
Hydralazine Revives Cellular and Ocular Lens Health-Span by Ameliorating the Aging and Oxidative-Dependent Loss of the Nrf2-Activated Cellular Stress Response


Abstract
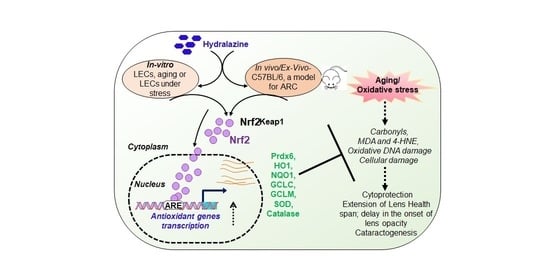
:
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.1.1. SRA-hLECs (Human Lens Epithelial Cells)
2.1.2. Isolation of LECs from the Lenses of Human Subjects and Maintenance
2.1.3. Generation of Mouse Lens Epithelial Cells (mLECs) and Primary mLECs
2.2. Cell Viability Assay
2.3. Measurement of Reactive Oxygen Species (ROS)
2.4. Measurement of the Oxidative Effect by Lipid Peroxidation Assay
2.5. Protein Carbonyl Assay
2.6. Measurement of 8-Hydroxydeoxyguanosine
2.7. Nrf2/ARE Driven Luciferase Reporter Assay
2.8. Protein Isolation and Expression Analysis
2.9. Quantitative Real-Time PCR
2.10. Assessment of Phospholipase A2 (PLA2) Activity
2.11. Estimation of Glutathione (GSH) Peroxidase Activity
2.12. Quantitation of Superoxide Dismutase (SOD) Activity
2.13. Quantitation of Catalase Activity
2.14. Isolation of Cytosol and Nuclear Protein Fractions
2.15. Determination of Nrf2-DNA (ARE) Binding Activity
2.16. Chromatin Immunoprecipitation (ChIP) Assay (In Vivo DNA Binding Assay)
2.17. Preparation of Prdx6 Promoter-Fused to Chloramphenicol Acetyltransferase (CAT) Reporter Vector
2.18. Nrf2 Knock down Experiment
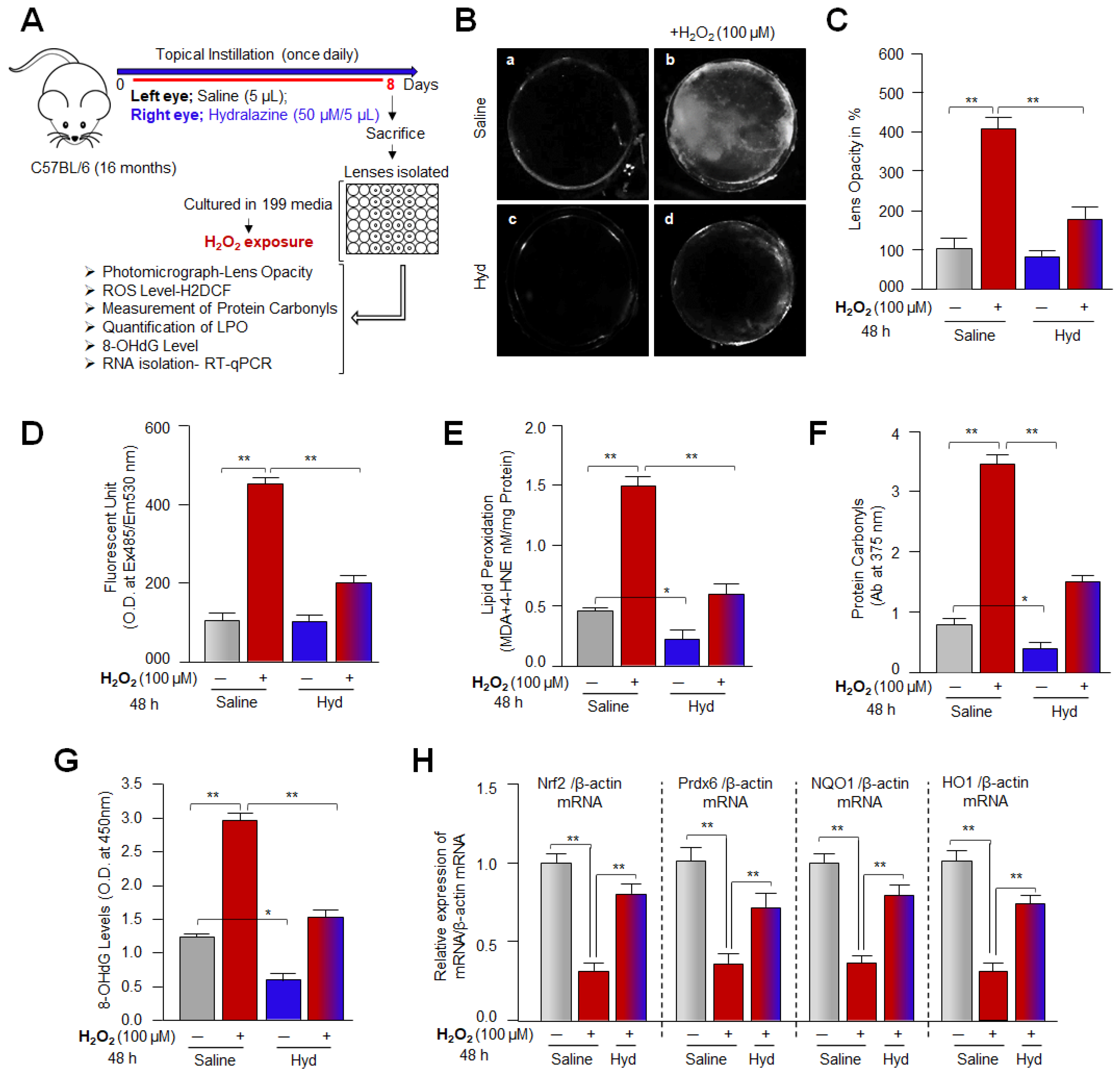
2.19. Lens Organ Culture and H2O2 Treatment
2.20. Statistical Analysis
3. Results
3.1. Hyd Efficaciously Rescued Lens Epithelial Cells under Oxidative Stress
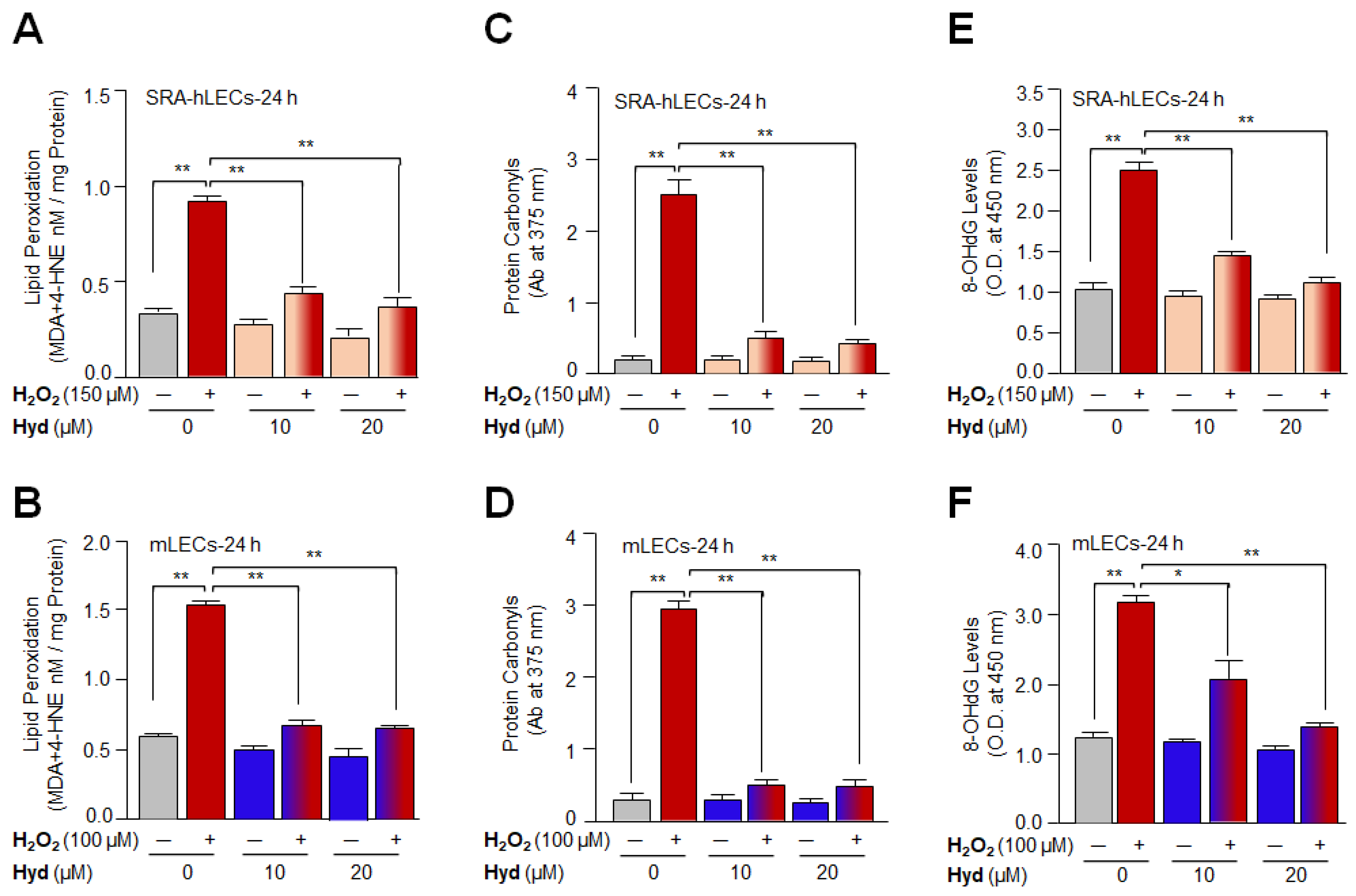
3.2. Hyd Efficaciously Blunted Oxidative Stress-Driven Aberrant Cellular Levels of Lipid Peroxidation and Rescued Oxidative Protein and DNA
3.3. Hyd Activated the Nrf2/ARE-Driven Luciferase Activity in LECs
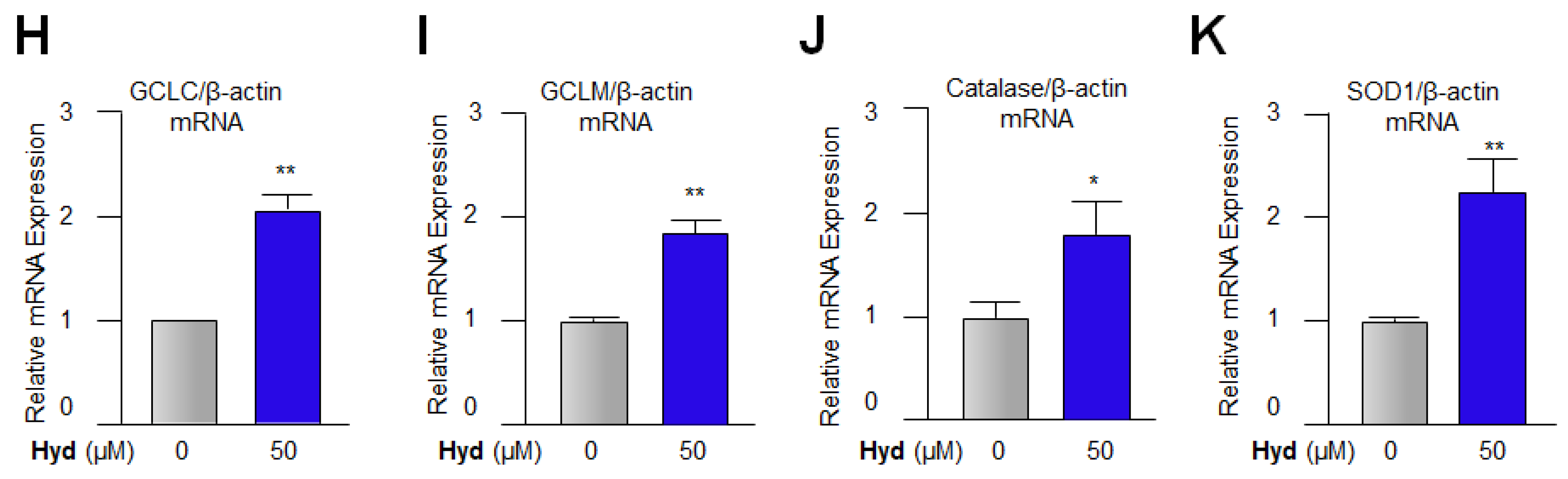
3.4. Hyd Amplified the Antioxidants Expression via Activating Nrf2/ARE Pathways in LECs
3.5. Hyd Treatment Enhanced the Enzymatic Activities of Antioxidant Genes
3.6. Hyd Triggered the Nuclear Accumulation of Nrf2 and Augmented the Nrf2 Activity in Concentration-Dependent Fashion
3.7. In Vivo DNA Binding and Transactivation Assays Disclosed That Hyd Activated Antioxidant Transcription via Nrf2/ARE Mechanism
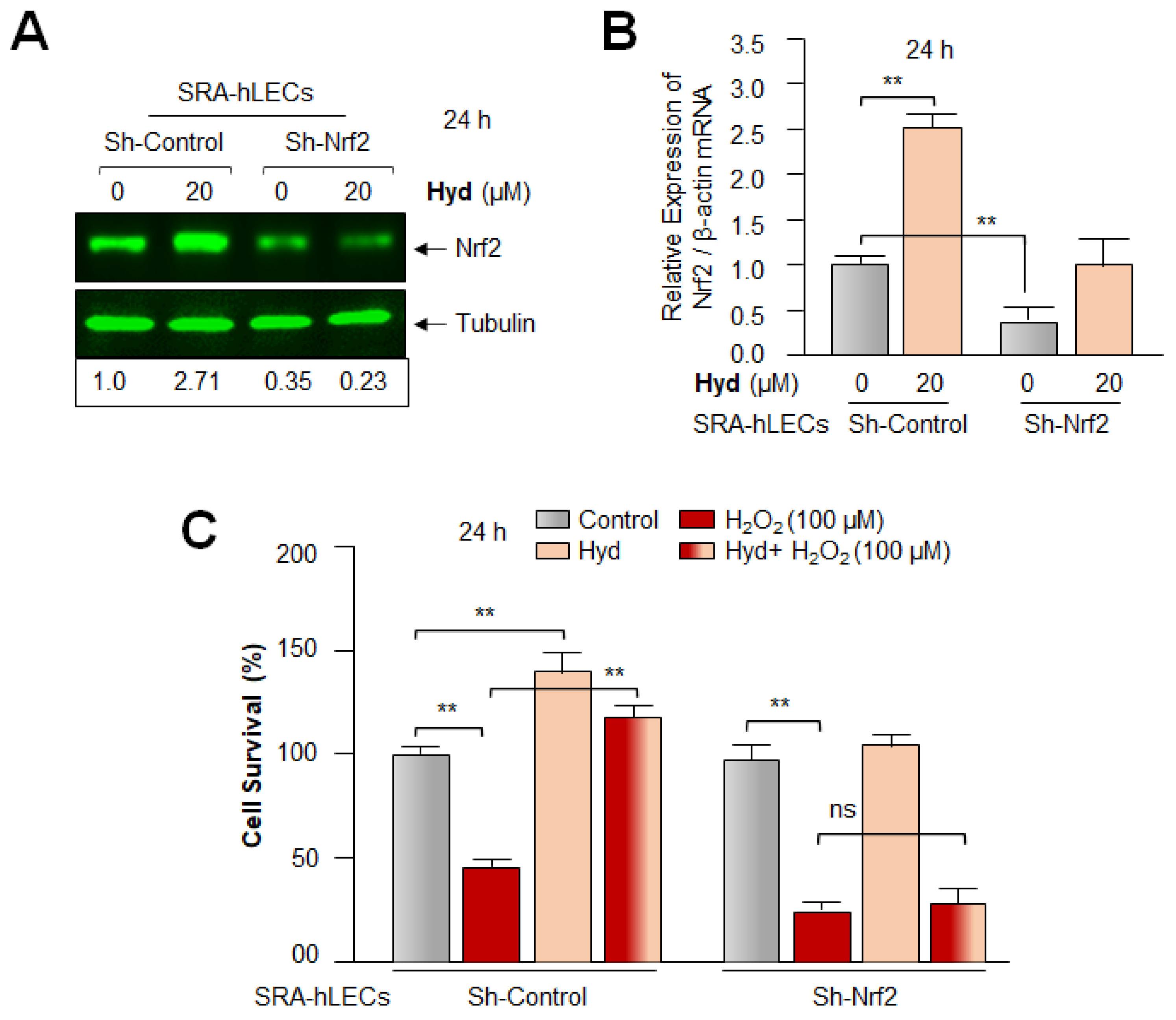
3.8. Hyd Failed to Rescue the Nrf2-Deficient SRA-hLECs against H2O2-Induced Toxicity
3.9. Hyd Amplified Nrf2 and Its Target Expression and Protected the Primary mLECs against H2O2-Induced Death by Mitigating ROS Accumulation
3.10. Hyd, When Instilled Topically in Mouse Eye, Enhanced Nrf2/ARE-Mediated Antioxidant Gene In Vivo
3.11. Topical Application of Hyd Reduced the ROS Generation and Augmented Nrf2 and Its Antioxidant Genes Expression in Aged Mouse Lenses In Vivo
3.12. Hyd’s Topical Instillation in Mouse Eye Optimized H2O2-Induced ROS Levels and Extended Lenses Health Span by Preventing/Delaying Lens Opacity against Oxidative Stress Ex-Vivo
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Finkel, T.; Holbrook, N.J. Oxidants, oxidative stress and the biology of ageing. Nature 2000, 408, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Chhunchha, B.; Kubo, E.; Singh, D.P. Switching of Redox Signaling by Prdx6 Expression Decides Cellular Fate by Hormetic Phenomena Involving Nrf2 and Reactive Oxygen Species. Cells 2022, 11, 1266. [Google Scholar] [CrossRef] [PubMed]
- Chhunchha, B.; Kubo, E.; Singh, D.P. Obligatory Role of AMPK Activation and Antioxidant Defense Pathway in the Regulatory Effects of Metformin on Cellular Protection and Prevention of Lens Opacity. Cells 2022, 11, 3021. [Google Scholar] [CrossRef]
- Zucker, S.N.; Fink, E.E.; Bagati, A.; Mannava, S.; Bianchi-Smiraglia, A.; Bogner, P.N.; Wawrzyniak, J.A.; Foley, C.; Leonova, K.I.; Grimm, M.J.; et al. Nrf2 amplifies oxidative stress via induction of Klf9. Mol. Cell 2014, 53, 916–928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chhunchha, B.; Fatma, N.; Kubo, E.; Singh, D.P. Aberrant sumoylation signaling evoked by reactive oxygen species impairs protective function of Prdx6 by destabilization and repression of its transcription. FEBS J. 2014, 281, 3357–3381. [Google Scholar] [CrossRef] [Green Version]
- Fatma, N.; Singh, P.; Chhunchha, B.; Kubo, E.; Shinohara, T.; Bhargavan, B.; Singh, D.P. Deficiency of Prdx6 in lens epithelial cells induces ER stress response-mediated impaired homeostasis and apoptosis. Am. J. Physiol. Cell Physiol. 2011, 301, C954–C967. [Google Scholar] [CrossRef]
- Jacob, K.D.; Noren Hooten, N.; Tadokoro, T.; Lohani, A.; Barnes, J.; Evans, M.K. Alzheimer’s disease-associated polymorphisms in human OGG1 alter catalytic activity and sensitize cells to DNA damage. Free Radic. Biol. Med. 2013, 63, 115–125. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Davies, K.J.A.; Forman, H.J. Oxidative stress response and Nrf2 signaling in aging. Free Radic. Biol. Med. 2015, 88, 314–336. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Unnikrishnan, A.; Deepa, S.S.; Liu, Y.; Li, Y.; Ikeno, Y.; Sosnowska, D.; Van Remmen, H.; Richardson, A. A new role for oxidative stress in aging: The accelerated aging phenotype in Sod1(-/)(-) mice is correlated to increased cellular senescence. Redox Biol. 2017, 11, 30–37. [Google Scholar] [CrossRef] [Green Version]
