
Nrf2 Pathway and Autophagy Crosstalk: New Insights into Therapeutic Strategies for Ischemic Cerebral Vascular Diseases

Abstract
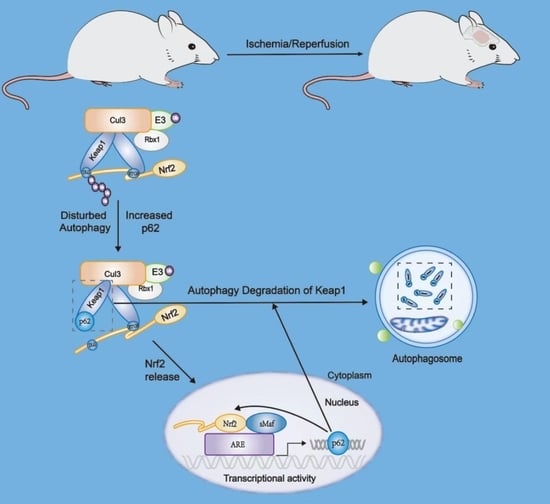
:
1. Introduction
2. Nrf2 Pathway
2.1. Structure of Nrf2
2.2. Nrf2-Involved in Ischemic Cerebrovascular Diseases
3. Autophagy
3.1. Process of Autophagy
3.2. Functions of Autophagy
3.3. Autophagy-Involved in Ischemic Cerebrovascular Diseases
4. Crosstalk between Nrf2 Pathway and Autophagy
5. Crosstalk between Nrf2 and Autophagy in Ischemic Cerebrovascular Disease
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ajoolabady, A.; Wang, S.; Kroemer, G.; Penninger, J.M.; Uversky, V.N.; Pratico, D.; Henninger, N.; Reiter, R.J.; Bruno, A.; Joshipura, K.; et al. Targeting autophagy in ischemic stroke: From molecular mechanisms to clinical therapeutics. Pharmacol. Ther. 2021, 225, 107848. [Google Scholar] [CrossRef] [PubMed]
- Feske, S.K. Ischemic Stroke. Am. J. Med. 2021, 134, 1457–1464. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, E.J.; Blaha, M.J.; Chiuve, S.E.; Cushman, M.; Das, S.R.; Deo, R.; de Ferranti, S.D.; Floyd, J.; Fornage, M.; Gillespie, C.; et al. Heart Disease and Stroke Statistics-2017 Update: A Report From the American Heart Association. Circulation 2017, 135, e146–e603. [Google Scholar] [CrossRef] [PubMed]
- Iadecola, C.; Buckwalter, M.S.; Anrather, J. Immune responses to stroke: Mechanisms, modulation, and therapeutic potential. J. Clin. Investig. 2020, 130, 2777–2788. [Google Scholar] [CrossRef] [PubMed]
- Reeves, M.J.; Arora, S.; Broderick, J.P.; Frankel, M.; Heinrich, J.P.; Hickenbottom, S.; Karp, H.; LaBresh, K.A.; Malarcher, A.; Mensah, G.; et al. Acute stroke care in the US: Results from 4 pilot prototypes of the Paul Coverdell National Acute Stroke Registry. Stroke 2005, 36, 1232–1240. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Zheng, Y.; Wang, T.; Jiao, L.; Luo, Y. VEGF, a Key Factor for Blood Brain Barrier Injury After Cerebral Ischemic Stroke. Aging Dis. 2022, 13, 647–654. [Google Scholar] [CrossRef] [PubMed]
- Dong, Q.; Lin, X.; Shen, L.; Feng, Y. The protective effect of herbal polysaccharides on ischemia-reperfusion injury. Int. J. Biol. Macromol. 2016, 92, 431–440. [Google Scholar] [CrossRef]
- Meng, H.; Jin, W.; Yu, L.; Xu, S.; Wan, H.; He, Y. Protective effects of polysaccharides on cerebral ischemia: A mini-review of the mechanisms. Int. J. Biol. Macromol. 2021, 169, 463–472. [Google Scholar] [CrossRef]
- Sachdev, S.; Ansari, S.A.; Ansari, M.I.; Fujita, M.; Hasanuzzaman, M. Abiotic Stress and Reactive Oxygen Species: Generation, Signaling, and Defense Mechanisms. Antioxidants 2021, 10, 277. [Google Scholar] [CrossRef]
- Chen, H.; Yoshioka, H.; Kim, G.S.; Jung, J.E.; Okami, N.; Sakata, H.; Maier, C.M.; Narasimhan, P.; Goeders, C.E.; Chan, P.H. Oxidative stress in ischemic brain damage: Mechanisms of cell death and potential molecular targets for neuroprotection. Antioxid. Redox Signal. 2011, 14, 1505–1517. [Google Scholar] [CrossRef] [Green Version]
