2 deficiency causes lipid oxidation, inflammation, and matrix-protease expression in DHA-supplemented and UVA-irradiated skin fibroblasts. Free Radic. Biol. Med. 2015, 88, 439–451. [Google Scholar] [CrossRef]
- Koyama, Y.; Brenner, D.A. Liver inflammation and fibrosis. J. Clin. Investig. 2017, 127, 55–64. [Google Scholar] [CrossRef]
- Parola, M.; Pinzani, M. Liver fibrosis: Pathophysiology, pathogenetic targets and clinical issues. Mol. Aspects Med. 2019, 65, 37–55. [Google Scholar] [CrossRef] [PubMed]
- Bataller, R.; Brenner, D.A. Liver fibrosis. J. Clin. Investig. 2005, 115, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Hao, W.; Li, M.; Cai, Q.; Wu, S.; Li, X.; He, Q.; Hu, Y. Roles of NRF2 in Fibrotic Diseases: From Mechanisms to Therapeutic Approaches. Front. Physiol. 2022, 13, 889792. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.; Yang, Y. Activation of Nrf2/AREs-mediated antioxidant signalling, and suppression of profibrotic TGF-beta1/Smad3 pathway: A promising therapeutic strategy for hepatic fibrosis—A review. Life Sci. 2020, 256, 117909. [Google Scholar] [CrossRef]
- Yang, H.; Luo, F.; Wei, Y.; Jiao, Y.; Qian, J.; Chen, S.; Gong, Y.; Tang, L. TGR5 protects against cholestatic liver disease via suppressing the NF-κB pathway and activating the Nrf2/HO-1 pathway. Ann. Transl. Med. 2021, 9, 1158. [Google Scholar] [CrossRef]
- Thimmulappa, R.K.; Lee, H.; Rangasamy, T.; Reddy, S.P.; Yamamoto, M.; Kensler, T.W.; Biswal, S. Nrf2 is a critical regulator of the innate immune response and survival during experimental sepsis. J. Clin. Investig. 2006, 116, 984–995. [Google Scholar] [CrossRef]
- Xue, R.; Qiu, J.; Wei, S.; Liu, M.; Wang, Q.; Wang, P.; Sha, B.; Wang, H.; Shi, Y.; Zhou, J.; et al. Lycopene alleviates hepatic ischemia reperfusion injury via the Nrf2/HO-1 pathway mediated NLRP3 inflammasome inhibition in Kupffer cells. Ann. Transl. Med. 2021, 9, 631. [Google Scholar] [CrossRef]
- Liu, X.; Wang, T.; Liu, X.; Cai, L.; Qi, J.; Zhang, P.; Li, Y. Biochanin A protects lipopolysaccharide/D-galactosamine-induced acute liver injury in mice by activating the Nrf2 pathway and inhibiting NLRP3 inflammasome activation. Int. Immunopharmacol. 2016, 38, 324–331. [Google Scholar] [CrossRef] [PubMed]
- Rushworth, S.A.; Chen, X.L.; Mackman, N.; Ogborne, R.M.; O’Connell, M.A. Lipopolysaccharide-induced heme oxygenase-1 expression in human monocytic cells is mediated via Nrf2 and protein kinase C. J. Immunol. 2005, 175, 4408–4415. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.; Greven, J.; Qin, K.; Fragoulis, A.; Horst, K.; Blasius, F.; Wruck, C.; Pufe, T.; Kobbe, P.; Hildebrand, F.; et al. Sulforaphane Exerts Beneficial Immunomodulatory Effects on Liver Tissue via a Nrf2 Pathway-Related Mechanism in a Murine Model of Hemorrhagic Shock and Resuscitation. Front. Immunol. 2022, 13, 822895. [Google Scholar] [CrossRef]
- Sierra-Filardi, E.; Vega, M.A.; Sanchez-Mateos, P.; Corbi, A.L.; Puig-Kroger, A. Heme Oxygenase-1 expression in M-CSF-polarized M2 macrophages contributes to LPS-induced IL-10 release. Immunobiology 2010, 215, 788–795. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; He, C. Nrf2-mediated anti-inflammatory polarization of macrophages as therapeutic targets for osteoarthritis. Front. Immunol. 2022, 13, 967193. [Google Scholar] [CrossRef]
- Chen, H.; Qin, J.; Shi, H.; Li, Q.; Zhou, S.; Chen, L. Rhoifolin ameliorates osteoarthritis via the Nrf2/NF-κB axis: In Vitro and in vivo experiments. Osteoarthr. Cartil. 2022, 30, 735–745. [Google Scholar] [CrossRef]