- Chhunchha, B.; Kubo, E.; Singh, D.P. Sulforaphane-Induced Klf9/Prdx6 Axis Acts as a Molecular Switch to Control Redox Signaling and Determines Fate of Cells. Cells 2019, 8, 1159. [Google Scholar] [CrossRef]
- Chhunchha, B.; Singh, P.; Stamer, W.D.; Singh, D.P. Prdx6 retards senescence and restores trabecular meshwork cell health by regulating reactive oxygen species. Cell Death Discov. 2017, 3, 17060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Liu, H.; Davies, K.J.; Sioutas, C.; Finch, C.E.; Morgan, T.E.; Forman, H.J. Nrf2-regulated phase II enzymes are induced by chronic ambient nanoparticle exposure in young mice with age-related impairments. Free Radic. Biol. Med. 2012, 52, 2038–2046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaspar, J.W.; Jaiswal, A.K. Antioxidant-induced phosphorylation of tyrosine 486 leads to rapid nuclear export of Bach1 that allows Nrf2 to bind to the antioxidant response element and activate defensive gene expression. J. Biol. Chem. 2010, 285, 153–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niture, S.K.; Kaspar, J.W.; Shen, J.; Jaiswal, A.K. Nrf2 signaling and cell survival. Toxicol. Appl. Pharmacol. 2010, 244, 37–42. [Google Scholar] [CrossRef] [Green Version]
- Bokov, A.; Chaudhuri, A.; Richardson, A. The role of oxidative damage and stress in aging. Mech. Ageing Dev. 2004, 125, 811–826. [Google Scholar] [CrossRef]
- Itoh, K.; Ishii, T.; Wakabayashi, N.; Yamamoto, M. Regulatory mechanisms of cellular response to oxidative stress. Free Radic Res 1999, 31, 319–324. [Google Scholar] [CrossRef]
- Chhunchha, B.; Fatma, N.; Kubo, E.; Rai, P.; Singh, S.P.; Singh, D.P. Curcumin abates hypoxia-induced oxidative stress based-ER stress-mediated cell death in mouse hippocampal cells (HT22) by controlling Prdx6 and NF-kappaB regulation. Am. J. Physiol. Cell Physiol. 2013, 304, C636–C655. [Google Scholar] [CrossRef] [Green Version]
- Kubben, N.; Zhang, W.; Wang, L.; Voss, T.C.; Yang, J.; Qu, J.; Liu, G.H.; Misteli, T. Repression of the Antioxidant NRF2 Pathway in Premature Aging. Cell 2016, 165, 1361–1374. [Google Scholar] [CrossRef] [Green Version]
- Kubo, E.; Chhunchha, B.; Singh, P.; Sasaki, H.; Singh, D.P. Sulforaphane reactivates cellular antioxidant defense by inducing Nrf2/ARE/Prdx6 activity during aging and oxidative stress. Sci. Rep. 2017, 7, 14130. [Google Scholar] [CrossRef] [Green Version]
- Chang, T.T.; Chen, J.W. Potential Impacts of Hydralazine as a Novel Antioxidant on Cardiovascular and Renal Disease-Beyond Vasodilation and Blood Pressure Lowering. Antioxidants 2022, 11, 2224. [Google Scholar] [CrossRef]
- Guo, X.; Han, C.; Ma, K.; Xia, Y.; Wan, F.; Yin, S.; Kou, L.; Sun, Y.; Wu, J.; Hu, J.; et al. Hydralazine Protects Nigrostriatal Dopaminergic Neurons From MPP(+) and MPTP Induced Neurotoxicity: Roles of Nrf2-ARE Signaling Pathway. Front. Neurol. 2019, 10, 271. [Google Scholar] [CrossRef] [PubMed]
- Dehghan, E.; Zhang, Y.; Saremi, B.; Yadavali, S.; Hakimi, A.; Dehghani, M.; Goodarzi, M.; Tu, X.; Robertson, S.; Lin, R.; et al. Hydralazine induces stress resistance and extends C. elegans lifespan by activating the NRF2/SKN-1 signalling pathway. Nat. Commun. 2017, 8, 2223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanninen, K.M.; Pomeshchik, Y.; Leinonen, H.; Malm, T.; Koistinaho, J.; Levonen, A.L. Applications of the Keap1-Nrf2 system for gene and cell therapy. Free Radic. Biol. Med. 2015, 88, 350–361. [Google Scholar] [CrossRef] [PubMed]
- Ziehm, M.; Kaur, S.; Ivanov, D.K.; Ballester, P.J.; Marcus, D.; Partridge, L.; Thornton, J.M. Drug repurposing for aging research using model organisms. Aging Cell 2017, 16, 1006–1015. [Google Scholar] [CrossRef] [PubMed]
- Davies, K.J.; Shringarpure, R. Preferential degradation of oxidized proteins by the 20S proteasome may be inhibited in aging and in inflammatory neuromuscular diseases. Neurology 2006, 66, S93–S96. [Google Scholar] [CrossRef] [PubMed]
- Fatma, N.; Kubo, E.; Toris, C.B.; Stamer, W.D.; Camras, C.B.; Singh, D.P. PRDX6 attenuates oxidative stress- and TGFbeta-induced abnormalities of human trabecular meshwork cells. Free Radic. Res. 2009, 43, 783–795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Assar, M.; Angulo, J.; Rodriguez-Manas, L. Oxidative stress and vascular inflammation in aging. Free Radic. Biol. Med. 2013, 65, 380–401. [Google Scholar] [CrossRef]
- Gounder, S.S.; Kannan, S.; Devadoss, D.; Miller, C.J.; Whitehead, K.J.; Odelberg, S.J.; Firpo, M.A.; Paine, R., 3rd; Hoidal, J.R.; Abel, E.D.; et al. Impaired transcriptional activity of Nrf2 in age-related myocardial oxidative stress is reversible by moderate exercise training. PLoS ONE 2012, 7, e45697. [Google Scholar] [CrossRef]
- Miller, C.J.; Gounder, S.S.; Kannan, S.; Goutam, K.; Muthusamy, V.R.; Firpo, M.A.; Symons, J.D.; Paine, R., 3rd; Hoidal, J.R.; Rajasekaran, N.S. Disruption of Nrf2/ARE signaling impairs antioxidant mechanisms and promotes cell degradation pathways in aged skeletal muscle. Biochim. Biophys. Acta 2012, 1822, 1038–1050. [Google Scholar] [CrossRef] [Green Version]