- Fabian, E.; Bogner, M.; Elmadfa, I. Age-related modification of antioxidant enzyme activities in relation to cardiovascular risk factors. Eur. J. Clin. Investig. 2012, 42, 42–48. [Google Scholar] [CrossRef]
- Wang, P.; Guan, Y.F.; Du, H.; Zhai, Q.W.; Su, D.F.; Miao, C.Y. Induction of autophagy contributes to the neuroprotection of nicotinamide phosphoribosyltransferase in cerebral ischemia. Autophagy 2012, 8, 77–87. [Google Scholar] [CrossRef]
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T.; Kominami, E.; Ohsumi, Y.; Yoshimori, T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000, 19, 5720–5728. [Google Scholar] [CrossRef] [PubMed]
- Button, R.W.; Luo, S.; Rubinsztein, D.C. Autophagic activity in neuronal cell death. Neurosci. Bull. 2015, 31, 382–394. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.W.; Huang, B.S.; Han, Y.; Deng, L.H.; Wu, L.X. Sodium hydrosulfide attenuates cerebral ischemia/reperfusion injury by suppressing overactivated autophagy in rats. FEBS Open Bio 2017, 7, 1686–1695. [Google Scholar] [CrossRef] [PubMed]
- Tonelli, C.; Chio, I.I.C.; Tuveson, D.A. Transcriptional Regulation by Nrf2. Antioxid. Redox Signal. 2018, 29, 1727–1745. [Google Scholar] [CrossRef] [PubMed]
- Eggler, A.L.; Liu, G.; Pezzuto, J.M.; van Breemen, R.B.; Mesecar, A.D. Modifying specific cysteines of the electrophile-sensing human Keap1 protein is insufficient to disrupt binding to the Nrf2 domain Neh2. Proc. Natl. Acad. Sci. USA 2005, 102, 10070–10075. [Google Scholar] [CrossRef]
- Fukutomi, T.; Takagi, K.; Mizushima, T.; Ohuchi, N.; Yamamoto, M. Kinetic, thermodynamic, and structural characterizations of the association between Nrf2-DLGex degron and Keap1. Mol. Cell. Biol. 2014, 34, 832–846. [Google Scholar] [CrossRef]
- Lo, S.C.; Li, X.; Henzl, M.T.; Beamer, L.J.; Hannink, M. Structure of the Keap1:Nrf2 interface provides mechanistic insight into Nrf2 signaling. EMBO J. 2006, 25, 3605–3617. [Google Scholar] [CrossRef]
- Bellezza, I.; Giambanco, I.; Minelli, A.; Donato, R. Nrf2-Keap1 signaling in oxidative and reductive stress. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 721–733. [Google Scholar] [CrossRef]
- Nioi, P.; Nguyen, T.; Sherratt, P.J.; Pickett, C.B. The carboxy-terminal Neh3 domain of Nrf2 is required for transcriptional activation. Mol. Cell. Biol. 2005, 25, 10895–10906. [Google Scholar] [CrossRef] [PubMed]
- Katoh, Y.; Itoh, K.; Yoshida, E.; Miyagishi, M.; Fukamizu, A.; Yamamoto, M. Two domains of Nrf2 cooperatively bind CBP, a CREB binding protein, and synergistically activate transcription. Genes Cells 2001, 6, 857–868. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Yu, S.; Chen, J.D.; Kong, A.N. The nuclear cofactor RAC3/AIB1/SRC-3 enhances Nrf2 signaling by interacting with transactivation domains. Oncogene 2013, 32, 514–527. [Google Scholar] [CrossRef]