- Liu, G.H.; Qu, J.; Shen, X. NF-κB/p65 antagonizes Nrf2-ARE pathway by depriving CBP from Nrf2 and facilitating recruitment of HDAC3 to MafK. Biochim. Biophys. Acta 2008, 1783, 713–727. [Google Scholar] [CrossRef]
- Bellezza, I.; Mierla, A.L.; Minelli, A. Nrf2 and NF-κB and Their Concerted Modulation in Cancer Pathogenesis and Progression. Cancers 2010, 2, 483–497. [Google Scholar] [CrossRef]
- Nair, S.; Doh, S.T.; Chan, J.Y.; Kong, A.N.; Cai, L. Regulatory potential for concerted modulation of Nrf2- and Nfkb1-mediated gene expression in inflammation and carcinogenesis. Br. J. Cancer 2008, 99, 2070–2082. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Tian, F.; Zheng, H.; Whitman, S.A.; Lin, Y.; Zhang, Z.; Zhang, N.; Zhang, D.D. Nrf2 suppresses lupus nephritis through inhibition of oxidative injury and the NF-κB-mediated inflammatory response. Kidney Int. 2014, 85, 333–343. [Google Scholar] [CrossRef]
- Heiss, E.; Herhaus, C.; Klimo, K.; Bartsch, H.; Gerhauser, C. Nuclear factor kappa B is a molecular target for sulforaphane-mediated anti-inflammatory mechanisms. J. Biol. Chem. 2001, 276, 32008–32015. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.E.; You, D.J.; Lee, C.; Ahn, C.; Seong, J.Y.; Hwang, J.I. Suppression of NF-κB signaling by KEAP1 regulation of IKKbeta activity through autophagic degradation and inhibition of phosphorylation. Cell. Signal. 2010, 22, 1645–1654. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.S.; Harrison, D.J.; Kisielewski, D.; Cassidy, D.M.; McNeilly, A.D.; Gallagher, J.R.; Walsh, S.V.; Honda, T.; McCrimmon, R.J.; Dinkova-Kostova, A.T.; et al. Experimental Nonalcoholic Steatohepatitis and Liver Fibrosis Are Ameliorated by Pharmacologic Activation of Nrf2 (NF-E2 p45-Related Factor 2). Cell. Mol. Gastroenterol. Hepatol. 2018, 5, 367–398. [Google Scholar] [CrossRef]
- Shi, Y.S.; Li, X.X.; Li, H.T.; Zhang, Y. Pelargonidin ameliorates CCl(4)-induced liver fibrosis by suppressing the ROS-NLRP3-IL-1beta axis via activating the Nrf2 pathway. Food Funct. 2020, 11, 5156–5165. [Google Scholar] [CrossRef]
- Prestigiacomo, V.; Suter-Dick, L. Nrf2 protects stellate cells from Smad-dependent cell activation. PLoS ONE 2018, 13, e0201044. [Google Scholar] [CrossRef]
- Qiu, L.; Hu, L.; Liu, X.; Li, W.; Zhang, X.; Xia, H.; Zhang, C. Physalin B inhibits PDGF-BB-induced VSMC proliferation, migration and phenotypic transformation by activating the Nrf2 pathway. Food Funct. 2021, 12, 10950–10966. [Google Scholar] [CrossRef]
- Del Campo, J.A.; Gallego, P.; Grande, L. Role of inflammatory response in liver diseases: Therapeutic strategies. World J. Hepatol. 2018, 10, 1–7. [Google Scholar] [CrossRef]
- Eisenstein, A.; Hilliard, B.K.; Pope, S.D.; Zhang, C.; Taskar, P.; Waizman, D.A.; Israni-Winger, K.; Tian, H.; Luan, H.H.; Wang, A. Activation of the transcription factor NRF2 mediates the anti-inflammatory properties of a subset of over-the-counter and prescription NSAIDs. Immunity 2022, 55, 1082–1095.e1085. [Google Scholar] [CrossRef]
- Kong, D.; Zhang, Z.; Chen, L.; Huang, W.; Zhang, F.; Wang, L.; Wang, Y.; Cao, P.; Zheng, S. Curcumin blunts epithelial-mesenchymal transition of hepatocytes to alleviate hepatic fibrosis through regulating oxidative stress and autophagy. Redox Biol. 2020, 36, 101600. [Google Scholar] [CrossRef]
- Yang, W.; Wang, Y.; Zhang, C.; Huang, Y.; Yu, J.; Shi, L.; Zhang, P.; Yin, Y.; Li, R.; Tao, K. Maresin1 Protect Against Ferroptosis-Induced Liver Injury Through ROS Inhibition and Nrf2/HO-1/GPX4 Activation. Front. Pharmacol. 2022, 13, 865689. [Google Scholar] [CrossRef] [PubMed]