- Batliwala, S.; Xavier, C.; Liu, Y.; Wu, H.; Pang, I.H. Involvement of Nrf2 in Ocular Diseases. Oxid. Med. Cell. Longev. 2017, 2017, 1703810. [Google Scholar] [CrossRef]
- Chhunchha, B.; Kubo, E.; Singh, D.P. Clock Protein Bmal1 and Nrf2 Cooperatively Control Aging or Oxidative Response and Redox Homeostasis by Regulating Rhythmic Expression of Prdx6. Cells 2020, 9, 1861. [Google Scholar] [CrossRef] [PubMed]
- Dodson, M.; de la Vega, M.R.; Cholanians, A.B.; Schmidlin, C.J.; Chapman, E.; Zhang, D.D. Modulating NRF2 in Disease: Timing Is Everything. Annu. Rev. Pharmacol. Toxicol. 2019, 59, 555–575. [Google Scholar] [CrossRef] [PubMed]
- Fulop, G.A.; Kiss, T.; Tarantini, S.; Balasubramanian, P.; Yabluchanskiy, A.; Farkas, E.; Bari, F.; Ungvari, Z.; Csiszar, A. Nrf2 deficiency in aged mice exacerbates cellular senescence promoting cerebrovascular inflammation. Geroscience 2018, 40, 513–521. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, T.; Tabuchi, K.; Nishimura, B.; Tanaka, S.; Nakayama, M.; Ishii, T.; Warabi, E.; Yanagawa, T.; Shimizu, R.; Yamamoto, M.; et al. Protective role of Nrf2 in age-related hearing loss and gentamicin ototoxicity. Biochem. Biophys. Res. Commun. 2011, 415, 94–98. [Google Scholar] [CrossRef] [Green Version]
- McMahon, M.; Itoh, K.; Yamamoto, M.; Chanas, S.A.; Henderson, C.J.; McLellan, L.I.; Wolf, C.R.; Cavin, C.; Hayes, J.D. The Cap’n’Collar basic leucine zipper transcription factor Nrf2 (NF-E2 p45-related factor 2) controls both constitutive and inducible expression of intestinal detoxification and glutathione biosynthetic enzymes. Cancer Res. 2001, 61, 3299–3307. [Google Scholar]
- Lee, J.M.; Li, J.; Johnson, D.A.; Stein, T.D.; Kraft, A.D.; Calkins, M.J.; Jakel, R.J.; Johnson, J.A. Nrf2, a multi-organ protector? FASEB J. 2005, 19, 1061–1066. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; An, C.; Gao, Y.; Leak, R.K.; Chen, J.; Zhang, F. Emerging roles of Nrf2 and phase II antioxidant enzymes in neuroprotection. Prog. Neurobiol. 2013, 100, 30–47. [Google Scholar] [CrossRef] [Green Version]
- Anisimov, V.N.; Berstein, L.M.; Egormin, P.A.; Piskunova, T.S.; Popovich, I.G.; Zabezhinski, M.A.; Tyndyk, M.L.; Yurova, M.V.; Kovalenko, I.G.; Poroshina, T.E.; et al. Metformin slows down aging and extends life span of female SHR mice. Cell Cycle 2008, 7, 2769–2773. [Google Scholar] [CrossRef] [Green Version]
- Ashabi, G.; Khalaj, L.; Khodagholi, F.; Goudarzvand, M.; Sarkaki, A. Pre-treatment with metformin activates Nrf2 antioxidant pathways and inhibits inflammatory responses through induction of AMPK after transient global cerebral ischemia. Metab. Brain Dis. 2015, 30, 747–754. [Google Scholar] [CrossRef]
- Barzilai, N.; Crandall, J.P.; Kritchevsky, S.B.; Espeland, M.A. Metformin as a Tool to Target Aging. Cell Metab. 2016, 23, 1060–1065. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.; Xia, D.; Pan, Z.; Xu, D.; Zhou, Y.; Wu, Y.; Cai, N.; Tang, Q.; Wang, C.; Yan, M.; et al. Metformin protects against apoptosis and senescence in nucleus pulposus cells and ameliorates disc degeneration in vivo. Cell Death Dis. 2016, 7, e2441. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Yang, J.; Wu, X.; Zhang, G.; Li, T.; Wang, X.; Zhang, H.; Wang, C.C.; Liu, G.H.; Wang, L. Metformin alleviates human cellular aging by upregulating the endoplasmic reticulum glutathione peroxidase 7. Aging Cell 2018, 17, e12765. [Google Scholar] [CrossRef] [PubMed]
- Martin-Montalvo, A.; Mercken, E.M.; Mitchell, S.J.; Palacios, H.H.; Mote, P.L.; Scheibye-Knudsen, M.; Gomes, A.P.; Ward, T.M.; Minor, R.K.; Blouin, M.J.; et al. Metformin improves healthspan and lifespan in mice. Nat. Commun. 2013, 4, 2192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canning, P.; Sorrell, F.J.; Bullock, A.N. Structural basis of Keap1 interactions with Nrf2. Free Radic. Biol. Med. 2015, 88, 101–107. [Google Scholar] [CrossRef] [Green Version]
- Chowdhry, S.; Zhang, Y.; McMahon, M.; Sutherland, C.; Cuadrado, A.; Hayes, J.D. Nrf2 is controlled by two distinct beta-TrCP recognition motifs in its Neh6 domain, one of which can be modulated by GSK-3 activity. Oncogene 2013, 32, 3765–3781. [Google Scholar] [CrossRef] [Green Version]
- Cuadrado, A. Structural and functional characterization of Nrf2 degradation by glycogen synthase kinase 3/beta-TrCP. Free Radic. Biol. Med. 2015, 88, 147–157. [Google Scholar] [CrossRef]
- Rada, P.; Rojo, A.I.; Chowdhry, S.; McMahon, M.; Hayes, J.D.; Cuadrado, A. SCF/{beta}-TrCP promotes glycogen synthase kinase 3-dependent degradation of the Nrf2 transcription factor in a Keap1-independent manner. Mol. Cell. Biol. 2011, 31, 1121–1133. [Google Scholar] [CrossRef] [Green Version]
- Chan, J.Y.; Kwong, M. Impaired expression of glutathione synthetic enzyme genes in mice with targeted deletion of the Nrf2 basic-leucine zipper protein. Biochim. Biophys. Acta 2000, 1517, 19–26. [Google Scholar] [CrossRef]
- Dai, D.F.; Chiao, Y.A.; Marcinek, D.J.; Szeto, H.H.; Rabinovitch, P.S. Mitochondrial oxidative stress in aging and healthspan. Longev. Healthspan 2014, 3, 6. [Google Scholar] [CrossRef] [Green Version]