- Chowdhry, S.; Zhang, Y.; McMahon, M.; Sutherland, C.; Cuadrado, A.; Hayes, J.D. Nrf2 is controlled by two distinct beta-TrCP recognition motifs in its Neh6 domain, one of which can be modulated by GSK-3 activity. Oncogene 2013, 32, 3765–3781. [Google Scholar] [CrossRef] [PubMed]
- Rada, P.; Rojo, A.I.; Chowdhry, S.; McMahon, M.; Hayes, J.D.; Cuadrado, A. SCF/{beta}-TrCP promotes glycogen synthase kinase 3-dependent degradation of the Nrf2 transcription factor in a Keap1-independent manner. Mol. Cell. Biol. 2011, 31, 1121–1133. [Google Scholar] [CrossRef]
- Wang, H.; Liu, K.; Geng, M.; Gao, P.; Wu, X.; Hai, Y.; Li, Y.; Li, Y.; Luo, L.; Hayes, J.D.; et al. RXRalpha inhibits the NRF2-ARE signaling pathway through a direct interaction with the Neh7 domain of NRF2. Cancer Res. 2013, 73, 3097–3108. [Google Scholar] [CrossRef] [PubMed]
- Zipper, L.M.; Mulcahy, R.T. The Keap1 BTB/POZ dimerization function is required to sequester Nrf2 in cytoplasm. J. Biol. Chem. 2002, 277, 36544–36552. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.D.; Lo, S.C.; Cross, J.V.; Templeton, D.J.; Hannink, M. Keap1 is a redox-regulated substrate adaptor protein for a Cul3-dependent ubiquitin ligase complex. Mol. Cell. Biol. 2004, 24, 10941–10953. [Google Scholar] [CrossRef]
- Yamamoto, T.; Suzuki, T.; Kobayashi, A.; Wakabayashi, J.; Maher, J.; Motohashi, H.; Yamamoto, M. Physiological significance of reactive cysteine residues of Keap1 in determining Nrf2 activity. Mol. Cell. Biol. 2008, 28, 2758–2770. [Google Scholar] [CrossRef]
- Canning, P.; Sorrell, F.J.; Bullock, A.N. Structural basis of Keap1 interactions with Nrf2. Free Radic. Biol. Med. 2015, 88, 101–107. [Google Scholar] [CrossRef] [Green Version]
- Miao, Z.Y.; Xia, X.; Che, L.; Song, Y.T. Genistein attenuates brain damage induced by transient cerebral ischemia through up-regulation of Nrf2 expression in ovariectomized rats. Neurol. Res. 2018, 40, 689–695. [Google Scholar] [CrossRef] [PubMed]
- Cai, M.; Guo, Y.; Wang, S.; Wei, H.; Sun, S.; Zhao, G.; Dong, H. Tanshinone IIA Elicits Neuroprotective Effect Through Activating the Nuclear Factor Erythroid 2-Related Factor-Dependent Antioxidant Response. Rejuvenation Res. 2017, 20, 286–297. [Google Scholar] [CrossRef]
- An, P.; Wu, T.; Yu, H.; Fang, K.; Ren, Z.; Tang, M. Hispidulin Protects Against Focal Cerebral Ischemia Reperfusion Injury in Rats. J. Mol. Neurosci. 2018, 65, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Fu, B.; Zhang, X.; Zhao, T.; Chen, L.; Zhang, J.; Wang, X. Paeonol pretreatment attenuates cerebral ischemic injury via upregulating expression of pAkt, Nrf2, HO-1 and ameliorating BBB permeability in mice. Brain Res. Bull. 2014, 109, 61–67. [Google Scholar] [CrossRef]
- Ya, B.L.; Li, H.F.; Wang, H.Y.; Wu, F.; Xin, Q.; Cheng, H.J.; Li, W.J.; Lin, N.; Ba, Z.H.; Zhang, R.J.; et al. 5-HMF attenuates striatum oxidative damage via Nrf2/ARE signaling pathway following transient global cerebral ischemia. Cell Stress Chaperones 2017, 22, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Song, J.K.; Yan, R.; Li, L.; Xiao, Z.Y.; Zhou, W.X.; Wang, Z.Z.; Xiao, W.; Du, G.H. Diterpene ginkgolides protect against cerebral ischemia/reperfusion damage in rats by activating Nrf2 and CREB through PI3K/Akt signaling. Acta Pharmacol. Sin. 2018, 39, 1259–1272. [Google Scholar] [CrossRef] [PubMed]