- Silva-Llanes, I.; Shin, C.H.; Jimenez-Villegas, J.; Gorospe, M.; Lastres-Becker, I. The Transcription Factor NRF2 Has Epigenetic Regulatory Functions Modulating HDACs, DNMTs, and miRNA Biogenesis. Antioxidants 2023, 12, 641. [Google Scholar] [CrossRef] [PubMed]
- Kowaltowski, A.J.; de Souza-Pinto, N.C.; Castilho, R.F.; Vercesi, A.E. Mitochondria and reactive oxygen species. Free Radic. Biol. Med. 2009, 47, 333–343. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.Z.; Jiang, S.; Zhang, L.; Yu, Z.B. Mitochondrial electron transport chain, ROS generation and uncoupling (Review). Int. J. Mol. Med. 2019, 44, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Suski, J.M.; Lebiedzinska, M.; Bonora, M.; Pinton, P.; Duszynski, J.; Wieckowski, M.R. Relation between mitochondrial membrane potential and ROS formation. Methods Mol. Biol. 2012, 810, 183–205. [Google Scholar] [CrossRef] [PubMed]
- Bhatti, J.S.; Bhatti, G.K.; Reddy, P.H. Mitochondrial dysfunction and oxidative stress in metabolic disorders—A step towards mitochondria based therapeutic strategies. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1066–1077. [Google Scholar] [CrossRef]
- Xiang, L.; Shao, Y.; Chen, Y. Mitochondrial dysfunction and mitochondrion-targeted therapeutics in liver diseases. J. Drug Target. 2021, 29, 1080–1093. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Zhao, Y.; Yu, M.; Qin, J.; Ye, B.; Wang, Q. Mitochondrial Dysfunction and Chronic Liver Disease. Curr. Issues Mol. Biol. 2022, 44, 3156–3165. [Google Scholar] [CrossRef]
- Koliaki, C.; Roden, M. Hepatic energy metabolism in human diabetes mellitus, obesity and non-alcoholic fatty liver disease. Mol. Cell. Endocrinol. 2013, 379, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Passarella, S.; Schurr, A.; Portincasa, P. Mitochondrial Transport in Glycolysis and Gluconeogenesis: Achievements and Perspectives. Int. J. Mol. Sci. 2021, 22, 12620. [Google Scholar] [CrossRef]
- Stark, R.; Guebre-Egziabher, F.; Zhao, X.; Feriod, C.; Dong, J.; Alves, T.C.; Ioja, S.; Pongratz, R.L.; Bhanot, S.; Roden, M.; et al. A role for mitochondrial phosphoenolpyruvate carboxykinase (PEPCK-M) in the regulation of hepatic gluconeogenesis. J. Biol. Chem. 2014, 289, 7257–7263. [Google Scholar] [CrossRef]
- Sunny, N.E.; Parks, E.J.; Browning, J.D.; Burgess, S.C. Excessive hepatic mitochondrial TCA cycle and gluconeogenesis in humans with nonalcoholic fatty liver disease. Cell Metab. 2011, 14, 804–810. [Google Scholar] [CrossRef] [PubMed]
- Moore, M.P.; Cunningham, R.P.; Meers, G.M.; Johnson, S.A.; Wheeler, A.A.; Ganga, R.R.; Spencer, N.M.; Pitt, J.B.; Diaz-Arias, A.; Swi, A.I.A.; et al. Compromised hepatic mitochondrial fatty acid oxidation and reduced markers of mitochondrial turnover in human NAFLD. Hepatology 2022, 76, 1452–1465. [Google Scholar] [CrossRef]
- Guerra, I.M.S.; Ferreira, H.B.; Melo, T.; Rocha, H.; Moreira, S.; Diogo, L.; Domingues, M.R.; Moreira, A.S.P. Mitochondrial Fatty Acid beta-Oxidation Disorders: From Disease to Lipidomic Studies-A Critical Review. Int. J. Mol. Sci. 2022, 23, 13933. [Google Scholar] [CrossRef] [PubMed]
- Barbier-Torres, L.; Fortner, K.A.; Iruzubieta, P.; Delgado, T.C.; Giddings, E.; Chen, Y.; Champagne, D.; Fernandez-Ramos, D.; Mestre, D.; Gomez-Santos, B.; et al. Silencing hepatic MCJ attenuates non-alcoholic fatty liver disease (NAFLD) by increasing mitochondrial fatty acid oxidation. Nat. Commun. 2020, 11, 3360. [Google Scholar] [CrossRef] [PubMed]