- Ferrucci, L.; Corsi, A.; Lauretani, F.; Bandinelli, S.; Bartali, B.; Taub, D.D.; Guralnik, J.M.; Longo, D.L. The origins of age-related proinflammatory state. Blood 2005, 105, 2294–2299. [Google Scholar] [CrossRef] [Green Version]
- Fatma, N.; Kubo, E.; Sharma, P.; Beier, D.R.; Singh, D.P. Impaired homeostasis and phenotypic abnormalities in Prdx6-/-mice lens epithelial cells by reactive oxygen species: Increased expression and activation of TGFbeta. Cell Death Differ. 2005, 12, 734–750. [Google Scholar] [CrossRef] [PubMed]
- Hodge, B.A.; Meyerhof, G.T.; Katewa, S.D.; Lian, T.; Lau, C.; Bar, S.; Leung, N.Y.; Li, M.; Li-Kroeger, D.; Melov, S.; et al. Dietary restriction and the transcription factor clock delay eye aging to extend lifespan in Drosophila Melanogaster. Nat. Commun. 2022, 13, 3156. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Cai, L.; Zheng, T.; Ye, H.; Rong, X.; Rao, J.; Lu, Y. The mechanism of UVB irradiation induced-apoptosis in cataract. Mol. Cell. Biochem. 2015, 401, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Kubo, E.; Fatma, N.; Akagi, Y.; Beier, D.R.; Singh, S.P.; Singh, D.P. TAT-mediated PRDX6 protein transduction protects against eye lens epithelial cell death and delays lens opacity. Am. J. Physiol. Cell Physiol. 2008, 294, C842–C855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neff, F.; Flores-Dominguez, D.; Ryan, D.P.; Horsch, M.; Schroder, S.; Adler, T.; Afonso, L.C.; Aguilar-Pimentel, J.A.; Becker, L.; Garrett, L.; et al. Rapamycin extends murine lifespan but has limited effects on aging. J. Clin. Investig. 2013, 123, 3272–3291. [Google Scholar] [CrossRef] [Green Version]
- Pearson, K.J.; Baur, J.A.; Lewis, K.N.; Peshkin, L.; Price, N.L.; Labinskyy, N.; Swindell, W.R.; Kamara, D.; Minor, R.K.; Perez, E.; et al. Resveratrol delays age-related deterioration and mimics transcriptional aspects of dietary restriction without extending life span. Cell Metab. 2008, 8, 157–168. [Google Scholar] [CrossRef] [Green Version]
- Shibata, S.; Shibata, N.; Shibata, T.; Sasaki, H.; Singh, D.P.; Kubo, E. The role of Prdx6 in the protection of cells of the crystalline lens from oxidative stress induced by UV exposure. Jpn. J. Ophthalmol. 2016, 60, 408–418. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Yan, H.; Lofgren, S.; Tian, X.; Lou, M.F. Ultraviolet radiation-induced cataract in mice: The effect of age and the potential biochemical mechanism. Investig. Ophthalmol. Vis. Sci. 2012, 53, 7276–7285. [Google Scholar] [CrossRef]
- Dubrovsky, Y.V.; Samsa, W.E.; Kondratov, R.V. Deficiency of circadian protein CLOCK reduces lifespan and increases age-related cataract development in mice. Aging 2010, 2, 936–944. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.B.; Tsubota, K.; Apte, R.S. A glimpse at the aging eye. NPJ Aging Mech. Dis. 2016, 2, 16003. [Google Scholar] [CrossRef] [Green Version]
- Quinlan, R.A.; Giblin, F. The eye lens as an aging paradigm par excellence. Exp. Eye Res. 2022, 218, 109003. [Google Scholar] [CrossRef] [PubMed]
- Rowan, S.; Jiang, S.; Francisco, S.G.; Pomatto, L.C.D.; Ma, Z.; Jiao, X.; Campos, M.M.; Aryal, S.; Patel, S.D.; Mahaling, B.; et al. Aged Nrf2-Null Mice Develop All Major Types of Age-Related Cataracts. Investig. Ophthalmol. Vis. Sci. 2021, 62, 10. [Google Scholar] [CrossRef] [PubMed]
- Ansari, N.H.; Wang, L.; Srivastava, S.K. Role of lipid aldehydes in cataractogenesis: 4-hydroxynonenal-induced cataract. Biochem. Mol. Med. 1996, 58, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Ates, N.A.; Yildirim, O.; Tamer, L.; Unlu, A.; Ercan, B.; Muslu, N.; Kanik, A.; Hatungil, R.; Atik, U. Plasma catalase activity and malondialdehyde level in patients with cataract. Eye 2004, 18, 785–788. [Google Scholar] [CrossRef] [Green Version]
- Lassen, N.; Bateman, J.B.; Estey, T.; Kuszak, J.R.; Nees, D.W.; Piatigorsky, J.; Duester, G.; Day, B.J.; Huang, J.; Hines, L.M.; et al. Multiple and additive functions of ALDH3A1 and ALDH1A1: Cataract phenotype and ocular oxidative damage in Aldh3a1(-/-)/Aldh1a1(-/-) knock-out mice. J. Biol. Chem. 2007, 282, 25668–25676. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.M.; Koh, H.J.; Park, D.C.; Song, B.J.; Huh, T.L.; Park, J.W. Cytosolic NADP(+)-dependent isocitrate dehydrogenase status modulates oxidative damage to cells. Free Radic. Biol. Med. 2002, 32, 1185–1196. [Google Scholar] [CrossRef]
- Park, J.; Zheng, L.; Marquis, A.; Walls, M.; Duerstock, B.; Pond, A.; Vega-Alvarez, S.; Wang, H.; Ouyang, Z.; Shi, R. Neuroprotective role of hydralazine in rat spinal cord injury-attenuation of acrolein-mediated damage. J. Neurochem. 2014, 129, 339–349. [Google Scholar] [CrossRef] [Green Version]
- Dulce, R.A.; Yiginer, O.; Gonzalez, D.R.; Goss, G.; Feng, N.; Zheng, M.; Hare, J.M. Hydralazine and organic nitrates restore impaired excitation-contraction coupling by reducing calcium leak associated with nitroso-redox imbalance. J. Biol. Chem. 2013, 288, 6522–6533. [Google Scholar] [CrossRef] [Green Version]
- Burcham, P.C.; Kaminskas, L.M.; Tan, D.; Pyke, S.M. Carbonyl-scavenging drugs & protection against carbonyl stress-associated cell injury. Mini. Rev. Med. Chem. 2008, 8, 319–330. [Google Scholar] [CrossRef]