- Clausen, B.H.; Lundberg, L.; Yli-Karjanmaa, M.; Martin, N.A.; Svensson, M.; Alfsen, M.Z.; Flaeng, S.B.; Lyngso, K.; Boza-Serrano, A.; Nielsen, H.H.; et al. Fumarate decreases edema volume and improves functional outcome after experimental stroke. Exp. Neurol. 2017, 295, 144–154. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Locascio, L.M.; Dore, S. Critical Role of Nrf2 in Experimental Ischemic Stroke. Front. Pharmacol. 2019, 10, 153. [Google Scholar] [CrossRef]
- Dore, S. Neuroprotective effert of carbon monoxide and Nrf2 in cerebral ischemia. Springerplus 2015, 4, L44. [Google Scholar] [CrossRef]
- Liu, L.; Vollmer, M.K.; Fernandez, V.M.; Dweik, Y.; Kim, H.; Dore, S. Korean Red Ginseng Pretreatment Protects Against Long-Term Sensorimotor Deficits After Ischemic Stroke Likely Through Nrf2. Front. Cell. Neurosci. 2018, 12, 74. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Wei, R.; Zhang, L.; Tan, Y.; Qian, C. Sirtuin 6 protects the brain from cerebral ischemia/reperfusion injury through NRF2 activation. Neuroscience 2017, 366, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, S.V.; Dave, K.R.; Saul, I.; Perez-Pinzon, M.A. Resveratrol Preconditioning Protects Against Cerebral Ischemic Injury via Nuclear Erythroid 2-Related Factor 2. Stroke 2015, 46, 1626–1632. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Miao, W.; Liu, Z.; Han, W.; Shi, K.; Shen, Y.; Li, H.; Liu, Q.; Fu, Y.; Huang, D.; et al. Dimethyl Fumarate and Monomethyl Fumarate Promote Post-Ischemic Recovery in Mice. Transl. Stroke Res. 2016, 7, 535–547. [Google Scholar] [CrossRef] [PubMed]
- Alfieri, A.; Srivastava, S.; Siow, R.C.M.; Cash, D.; Modo, M.; Duchen, M.R.; Fraser, P.A.; Williams, S.C.R.; Mann, G.E. Sulforaphane preconditioning of the Nrf2/HO-1 defense pathway protects the cerebral vasculature against blood-brain barrier disruption and neurological deficits in stroke. Free Radic. Biol. Med. 2013, 65, 1012–1022. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Wang, Y.; He, Q.; Li, L.; Xie, H.; Zhao, Y.; Zhao, J. Nrf2 inhibits NLRP3 inflammasome activation through regulating Trx1/TXNIP complex in cerebral ischemia reperfusion injury. Behav. Brain Res. 2018, 336, 32–39. [Google Scholar] [CrossRef]
- Ucar, B.I.; Ucar, G.; Saha, S.; Buttari, B.; Profumo, E.; Saso, L. Pharmacological Protection against Ischemia-Reperfusion Injury by Regulating the Nrf2-Keap1-ARE Signaling Pathway. Antioxidants 2021, 10, 823. [Google Scholar] [CrossRef]
- Shen, Y.; Liu, X.; Shi, J.; Wu, X. Involvement of Nrf2 in myocardial ischemia and reperfusion injury. Int. J. Biol. Macromol. 2019, 125, 496–502. [Google Scholar] [CrossRef]
- Oh, Y.S.; Jun, H.S. Effects of Glucagon-Like Peptide-1 on Oxidative Stress and Nrf2 Signaling. Int. J. Mol. Sci. 2017, 19, 26. [Google Scholar] [CrossRef]