- Fang, E.F.; Waltz, T.B.; Kassahun, H.; Lu, Q.; Kerr, J.S.; Morevati, M.; Fivenson, E.M.; Wollman, B.N.; Marosi, K.; Wilson, M.A.; et al. Tomatidine enhances lifespan and healthspan in C. elegans through mitophagy induction via the SKN-1/Nrf2 pathway. Sci. Rep. 2017, 7, 46208. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Feng, Y.F.; Liu, X.T.; Li, Y.C.; Zhu, H.M.; Sun, M.R.; Li, P.; Liu, B.; Yang, H. Songorine promotes cardiac mitochondrial biogenesis via Nrf2 induction during sepsis. Redox Biol. 2021, 38, 101771. [Google Scholar] [CrossRef]
- Schofield, J.H.; Schafer, Z.T. Mitochondrial Reactive Oxygen Species and Mitophagy: A Complex and Nuanced Relationship. Antioxid. Redox Signal. 2021, 34, 517–530. [Google Scholar] [CrossRef]
- Mao, Y.; Du, J.; Chen, X.; Al Mamun, A.; Cao, L.; Yang, Y.; Mubwandarikwa, J.; Zaeem, M.; Zhang, W.; Chen, Y.; et al. Maltol Promotes Mitophagy and Inhibits Oxidative Stress via the Nrf2/PINK1/Parkin Pathway after Spinal Cord Injury. Oxid. Med. Cell. Longev. 2022, 2022, 1337630. [Google Scholar] [CrossRef]
- Xiao, L.; Xu, X.; Zhang, F.; Wang, M.; Xu, Y.; Tang, D.; Wang, J.; Qin, Y.; Liu, Y.; Tang, C.; et al. The mitochondria-targeted antioxidant MitoQ ameliorated tubular injury mediated by mitophagy in diabetic kidney disease via Nrf2/PINK1. Redox Biol. 2017, 11, 297–311. [Google Scholar] [CrossRef] [PubMed]
- Gureev, A.P.; Shaforostova, E.A.; Popov, V.N. Regulation of Mitochondrial Biogenesis as a Way for Active Longevity: Interaction Between the Nrf2 and PGC-1alpha Signaling Pathways. Front. Genet. 2019, 10, 435. [Google Scholar] [CrossRef]
- Hayashi, G.; Jasoliya, M.; Sahdeo, S.; Sacca, F.; Pane, C.; Filla, A.; Marsili, A.; Puorro, G.; Lanzillo, R.; Brescia Morra, V.; et al. Dimethyl fumarate mediates Nrf2-dependent mitochondrial biogenesis in mice and humans. Hum. Mol. Genet. 2017, 26, 2864–2873. [Google Scholar] [CrossRef]
- Palikaras, K.; Tavernarakis, N. Mitochondrial homeostasis: The interplay between mitophagy and mitochondrial biogenesis. Exp. Gerontol. 2014, 56, 182–188. [Google Scholar] [CrossRef]
- Grattagliano, I.; Montezinho, L.P.; Oliveira, P.J.; Fruhbeck, G.; Gomez-Ambrosi, J.; Montecucco, F.; Carbone, F.; Wieckowski, M.R.; Wang, D.Q.; Portincasa, P. Targeting mitochondria to oppose the progression of nonalcoholic fatty liver disease. Biochem. Pharmacol. 2019, 160, 34–45. [Google Scholar] [CrossRef]
- Serviddio, G.; Bellanti, F.; Sastre, J.; Vendemiale, G.; Altomare, E. Targeting mitochondria: A new promising approach for the treatment of liver diseases. Curr. Med. Chem. 2010, 17, 2325–2337. [Google Scholar] [CrossRef] [PubMed]
- Nassir, F.; Ibdah, J.A. Role of mitochondria in alcoholic liver disease. World J. Gastroenterol. 2014, 20, 2136–2142. [Google Scholar] [CrossRef] [PubMed]
- Yu, A.; Zhou, R.; Xia, B.; Dang, W.; Yang, Z.; Chen, X. NAMPT maintains mitochondria content via NRF2-PPARalpha/AMPKalpha pathway to promote cell survival under oxidative stress. Cell. Signal. 2020, 66, 109496. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Zhu, T.; Zhou, P.; Xu, H.; Meng, X.; Ding, T.; Nan, F.; Sun, G.; Sun, X. Notoginseng leaf triterpenes ameliorates mitochondrial oxidative injury via the NAMPT-SIRT1/2/3 signaling pathways in cerebral ischemic model rats. J. Ginseng Res. 2023, 47, 199–209. [Google Scholar] [CrossRef]
- Gabande-Rodriguez, E.; Gomez de Las Heras, M.M.; Mittelbrunn, M. Control of Inflammation by Calorie Restriction Mimetics: On the Crossroad of Autophagy and Mitochondria. Cells 2019, 9, 82. [Google Scholar] [CrossRef]
- Wang, S.; Wan, T.; Ye, M.; Qiu, Y.; Pei, L.; Jiang, R.; Pang, N.; Huang, Y.; Liang, B.; Ling, W.; et al. Nicotinamide riboside attenuates alcohol induced liver injuries via activation of SirT1/PGC-1alpha/mitochondrial biosynthesis pathway. Redox Biol. 2018, 17, 89–98. [Google Scholar] [CrossRef]