- Hamann, K.; Nehrt, G.; Ouyang, H.; Duerstock, B.; Shi, R. Hydralazine inhibits compression and acrolein-mediated injuries in ex vivo spinal cord. J. Neurochem. 2008, 104, 708–718. [Google Scholar] [CrossRef]
- Karna, E.; Szoka, L.; Palka, J.A. The mechanism of hydralazine-induced collagen biosynthesis in cultured fibroblasts. Naunyn Schmiedebergs Arch. Pharm. 2013, 386, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Maheshwari, M.; Roberts, J.K.; Desutter, B.; Duong, K.T.; Tingling, J.; Fawver, J.N.; Schall, H.E.; Kahle, M.; Murray, I.V. Hydralazine modifies Abeta fibril formation and prevents modification by lipids in vitro. Biochemistry 2010, 49, 10371–10380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibaraki, N.; Chen, S.C.; Lin, L.R.; Okamoto, H.; Pipas, J.M.; Reddy, V.N. Human lens epithelial cell line. Exp. Eye Res. 1998, 67, 577–585. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.P.; Kubo, E.; Takamura, Y.; Shinohara, T.; Kumar, A.; Chylack, L.T.; Fatma, N. DNA binding domains and nuclear localization signal of LEDGF: Contribution of two helix-turn-helix (HTH)-like domains and a stretch of 58 amino acids of the N-terminal to the trans-activation potential of LEDGF. J. Mol. Biol. 2006, 355, 379–394. [Google Scholar] [CrossRef]
- McAvoy, J.W.; Chamberlain, C.G.; de Iongh, R.U.; Hales, A.M.; Lovicu, F.J. Lens development. Eye 1999, 13 Pt 3, 425–437. [Google Scholar] [CrossRef] [Green Version]
- Piatigorsky, J.; Rothschild, S.S. Loss during development of the ability of chick embryonic lens cells to elongate in culture: Inverse relationship between cell division and elongation. Dev. Biol. 1972, 28, 382–389. [Google Scholar] [CrossRef]
- Singh, D.P.; Bhargavan, B.; Chhunchha, B.; Kubo, E.; Kumar, A.; Fatma, N. Transcriptional protein Sp1 regulates LEDGF transcription by directly interacting with its cis-elements in GC-rich region of TATA-less gene promoter. PLoS ONE 2012, 7, e37012. [Google Scholar] [CrossRef] [Green Version]
- Singh, D.P.; Ohguro, N.; Kikuchi, T.; Sueno, T.; Reddy, V.N.; Yuge, K.; Chylack, L.T.; Shinohara, T. Lens epithelium-derived growth factor: Effects on growth and survival of lens epithelial cells, keratinocytes, and fibroblasts. Biochem. Biophys. Res. Commun. 2000, 267, 373–381. [Google Scholar] [CrossRef]
- Cong, L.; Pakala, S.B.; Ohshiro, K.; Li, D.Q.; Kumar, R. SUMOylation and SUMO-interacting motif (SIM) of metastasis tumor antigen 1 (MTA1) synergistically regulate its transcriptional repressor function. J. Biol. Chem. 2011, 286, 43793–43808. [Google Scholar] [CrossRef] [Green Version]
- Chhunchha, B.; Fatma, N.; Bhargavan, B.; Kubo, E.; Kumar, A.; Singh, D.P. Specificity protein, Sp1-mediated increased expression of Prdx6 as a curcumin-induced antioxidant defense in lens epithelial cells against oxidative stress. Cell Death Dis. 2011, 2, e234. [Google Scholar] [CrossRef] [Green Version]
- Chhunchha, B.; Kubo, E.; Fatma, N.; Singh, D.P. Sumoylation-deficient Prdx6 gains protective function by amplifying enzymatic activity and stability and escapes oxidative stress-induced aberrant Sumoylation. Cell Death Dis. 2017, 8, e2525. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.M.; Okazaki, K.; Nguyen, L.T.T.; Ota, N.; Kitamura, H.; Murakami, S.; Shima, H.; Igarashi, K.; Sekine, H.; Motohashi, H. Glucocorticoid receptor signaling represses the antioxidant response by inhibiting histone acetylation mediated by the transcriptional activator NRF2. J. Biol. Chem. 2017, 292, 7519–7530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chhunchha, B.; Kubo, E.; Kompella, U.B.; Singh, D.P. Engineered Sumoylation-Deficient Prdx6 Mutant Protein-Loaded Nanoparticles Provide Increased Cellular Defense and Prevent Lens Opacity. Antioxidants 2021, 10, 1245. [Google Scholar] [CrossRef] [PubMed]
- Fatma, N.; Singh, D.P.; Shinohara, T.; Chylack, L.T., Jr. Transcriptional regulation of the antioxidant protein 2 gene, a thiol-specific antioxidant, by lens epithelium-derived growth factor to protect cells from oxidative stress. J. Biol. Chem. 2001, 276, 48899–48907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chhunchha, B.; Kubo, E.; Singh, P.; Singh, D.P. Sumoylation-deficient Prdx6 repairs aberrant Sumoylation-mediated Sp1 dysregulation-dependent Prdx6 repression and cell injury in aging and oxidative stress. Aging 2018, 10, 2284–2315. [Google Scholar] [CrossRef]
- Kubo, E.; Singh, D.P.; Fatma, N.; Akagi, Y. TAT-mediated peroxiredoxin 5 and 6 protein transduction protects against high-glucose-induced cytotoxicity in retinal pericytes. Life Sci. 2009, 84, 857–864. [Google Scholar] [CrossRef] [Green Version]
- Kubo, E.; Miyazawa, T.; Fatma, N.; Akagi, Y.; Singh, D.P. Development- and age-associated expression pattern of peroxiredoxin 6, and its regulation in murine ocular lens. Mech. Ageing Dev. 2006, 127, 249–256. [Google Scholar] [CrossRef]
- Burcham, P.C. Carbonyl scavengers as pharmacotherapies in degenerative disease: Hydralazine repurposing and challenges in clinical translation. Biochem. Pharmacol. 2018, 154, 397–406. [Google Scholar] [CrossRef] [Green Version]
- Dehghan, E.; Goodarzi, M.; Saremi, B.; Lin, R.; Mirzaei, H. Hydralazine targets cAMP-dependent protein kinase leading to sirtuin1/5 activation and lifespan extension in C. elegans. Nat. Commun. 2019, 10, 4905. [Google Scholar] [CrossRef] [Green Version]