- Hayes, J.D.; Dinkova-Kostova, A.T. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 2014, 39, 199–218. [Google Scholar] [CrossRef]
- Saha, S.; Buttari, B.; Panieri, E.; Profumo, E.; Saso, L. An Overview of Nrf2 Signaling Pathway and Its Role in Inflammation. Molecules 2020, 25, 5474. [Google Scholar] [CrossRef]
- Li, L.; Tan, J.; Miao, Y.; Lei, P.; Zhang, Q. ROS and Autophagy: Interactions and Molecular Regulatory Mechanisms. Cell. Mol. Neurobiol. 2015, 35, 615–621. [Google Scholar] [CrossRef] [PubMed]
- Culbreth, M.; Aschner, M. GSK-3beta, a double-edged sword in Nrf2 regulation: Implications for neurological dysfunction and disease. F1000Research 2018, 7, 1043. [Google Scholar] [CrossRef] [PubMed]
- Prema, A.; Janakiraman, U.; Manivasagam, T.; Thenmozhi, A.J. Neuroprotective effect of lycopene against MPTP induced experimental Parkinson’s disease in mice. Neurosci. Lett. 2015, 599, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.H.; Yen, T.L.; Hsu, C.Y.; Thomas, P.A.; Sheu, J.R.; Jayakumar, T. Multi-Targeting Andrographolide, a Novel NF-kappaB Inhibitor, as a Potential Therapeutic Agent for Stroke. Int. J. Mol. Sci. 2017, 18, 1638. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Mao, Y.; Li, H.; Shen, G.; Nan, G. Knockdown of Nrf2 inhibits angiogenesis by downregulating VEGF expression through PI3K/Akt signaling pathway in cerebral microvascular endothelial cells under hypoxic conditions. Biochem. Cell. Biol. 2018, 96, 475–482. [Google Scholar] [CrossRef] [PubMed]
- Bai, J.; Yu, X.J.; Liu, K.L.; Wang, F.F.; Jing, G.X.; Li, H.B.; Zhang, Y.; Huo, C.J.; Li, X.; Gao, H.L.; et al. Central administration of tert-butylhydroquinone attenuates hypertension via regulating Nrf2 signaling in the hypothalamic paraventricular nucleus of hypertensive rats. Toxicol. Appl. Pharmacol. 2017, 333, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Li, M.T.; Ke, J.; Guo, S.F.; Wu, Y.; Bian, Y.F.; Shan, L.L.; Liu, Q.Y.; Huo, Y.J.; Guo, C.; Liu, M.Y.; et al. The Protective Effect of Quercetin on Endothelial Cells Injured by Hypoxia and Reoxygenation. Front. Pharmacol. 2021, 12, 732874. [Google Scholar] [CrossRef]
- Sanders, O.; Rajagopal, L. Phosphodiesterase Inhibitors for Alzheimer’s Disease: A Systematic Review of Clinical Trials and Epidemiology with a Mechanistic Rationale. J. Alzheimer’s Dis. Rep. 2020, 4, 185–215. [Google Scholar] [CrossRef]
- Morishita, H.; Mizushima, N. Diverse Cellular Roles of Autophagy. Annu. Rev. Cell Dev. Biol. 2019, 35, 453–475. [Google Scholar] [CrossRef]
- Boya, P.; Reggiori, F.; Codogno, P. Emerging regulation and functions of autophagy. Nat. Cell Biol. 2013, 15, 713–720. [Google Scholar] [CrossRef]
- Kopitz, J.; Kisen, G.O.; Gordon, P.B.; Bohley, P.; Seglen, P.O. Nonselective autophagy of cytosolic enzymes by isolated rat hepatocytes. J. Cell Biol. 1990, 111, 941–953. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J. Autophagy: From phenomenology to molecular understanding in less than a decade. Nat. Rev. Mol. Cell Biol. 2007, 8, 931–937. [Google Scholar] [CrossRef] [PubMed]