- Ryoo, I.G.; Kwak, M.K. Regulatory crosstalk between the oxidative stress-related transcription factor Nfe2l2/Nrf2 and mitochondria. Toxicol. Appl. Pharmacol. 2018, 359, 24–33. [Google Scholar] [CrossRef]
- Cardin, R.; Piciocchi, M.; Bortolami, M.; Kotsafti, A.; Barzon, L.; Lavezzo, E.; Sinigaglia, A.; Rodriguez-Castro, K.I.; Rugge, M.; Farinati, F. Oxidative damage in the progression of chronic liver disease to hepatocellular carcinoma: An intricate pathway. World J. Gastroenterol. 2014, 20, 3078–3086. [Google Scholar] [CrossRef]
- Li, S.; Tan, H.Y.; Wang, N.; Zhang, Z.J.; Lao, L.; Wong, C.W.; Feng, Y. The Role of Oxidative Stress and Antioxidants in Liver Diseases. Int. J. Mol. Sci. 2015, 16, 26087–26124. [Google Scholar] [CrossRef]
- Bataille, A.M.; Manautou, J.E. Nrf2: A potential target for new therapeutics in liver disease. Clin. Pharmacol. Ther. 2012, 92, 340–348. [Google Scholar] [CrossRef] [PubMed]
- Okada, K.; Shoda, J.; Taguchi, K.; Maher, J.M.; Ishizaki, K.; Inoue, Y.; Ohtsuki, M.; Goto, N.; Takeda, K.; Utsunomiya, H.; et al. Ursodeoxycholic acid stimulates Nrf2-mediated hepatocellular transport, detoxification, and antioxidative stress systems in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 295, G735–G747. [Google Scholar] [CrossRef]
- Cuadrado, A.; Rojo, A.I.; Wells, G.; Hayes, J.D.; Cousin, S.P.; Rumsey, W.L.; Attucks, O.C.; Franklin, S.; Levonen, A.L.; Kensler, T.W.; et al. Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases. Nat. Rev. Drug Discov. 2019, 18, 295–317. [Google Scholar] [CrossRef] [PubMed]
- Boyenle, I.D.; Divine, U.C.; Adeyemi, R.; Ayinde, K.S.; Olaoba, O.T.; Apu, C.; Du, L.; Lu, Q.; Yin, X.; Adelusi, T.I. Direct Keap1-kelch inhibitors as potential drug candidates for oxidative stress-orchestrated diseases: A review on In silico perspective. Pharmacol. Res. 2021, 167, 105577. [Google Scholar] [CrossRef] [PubMed]
- Madden, S.K.; Itzhaki, L.S. Structural and mechanistic insights into the Keap1-Nrf2 system as a route to drug discovery. Biochim. Biophys. Acta Proteins Proteom. 2020, 1868, 140405. [Google Scholar] [CrossRef]
- Dayalan Naidu, S.; Dinkova-Kostova, A.T. KEAP1, a cysteine-based sensor and a drug target for the prevention and treatment of chronic disease. Open Biol. 2020, 10, 200105. [Google Scholar] [CrossRef] [PubMed]
- Kawata, K.; Kobayashi, Y.; Souda, K.; Kawamura, K.; Sumiyoshi, S.; Takahashi, Y.; Noritake, H.; Watanabe, S.; Suehiro, T.; Nakamura, H. Enhanced hepatic Nrf2 activation after ursodeoxycholic acid treatment in patients with primary biliary cirrhosis. Antioxid. Redox Signal. 2010, 13, 259–268. [Google Scholar] [CrossRef]
- Angulo, P. Use of ursodeoxycholic acid in patients with liver disease. Curr. Gastroenterol. Rep. 2002, 4, 37–44. [Google Scholar] [CrossRef]
- Yu, Z.; Shao, W.; Chiang, Y.; Foltz, W.; Zhang, Z.; Ling, W.; Fantus, I.G.; Jin, T. Oltipraz upregulates the nuclear factor (erythroid-derived 2)-like 2 [corrected](NRF2) antioxidant system and prevents insulin resistance and obesity induced by a high-fat diet in C57BL/6J mice. Diabetologia 2011, 54, 922–934. [Google Scholar] [CrossRef]
- Kim, W.; Kim, B.G.; Lee, J.S.; Lee, C.K.; Yeon, J.E.; Chang, M.S.; Kim, J.H.; Kim, H.; Yi, S.; Lee, J.; et al. Randomised clinical trial: The efficacy and safety of oltipraz, a liver X receptor alpha-inhibitory dithiolethione in patients with non-alcoholic fatty liver disease. Aliment. Pharmacol. Ther. 2017, 45, 1073–1083. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.G.; Kim, Y.M.; Choi, J.Y.; Han, J.Y.; Jang, J.W.; Cho, S.H.; Um, S.H.; Chon, C.Y.; Lee, D.H.; Jang, J.J.; et al. Oltipraz therapy in patients with liver fibrosis or cirrhosis: A randomized, double-blind, placebo-controlled phase II trial. J. Pharm. Pharmacol. 2011, 63, 627–635. [Google Scholar] [CrossRef]