- Kwak, M.K.; Itoh, K.; Yamamoto, M.; Kensler, T.W. Enhanced expression of the transcription factor Nrf2 by cancer chemopreventive agents: Role of antioxidant response element-like sequences in the nrf2 promoter. Mol. Cell. Biol. 2002, 22, 2883–2892. [Google Scholar] [CrossRef] [Green Version]
- Almalki, S.G.; Agrawal, D.K. Key transcription factors in the differentiation of mesenchymal stem cells. Differentiation 2016, 92, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Nie, C.; Li, Y.; Li, R.; Yan, Y.; Zhang, D.; Li, T.; Li, Z.; Sun, Y.; Zhen, H.; Ding, J.; et al. Distinct biological ages of organs and systems identified from a multi-omics study. Cell Rep. 2022, 38, 110459. [Google Scholar] [CrossRef] [PubMed]
- Richard, G.; Stefanacci, D.; MGH; MBA. Changes in the Body with Aging. Healthy Living 2022. [Google Scholar]
- Schaum, N.; Lehallier, B.; Hahn, O.; Palovics, R.; Hosseinzadeh, S.; Lee, S.E.; Sit, R.; Lee, D.P.; Losada, P.M.; Zardeneta, M.E.; et al. Ageing hallmarks exhibit organ-specific temporal signatures. Nature 2020, 583, 596–602. [Google Scholar] [CrossRef] [PubMed]
- Ackert-Bicknell, C.L.; Anderson, L.C.; Sheehan, S.; Hill, W.G.; Chang, B.; Churchill, G.A.; Chesler, E.J.; Korstanje, R.; Peters, L.L. Aging Research Using Mouse Models. Curr. Protoc. Mouse Biol. 2015, 5, 95–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flurkey, C.; Harrison, D.E. The Mouse in Biomedical Research; Elsevier: Amsterdam, The Netherlands, 2007. [Google Scholar]
- Hauck, A.K.; Huang, Y.; Hertzel, A.V.; Bernlohr, D.A. Adipose oxidative stress and protein carbonylation. J. Biol. Chem. 2019, 294, 1083–1088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hybertson, B.M.; Gao, B. Role of the Nrf2 signaling system in health and disease. Clin. Genet. 2014, 86, 447–452. [Google Scholar] [CrossRef]
- Hybertson, B.M.; Gao, B.; Bose, S.K.; McCord, J.M. Oxidative stress in health and disease: The therapeutic potential of Nrf2 activation. Mol. Aspects Med. 2011, 32, 234–246. [Google Scholar] [CrossRef]
- Chapple, S.J.; Siow, R.C.; Mann, G.E. Crosstalk between Nrf2 and the proteasome: Therapeutic potential of Nrf2 inducers in vascular disease and aging. Int. J. Biochem. Cell Biol. 2012, 44, 1315–1320. [Google Scholar] [CrossRef]
- Chartoumpekis, D.V.; Kensler, T.W. New player on an old field; the keap1/Nrf2 pathway as a target for treatment of type 2 diabetes and metabolic syndrome. Curr. Diabetes Rev. 2013, 9, 137–145. [Google Scholar] [CrossRef]
- Sadowska-Bartosz, I.; Bartosz, G. Effect of antioxidants supplementation on aging and longevity. Biomed. Res. Int. 2014, 2014, 404680. [Google Scholar] [CrossRef]
- Shen, L.; Ji, H.F. Insights into the disappointing clinical trials of antioxidants in neurodegenerative diseases. J. Alzheimers Dis. 2010, 19, 1141–1142. [Google Scholar] [CrossRef] [PubMed]
- Ashrafian, H.; Czibik, G.; Bellahcene, M.; Aksentijevic, D.; Smith, A.C.; Mitchell, S.J.; Dodd, M.S.; Kirwan, J.; Byrne, J.J.; Ludwig, C.; et al. Fumarate is cardioprotective via activation of the Nrf2 antioxidant pathway. Cell Metab. 2012, 15, 361–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calkins, M.J.; Johnson, D.A.; Townsend, J.A.; Vargas, M.R.; Dowell, J.A.; Williamson, T.P.; Kraft, A.D.; Lee, J.M.; Li, J.; Johnson, J.A. The Nrf2/ARE pathway as a potential therapeutic target in neurodegenerative disease. Antioxid. Redox Signal. 2009, 11, 497–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, B.; Doan, A.; Hybertson, B.M. The clinical potential of influencing Nrf2 signaling in degenerative and immunological disorders. Clin. Pharmacol. 2014, 6, 19–34. [Google Scholar] [CrossRef] [Green Version]
- Magesh, S.; Chen, Y.; Hu, L. Small molecule modulators of Keap1-Nrf2-ARE pathway as potential preventive and therapeutic agents. Med. Res. Rev. 2012, 32, 687–726. [Google Scholar] [CrossRef] [Green Version]
- Arnold, P.; Mojumder, D.; Detoledo, J.; Lucius, R.; Wilms, H. Pathophysiological processes in multiple sclerosis: Focus on nuclear factor erythroid-2-related factor 2 and emerging pathways. Clin. Pharmacol. 2014, 6, 35–42. [Google Scholar] [CrossRef] [Green Version]
- Bar-Or, A.; Gold, R.; Kappos, L.; Arnold, D.L.; Giovannoni, G.; Selmaj, K.; O’Gorman, J.; Stephan, M.; Dawson, K.T. Clinical efficacy of BG-12 (dimethyl fumarate) in patients with relapsing-remitting multiple sclerosis: Subgroup analyses of the DEFINE study. J. Neurol. 2013, 260, 2297–2305. [Google Scholar] [CrossRef]
- Rojo de la Vega, M.; Dodson, M.; Chapman, E.; Zhang, D.D. NRF2-targeted therapeutics: New targets and modes of NRF2 regulation. Curr. Opin. Toxicol. 2016, 1, 62–70. [Google Scholar] [CrossRef] [Green Version]
- Colombo, G.; Garavaglia, M.L.; Astori, E.; Giustarini, D.; Rossi, R.; Milzani, A.; Dalle-Donne, I. Protein carbonylation in human bronchial epithelial cells exposed to cigarette smoke extract. Cell Biol. Toxicol. 2019, 35, 345–360. [Google Scholar] [CrossRef]
- Dalle-Donne, I.; Aldini, G.; Carini, M.; Colombo, R.; Rossi, R.; Milzani, A. Protein carbonylation, cellular dysfunction, and disease progression. J. Cell Mol. Med. 2006, 10, 389–406. [Google Scholar] [CrossRef] [PubMed]