- Dikic, I.; Elazar, Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 349–364. [Google Scholar] [CrossRef]
- Wu, M.; Lao, Y.Z.; Tan, H.S.; Lu, G.; Ren, Y.; Zheng, Z.Q.; Yi, J.; Fu, W.W.; Shen, H.M.; Xu, H.X. Oblongifolin C suppresses lysosomal function independently of TFEB nuclear translocation. Acta Pharmacol. Sin. 2019, 40, 929–937. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Zou, Z.; Becker, N.; Anderson, M.; Sumpter, R.; Xiao, G.; Kinch, L.; Koduru, P.; Christudass, C.S.; Veltri, R.W.; et al. EGFR-mediated Beclin 1 phosphorylation in autophagy suppression, tumor progression, and tumor chemoresistance. Cell 2013, 154, 1269–1284. [Google Scholar] [CrossRef] [PubMed]
- Sousa, C.M.; Biancur, D.E.; Wang, X.; Halbrook, C.J.; Sherman, M.H.; Zhang, L.; Kremer, D.; Hwang, R.F.; Witkiewicz, A.K.; Ying, H.; et al. Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature 2016, 536, 479–483. [Google Scholar] [CrossRef]
- He, C.; Bassik, M.C.; Moresi, V.; Sun, K.; Wei, Y.; Zou, Z.; An, Z.; Loh, J.; Fisher, J.; Sun, Q.; et al. Exercise-induced BCL2-regulated autophagy is required for muscle glucose homeostasis. Nature 2012, 481, 511–515. [Google Scholar] [CrossRef]
- Fernandez, A.F.; Sebti, S.; Wei, Y.; Zou, Z.; Shi, M.; McMillan, K.L.; He, C.; Ting, T.; Liu, Y.; Chiang, W.C.; et al. Disruption of the beclin 1-BCL2 autophagy regulatory complex promotes longevity in mice. Nature 2018, 558, 136–140. [Google Scholar] [CrossRef]
- Green, D.R.; Galluzzi, L.; Kroemer, G. Cell biology. Metabolic control of cell death. Science 2014, 345, 1250256. [Google Scholar] [CrossRef]
- Khaminets, A.; Heinrich, T.; Mari, M.; Grumati, P.; Huebner, A.K.; Akutsu, M.; Liebmann, L.; Stolz, A.; Nietzsche, S.; Koch, N.; et al. Regulation of endoplasmic reticulum turnover by selective autophagy. Nature 2015, 522, 354–358. [Google Scholar] [CrossRef]
- Jiang, T.; Harder, B.; Rojo de la Vega, M.; Wong, P.K.; Chapman, E.; Zhang, D.D. p62 links autophagy and Nrf2 signaling. Free Radic. Biol. Med. 2015, 88, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Amaravadi, R.; Kimmelman, A.C.; White, E. Recent insights into the function of autophagy in cancer. Genes Dev. 2016, 30, 1913–1930. [Google Scholar] [CrossRef] [PubMed]
- Marino, G.; Niso-Santano, M.; Baehrecke, E.H.; Kroemer, G. Self-consumption: The interplay of autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2014, 15, 81–94. [Google Scholar] [CrossRef]