- Faghihzadeh, F.; Adibi, P.; Hekmatdoost, A. The effects of resveratrol supplementation on cardiovascular risk factors in patients with non-alcoholic fatty liver disease: A randomised, double-blind, placebo-controlled study. Br. J. Nutr. 2015, 114, 796–803. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhao, X.; Ran, L.; Wan, J.; Wang, X.; Qin, Y.; Shu, F.; Gao, Y.; Yuan, L.; Zhang, Q.; et al. Resveratrol improves insulin resistance, glucose and lipid metabolism in patients with non-alcoholic fatty liver disease: A randomized controlled trial. Dig. Liver Dis. 2015, 47, 226–232. [Google Scholar] [CrossRef]
- Heeboll, S.; Kreuzfeldt, M.; Hamilton-Dutoit, S.; Kjaer Poulsen, M.; Stodkilde-Jorgensen, H.; Moller, H.J.; Jessen, N.; Thorsen, K.; Kristina Hellberg, Y.; Bonlokke Pedersen, S.; et al. Placebo-controlled, randomised clinical trial: High-dose resveratrol treatment for non-alcoholic fatty liver disease. Scand. J. Gastroenterol. 2016, 51, 456–464. [Google Scholar] [CrossRef]
- Chachay, V.S.; Macdonald, G.A.; Martin, J.H.; Whitehead, J.P.; O’Moore-Sullivan, T.M.; Lee, P.; Franklin, M.; Klein, K.; Taylor, P.J.; Ferguson, M.; et al. Resveratrol does not benefit patients with nonalcoholic fatty liver disease. Clin. Gastroenterol. Hepatol. 2014, 12, 2092–2103. [Google Scholar] [CrossRef]
- Faghihzadeh, F.; Adibi, P.; Rafiei, R.; Hekmatdoost, A. Resveratrol supplementation improves inflammatory biomarkers in patients with nonalcoholic fatty liver disease. Nutr. Res. 2014, 34, 837–843. [Google Scholar] [CrossRef]
- Timmers, S.; Hesselink, M.K.; Schrauwen, P. Therapeutic potential of resveratrol in obesity and type 2 diabetes: New avenues for health benefits? Ann. N. Y. Acad. Sci. 2013, 1290, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Poulsen, M.M.; Vestergaard, P.F.; Clasen, B.F.; Radko, Y.; Christensen, L.P.; Stodkilde-Jorgensen, H.; Moller, N.; Jessen, N.; Pedersen, S.B.; Jorgensen, J.O. High-dose resveratrol supplementation in obese men: An investigator-initiated, randomized, placebo-controlled clinical trial of substrate metabolism, insulin sensitivity, and body composition. Diabetes 2013, 62, 1186–1195. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Yin, X.; Sui, S. Resveratrol inhibited the progression of human hepatocellular carcinoma by inducing autophagy via regulating p53 and the phosphoinositide 3-kinase/protein kinase B pathway. Oncol. Rep. 2018, 40, 2758–2765. [Google Scholar] [CrossRef]
- Farzaei, M.H.; Zobeiri, M.; Parvizi, F.; El-Senduny, F.F.; Marmouzi, I.; Coy-Barrera, E.; Naseri, R.; Nabavi, S.M.; Rahimi, R.; Abdollahi, M. Curcumin in Liver Diseases: A Systematic Review of the Cellular Mechanisms of Oxidative Stress and Clinical Perspective. Nutrients 2018, 10, 855. [Google Scholar] [CrossRef]
- Panahi, Y.; Kianpour, P.; Mohtashami, R.; Jafari, R.; Simental-Mendia, L.E.; Sahebkar, A. Efficacy and Safety of Phytosomal Curcumin in Non-Alcoholic Fatty Liver Disease: A Randomized Controlled Trial. Drug Res. 2017, 67, 244–251. [Google Scholar] [CrossRef]
- Rahmani, S.; Asgary, S.; Askari, G.; Keshvari, M.; Hatamipour, M.; Feizi, A.; Sahebkar, A. Treatment of Non-alcoholic Fatty Liver Disease with Curcumin: A Randomized Placebo-controlled Trial. Phytother. Res. 2016, 30, 1540–1548. [Google Scholar] [CrossRef]