- Dalle-Donne, I.; Giustarini, D.; Colombo, R.; Rossi, R.; Milzani, A. Protein carbonylation in human diseases. Trends Mol. Med. 2003, 9, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Dalle-Donne, I.; Rossi, R.; Colombo, R.; Giustarini, D.; Milzani, A. Biomarkers of oxidative damage in human disease. Clin. Chem. 2006, 52, 601–623. [Google Scholar] [CrossRef] [PubMed]
- Dalle-Donne, I.; Rossi, R.; Giustarini, D.; Milzani, A.; Colombo, R. Protein carbonyl groups as biomarkers of oxidative stress. Clin. Chim. Acta 2003, 329, 23–38. [Google Scholar] [CrossRef]
- Giustarini, D.; Galvagni, F.; Colombo, G.; Dalle-Donne, I.; Milzani, A.; Aloisi, A.M.; Rossi, R. Determination of protein thiolation index (PTI) as a biomarker of oxidative stress in human serum. Anal. Biochem. 2017, 538, 38–41. [Google Scholar] [CrossRef]
- Grune, T.; Reinheckel, T.; Davies, K.J. Degradation of oxidized proteins in mammalian cells. FASEB J. 1997, 11, 526–534. [Google Scholar] [CrossRef]
- Hernebring, M.; Adelof, J.; Wiseman, J.; Petersen, A.; Zetterberg, M. H2O2-induced cataract as a model of age-related cataract: Lessons learned from overexpressing the proteasome activator PA28alphabeta in mouse eye lens. Exp. Eye Res. 2021, 203, 108395. [Google Scholar] [CrossRef]
- Truscott, R.J. Age-related nuclear cataract: A lens transport problem. Ophthalmic Res. 2000, 32, 185–194. [Google Scholar] [CrossRef]
- Schmid, P.W.N.; Lim, N.C.H.; Peters, C.; Back, K.C.; Bourgeois, B.; Pirolt, F.; Richter, B.; Peschek, J.; Puk, O.; Amarie, O.V.; et al. Imbalances in the eye lens proteome are linked to cataract formation. Nat. Struct. Mol. Biol. 2021, 28, 143–151. [Google Scholar] [CrossRef]
- Adams, S.; Green, P.; Claxton, R.; Simcox, S.; Williams, M.V.; Walsh, K.; Leeuwenburgh, C. Reactive carbonyl formation by oxidative and non-oxidative pathways. Front. Biosci. 2001, 6, A17–A24. [Google Scholar] [CrossRef] [Green Version]
- Di Domenico, F.; Tramutola, A.; Butterfield, D.A. Role of 4-hydroxy-2-nonenal (HNE) in the pathogenesis of alzheimer disease and other selected age-related neurodegenerative disorders. Free Radic. Biol. Med. 2017, 111, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Verdejo, C.; Marco, P.; Renau-Piqueras, J.; Pinazo-Duran, M.D. Lipid peroxidation in proliferative vitreoretinopathies. Eye 1999, 13 Pt 2, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Liu-Snyder, P.; Borgens, R.B.; Shi, R. Hydralazine rescues PC12 cells from acrolein-mediated death. J. Neurosci. Res. 2006, 84, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Lewis, K.N.; Wason, E.; Edrey, Y.H.; Kristan, D.M.; Nevo, E.; Buffenstein, R. Regulation of Nrf2 signaling and longevity in naturally long-lived rodents. Proc. Natl. Acad. Sci. USA 2015, 112, 3722–3727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knowles, H.J.; Tian, Y.M.; Mole, D.R.; Harris, A.L. Novel mechanism of action for hydralazine: Induction of hypoxia-inducible factor-1alpha, vascular endothelial growth factor, and angiogenesis by inhibition of prolyl hydroxylases. Circ. Res. 2004, 95, 162–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulkarni, S.R.; Donepudi, A.C.; Xu, J.; Wei, W.; Cheng, Q.C.; Driscoll, M.V.; Johnson, D.A.; Johnson, J.A.; Li, X.; Slitt, A.L. Fasting induces nuclear factor E2-related factor 2 and ATP-binding Cassette transporters via protein kinase A and Sirtuin-1 in mouse and human. Antioxid. Redox Signal. 2014, 20, 15–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcotte, D.; Zeng, W.; Hus, J.C.; McKenzie, A.; Hession, C.; Jin, P.; Bergeron, C.; Lugovskoy, A.; Enyedy, I.; Cuervo, H.; et al. Small molecules inhibit the interaction of Nrf2 and the Keap1 Kelch domain through a non-covalent mechanism. Bioorg. Med. Chem. 2013, 21, 4011–4019. [Google Scholar] [CrossRef]
- O’Connell, M.A.; Hayes, J.D. The Keap1/Nrf2 pathway in health and disease: From the bench to the clinic. Biochem. Soc. Trans. 2015, 43, 687–689. [Google Scholar] [CrossRef] [Green Version]
- Ma, Q. Role of nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef] [Green Version]
- Gao, M.; Hu, J.; Zhu, Y.; Wang, X.; Zeng, S.; Hong, Y.; Zhao, G. Ferroptosis and Apoptosis Are Involved in the Formation of L-Selenomethionine-Induced Ocular Defects in Zebrafish Embryos. Int. J. Mol. Sci. 2022, 23, 4783. [Google Scholar] [CrossRef]
- Li, W.C.; Kuszak, J.R.; Dunn, K.; Wang, R.R.; Ma, W.; Wang, G.M.; Spector, A.; Leib, M.; Cotliar, A.M.; Weiss, M.; et al. Lens epithelial cell apoptosis appears to be a common cellular basis for non-congenital cataract development in humans and animals. J. Cell Biol. 1995, 130, 169–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Eitan, L.N.; Almomani, B.A.; Nassar, A.M.; Elsaqa, B.Z.; Saadeh, N.A. Metformin Pharmacogenetics: Effects of SLC22A1, SLC22A2, and SLC22A3 Polymorphisms on Glycemic Control and HbA1c Levels. J. Pers. Med. 2019, 9, 17. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Jin, H.; Shi, Y.; Guo, Y.; Zhang, H. Pyroptosis, a novel mechanism implicated in cataracts. Mol. Med. Rep. 2018, 18, 2277–2285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, Z.; Hao, C.; Huangfu, J.; Srinivasagan, R.; Zhang, X.; Fan, X. Aging lens epithelium is susceptible to ferroptosis. Free Radic. Biol. Med. 2021, 167, 94–108. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Jiao, Y.; Li, X.; Gao, S.; Zhou, N.; Duan, J.; Zhang, M. Pyroptosis: A New Insight Into Eye Disease Therapy. Front. Pharmacol. 2021, 12, 797110. [Google Scholar] [CrossRef] [PubMed]