- Rong, S.; Zhao, Y.; Bao, W.; Xiao, X.; Wang, D.; Nussler, A.K.; Yan, H.; Yao, P.; Liu, L. Curcumin prevents chronic alcohol-induced liver disease involving decreasing ROS generation and enhancing antioxidative capacity. Phytomedicine 2012, 19, 545–550. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Ding, C.; Zhang, G.; Pan, R.; Liu, Y.; Huang, N.; Hou, N.; Han, F.; Xu, W.; Sun, X. Liraglutide ameliorates obesity-related nonalcoholic fatty liver disease by regulating Sestrin2-mediated Nrf2/HO-1 pathway. Biochem. Biophys. Res. Commun. 2020, 525, 895–901. [Google Scholar] [CrossRef]
- Petit, J.M.; Cercueil, J.P.; Loffroy, R.; Denimal, D.; Bouillet, B.; Fourmont, C.; Chevallier, O.; Duvillard, L.; Verges, B. Effect of Liraglutide Therapy on Liver Fat Content in Patients With Inadequately Controlled Type 2 Diabetes: The Lira-NAFLD Study. J. Clin. Endocrinol. Metab. 2017, 102, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Song, T.; Jia, Y.; Li, Z.; Wang, F.; Ren, L.; Chen, S. Effects of Liraglutide on Nonalcoholic Fatty Liver Disease in Patients with Type 2 Diabetes Mellitus: A Systematic Review and Meta-Analysis. Diabetes Ther. 2021, 12, 1735–1749. [Google Scholar] [CrossRef]
- Chan, B.K.Y.; Elmasry, M.; Forootan, S.S.; Russomanno, G.; Bunday, T.M.; Zhang, F.; Brillant, N.; Starkey Lewis, P.J.; Aird, R.; Ricci, E.; et al. Pharmacological Activation of Nrf2 Enhances Functional Liver Regeneration. Hepatology 2021, 74, 973–986. [Google Scholar] [CrossRef] [PubMed]
- Camer, D.; Yu, Y.; Szabo, A.; Dinh, C.H.; Wang, H.; Cheng, L.; Huang, X.F. Bardoxolone methyl prevents insulin resistance and the development of hepatic steatosis in mice fed a high-fat diet. Mol. Cell. Endocrinol. 2015, 412, 36–43. [Google Scholar] [CrossRef]
- Hong, D.S.; Kurzrock, R.; Supko, J.G.; He, X.; Naing, A.; Wheler, J.; Lawrence, D.; Eder, J.P.; Meyer, C.J.; Ferguson, D.A.; et al. A phase I first-in-human trial of bardoxolone methyl in patients with advanced solid tumors and lymphomas. Clin. Cancer Res. 2012, 18, 3396–3406. [Google Scholar] [CrossRef]
- Ruggenenti, P. The CARDINAL Trial of Bardoxolone Methyl in Alport Syndrome: When Marketing Interests Prevail over Patients Clinical Needs. Nephron 2023, 147, 465–469. [Google Scholar] [CrossRef] [PubMed]
- Reisman, S.A.; Ferguson, D.A.; Lee, C.I.; Proksch, J.W. Omaveloxolone and TX63682 are hepatoprotective in the STAM mouse model of nonalcoholic steatohepatitis. J. Biochem. Mol. Toxicol. 2020, 34, e22526. [Google Scholar] [CrossRef] [PubMed]
- Lynch, D.R.; Chin, M.P.; Delatycki, M.B.; Subramony, S.H.; Corti, M.; Hoyle, J.C.; Boesch, S.; Nachbauer, W.; Mariotti, C.; Mathews, K.D.; et al. Safety and Efficacy of Omaveloxolone in Friedreich Ataxia (MOXIe Study). Ann. Neurol. 2021, 89, 212–225. [Google Scholar] [CrossRef] [PubMed]
- Reisman, S.A.; Gahir, S.S.; Lee, C.I.; Proksch, J.W.; Sakamoto, M.; Ward, K.W. Pharmacokinetics and pharmacodynamics of the novel Nrf2 activator omaveloxolone in primates. Drug Des. Devel Ther. 2019, 13, 1259–1270. [Google Scholar] [CrossRef]
- Tang, W.; Jiang, Y.F.; Ponnusamy, M.; Diallo, M. Role of Nrf2 in chronic liver disease. World J. Gastroenterol. 2014, 20, 13079–13087. [Google Scholar] [CrossRef] [PubMed]
- Kamisako, T.; Tanaka, Y. Oltipraz ameliorates the progression of steatohepatitis in Nrf2-null mice fed a high-fat diet. J. Clin. Biochem. Nutr. 2022, 70, 147–153. [Google Scholar] [CrossRef]
- Lu, M.C.; Ji, J.A.; Jiang, Z.Y.; You, Q.D. The Keap1-Nrf2-ARE Pathway As a Potential Preventive and Therapeutic Target: An Update. Med. Res. Rev. 2016, 36, 924–963. [Google Scholar] [CrossRef]
- Pergola, P.E.; Raskin, P.; Toto, R.D.; Meyer, C.J.; Huff, J.W.; Grossman, E.B.; Krauth, M.; Ruiz, S.; Audhya, P.; Christ-Schmidt, H.; et al. Bardoxolone methyl and kidney function in CKD with type 2 diabetes. N. Engl. J. Med. 2011, 365, 327–336. [Google Scholar] [CrossRef]
- Poornashree, M.; Kumar, H.; Ajmeer, R.; Jain, R.; Jain, V. Dual role of Nrf2 in cancer: Molecular mechanisms, cellular functions and therapeutic interventions. Mol. Biol. Rep. 2023, 50, 1871–1883. [Google Scholar] [CrossRef]
- Wheeler, M.D. Endotoxin and Kupffer cell activation in alcoholic liver disease. Alcohol. Res. Health 2003, 27, 300–306. [Google Scholar]
- Ma-On, C.; Sanpavat, A.; Whongsiri, P.; Suwannasin, S.; Hirankarn, N.; Tangkijvanich, P.; Boonla, C. Oxidative stress indicated by elevated expression of Nrf2 and 8-OHdG promotes hepatocellular carcinoma progression. Med. Oncol. 2017, 34, 57. [Google Scholar] [CrossRef]
- Raghunath, A.; Sundarraj, K.; Arfuso, F.; Sethi, G.; Perumal, E. Dysregulation of Nrf2 in Hepatocellular Carcinoma: Role in Cancer Progression and Chemoresistance. Cancers 2018, 10, 481. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Gomez, M.; Kwak, M.K.; Dolan, P.M.; Itoh, K.; Yamamoto, M.; Talalay, P.; Kensler, T.W. Sensitivity to carcinogenesis is increased and chemoprotective efficacy of enzyme inducers is lost in nrf2 transcription factor-deficient mice. Proc. Natl. Acad. Sci. USA 2001, 98, 3410–3415. [Google Scholar] [CrossRef] [PubMed]
- Haque, E.; Karim, M.R.; Salam Teeli, A.; Smiech, M.; Leszczynski, P.; Winiarczyk, D.; Parvanov, E.D.; Atanasov, A.G.; Taniguchi, H. Molecular Mechanisms Underlying Hepatocellular Carcinoma Induction by Aberrant NRF2 Activation-Mediated Transcription Networks: Interaction of NRF2-KEAP1 Controls the Fate of Hepatocarcinogenesis. Int. J. Mol. Sci. 2020, 21, 5378. [Google Scholar] [CrossRef]
- Li, L.; Fu, J.; Liu, D.; Sun, J.; Hou, Y.; Chen, C.; Shao, J.; Wang, L.; Wang, X.; Zhao, R.; et al. Hepatocyte-specific Nrf2 deficiency mitigates high-fat diet-induced hepatic steatosis: Involvement of reduced PPARgamma expression. Redox Biol. 2020, 30, 101412. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Compound | Type | Mechanism of Action | Clinical Trial | Registration Number |
|---|---|---|---|---|
| Bardoxolone | Synthetic Triterpenoid | Keap1 Modification | Phase I | NCT01563562 |
| Omaveloxolone (RTA-408) | Synthetic Triterpenoid | Keap1 Modification | Phase I | NCT03902002 |
| Oltipraz | Synthetic Dithiolethione | Keap1 Modification | Phase II | NCT00956098 |
| Liraglutide | Synthetic Peptide | Keap1 Modification | Phase II | NCT01237119 |
| Ursodiol | Bile Acid | Keap1 Modification | Phase IV | NCT05849558 |
| Resveratrol | Nonflavonoid Polyphenol | Keap1 Modification | Phase II | NCT02216552 |
| Curcumin | Diarylheptanoid | Keap1 Modification | Phase II | NCT04109742 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, J.-S.; Rustamov, N.; Roh, Y.-S. The Roles of NFR2-Regulated Oxidative Stress and Mitochondrial Quality Control in Chronic Liver Diseases. Antioxidants 2023, 12, 1928. https://doi.org/10.3390/antiox12111928
Park J-S, Rustamov N, Roh Y-S. The Roles of NFR2-Regulated Oxidative Stress and Mitochondrial Quality Control in Chronic Liver Diseases. Antioxidants. 2023; 12(11):1928. https://doi.org/10.3390/antiox12111928
Chicago/Turabian StylePark, Jeong-Su, Nodir Rustamov, and Yoon-Seok Roh. 2023. "The Roles of NFR2-Regulated Oxidative Stress and Mitochondrial Quality Control in Chronic Liver Diseases" Antioxidants 12, no. 11: 1928. https://doi.org/10.3390/antiox12111928