- Onken, B.; Driscoll, M. Metformin induces a dietary restriction-like state and the oxidative stress response to extend C. elegans Healthspan via AMPK, LKB1, and SKN-1. PLoS ONE 2010, 5, e8758. [Google Scholar] [CrossRef]
- Cabreiro, F. Metformin Joins Forces with Microbes. Cell Host Microbe 2016, 19, 1–3. [Google Scholar] [CrossRef] [Green Version]
- Cabreiro, F.; Au, C.; Leung, K.Y.; Vergara-Irigaray, N.; Cocheme, H.M.; Noori, T.; Weinkove, D.; Schuster, E.; Greene, N.D.; Gems, D. Metformin retards aging in C. elegans by altering microbial folate and methionine metabolism. Cell 2013, 153, 228–239. [Google Scholar] [CrossRef] [Green Version]
- De Haes, W.; Frooninckx, L.; Van Assche, R.; Smolders, A.; Depuydt, G.; Billen, J.; Braeckman, B.P.; Schoofs, L.; Temmerman, L. Metformin promotes lifespan through mitohormesis via the peroxiredoxin PRDX-2. Proc. Natl. Acad. Sci. USA 2014, 111, E2501–E2509. [Google Scholar] [CrossRef] [Green Version]
- Altomare, E.; Grattagliano, I.; Vendemaile, G.; Micelli-Ferrari, T.; Signorile, A.; Cardia, L. Oxidative protein damage in human diabetic eye: Evidence of a retinal participation. Eur. J. Clin. Investig. 1997, 27, 141–147. [Google Scholar] [CrossRef]
- Boscia, F.; Grattagliano, I.; Vendemiale, G.; Micelli-Ferrari, T.; Altomare, E. Protein oxidation and lens opacity in humans. Investig. Ophthalmol. Vis. Sci. 2000, 41, 2461–2465. [Google Scholar] [PubMed]
- Chatard, M.; Puech, C.; Perek, N.; Roche, F. Hydralazine is a Suitable Mimetic Agent of Hypoxia to Study the Impact of Hypoxic Stress on In Vitro Blood-Brain Barrier Model. Cell. Physiol. Biochem. 2017, 42, 1592–1602. [Google Scholar] [CrossRef]
- Leiro, J.M.; Alvarez, E.; Arranz, J.A.; Siso, I.G.; Orallo, F. In vitro effects of mangiferin on superoxide concentrations and expression of the inducible nitric oxide synthase, tumour necrosis factor-alpha and transforming growth factor-beta genes. Biochem. Pharmacol. 2003, 65, 1361–1371. [Google Scholar] [CrossRef]
- Leu, J.G.; Su, W.H.; Chen, Y.C.; Liang, Y.J. Hydralazine attenuates renal inflammation in diabetic rats with ischemia/reperfusion acute kidney injury. Eur. J. Pharmacol. 2021, 910, 174468. [Google Scholar] [CrossRef]
- Shahzad Qamar, A.; Zamir, A.; Khalid, S.; Ashraf, W.; Imran, I.; Hussain, I.; Rehman, A.U.; Saeed, H.; Majeed, A.; Alqahtani, F.; et al. A review on the clinical pharmacokinetics of hydralazine. Expert Opin. Drug Metab. Toxicol. 2022, 18, 707–714. [Google Scholar] [CrossRef] [PubMed]
- Early, J.O.; Menon, D.; Wyse, C.A.; Cervantes-Silva, M.P.; Zaslona, Z.; Carroll, R.G.; Palsson-McDermott, E.M.; Angiari, S.; Ryan, D.G.; Corcoran, S.E.; et al. Circadian clock protein BMAL1 regulates IL-1beta in macrophages via NRF2. Proc. Natl. Acad. Sci. USA 2018, 115, E8460–E8468. [Google Scholar] [CrossRef] [PubMed]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward: 3′ to 5′ | Reverse: 3′ to 5′ |
|---|---|---|
| hNrf2 | TGCTTTATAGCGTGCAAACCTCGC | ATCCATGTCCCTTGACAGCACAGA |
| hPrdx6 | GCATCCGTTTCCACGACT | TGCACACTGGGGTAAAGTCC |
| hHO1 | GGCAGAGGGTGATAGAAGAGG | AGCTCCTGCAACTCCTCAAA |
| hNQO1 | ATGTATGACAAAGGACCCTTCC | TCCCTTGCAGAGAGTACATGG |
| hGCLC | ATGCCATGGGATTTGGAAT | GATCATAAAGGTATCTGGCCTCA |
| hGCLM | GACAAAACACAGTTGGAACAGC | CAGTCAAATCTGGTGGCATC |
| hβ-actin | CCAACCGCGAGAAGATGA | CCAGAGGCGTACAGGGATAG |
| mNrf2 | TCTCCTCGCTGGAAAAAGAA | AATGTGCTGGCTGTGCTTTA |
| mPrdx6 | TTCAATAGACAGTGTTGAGGATCA | CGTGGGTGTTTCACCATTG |
| mHO1 | AGGCTAAGACCGCCTTCCT | TGTGTTCCTCTGTCAGCATCA |
| mNQO1 | AGCGTTCGGTATTACGATCC | AGTACAATCAGGGCTCTTCTCG |
| mGCLC | AGATGATAGAACACGGGAGGAG | TGATCCTAAAGCGATTGTTCTTC |
| mGCLM | TGACTCACAATGACCCGAAA | TCAATGTCAGGGATGCTTTCT |
| mCatalase | CCTTCAAGTTGGTTAATGCAGA | CAAGTTTTTGATGCCCTGGT |
| mSOD1 | CAGGACCTCATTTTAATCCTCAC | TGCCCAGGTCTCCAACAT |
| mβ-actin | CTAAGGCCAACCGTGAAAAG- | ACCAGAGGCATACAGGGACA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chhunchha, B.; Kubo, E.; Krueger, R.R.; Singh, D.P. Hydralazine Revives Cellular and Ocular Lens Health-Span by Ameliorating the Aging and Oxidative-Dependent Loss of the Nrf2-Activated Cellular Stress Response. Antioxidants 2023, 12, 140. https://doi.org/10.3390/antiox12010140
Chhunchha B, Kubo E, Krueger RR, Singh DP. Hydralazine Revives Cellular and Ocular Lens Health-Span by Ameliorating the Aging and Oxidative-Dependent Loss of the Nrf2-Activated Cellular Stress Response. Antioxidants. 2023; 12(1):140. https://doi.org/10.3390/antiox12010140
Chicago/Turabian StyleChhunchha, Bhavana, Eri Kubo, Ronald R. Krueger, and Dhirendra P. Singh. 2023. "Hydralazine Revives Cellular and Ocular Lens Health-Span by Ameliorating the Aging and Oxidative-Dependent Loss of the Nrf2-Activated Cellular Stress Response" Antioxidants 12, no. 1: 140. https://doi.org/10.3390/antiox12010140