- Mauthe, M.; Langereis, M.; Jung, J.; Zhou, X.; Jones, A.; Omta, W.; Tooze, S.A.; Stork, B.; Paludan, S.R.; Ahola, T.; et al. An siRNA screen for ATG protein depletion reveals the extent of the unconventional functions of the autophagy proteome in virus replication. J. Cell Biol. 2016, 214, 619–635. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Ni, D.; Ma, B.; Lee, J.H.; Zhang, T.; Ghozalli, I.; Pirooz, S.D.; Zhao, Z.; Bharatham, N.; Li, B.; et al. PtdIns(3)P-bound UVRAG coordinates Golgi-ER retrograde and Atg9 transport by differential interactions with the ER tether and the beclin 1 complex. Nat. Cell Biol. 2013, 15, 1206–1219. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Iyengar, R.; Li-Harms, X.; Joo, J.H.; Wright, C.; Lavado, A.; Horner, L.; Yang, M.; Guan, J.L.; Frase, S.; et al. The autophagy-inducing kinases, ULK1 and ULK2, regulate axon guidance in the developing mouse forebrain via a noncanonical pathway. Autophagy 2018, 14, 796–811. [Google Scholar] [CrossRef]
- Wang, B.; Kundu, M. Canonical and noncanonical functions of ULK/Atg1. Curr. Opin. Cell Biol. 2017, 45, 47–54. [Google Scholar] [CrossRef]
- Imagawa, Y.; Saitoh, T.; Tsujimoto, Y. Vital staining for cell death identifies Atg9a-dependent necrosis in developmental bone formation in mouse. Nat. Commun. 2016, 7, 13391. [Google Scholar] [CrossRef]
- Park, J.M.; Tougeron, D.; Huang, S.; Okamoto, K.; Sinicrope, F.A. Beclin 1 and UVRAG confer protection from radiation-induced DNA damage and maintain centrosome stability in colorectal cancer cells. PLoS ONE 2014, 9, e100819. [Google Scholar] [CrossRef]
- Yang, Y.; He, S.; Wang, Q.; Li, F.; Kwak, M.J.; Chen, S.; O’Connell, D.; Zhang, T.; Pirooz, S.D.; Jeon, Y.H.; et al. Autophagic UVRAG Promotes UV-Induced Photolesion Repair by Activation of the CRL4(DDB2) E3 Ligase. Mol. Cell 2016, 62, 507–519. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Jang, G.B.; Yang, X.; Wang, Q.; He, S.; Li, S.; Quach, C.; Zhao, S.; Li, F.; Yuan, Z.; et al. Central role of autophagic UVRAG in melanogenesis and the suntan response. Proc. Natl. Acad. Sci. USA 2018, 115, E7728–E7737. [Google Scholar] [CrossRef] [PubMed]
- Thoresen, S.B.; Pedersen, N.M.; Liestol, K.; Stenmark, H. A phosphatidylinositol 3-kinase class III sub-complex containing VPS15, VPS34, Beclin 1, UVRAG and BIF-1 regulates cytokinesis and degradative endocytic traffic. Exp. Cell Res. 2010, 316, 3368–3378. [Google Scholar] [CrossRef] [PubMed]
- McKnight, N.C.; Zhong, Y.; Wold, M.S.; Gong, S.; Phillips, G.R.; Dou, Z.; Zhao, Y.; Heintz, N.; Zong, W.X.; Yue, Z. Beclin 1 is required for neuron viability and regulates endosome pathways via the UVRAG-VPS34 complex. PLoS Genet. 2014, 10, e1004626. [Google Scholar] [CrossRef] [PubMed]
- Martinez, J.; Malireddi, R.K.; Lu, Q.; Cunha, L.D.; Pelletier, S.; Gingras, S.; Orchard, R.; Guan, J.L.; Tan, H.; Peng, J.; et al. Molecular characterization of LC3-associated phagocytosis reveals distinct roles for Rubicon, NOX2 and autophagy proteins. Nat. Cell Biol. 2015, 17, 893–906. [Google Scholar] [CrossRef]
- Zhao, Z.; Oh, S.; Li, D.; Ni, D.; Pirooz, S.D.; Lee, J.H.; Yang, S.; Lee, J.Y.; Ghozalli, I.; Costanzo, V.; et al. A dual role for UVRAG in maintaining chromosomal stability independent of autophagy. Dev. Cell 2012, 22, 1001–1016. [Google Scholar] [CrossRef]
- Song, Y.; Quach, C.; Liang, C. UVRAG in autophagy, inflammation, and cancer. Autophagy 2020, 16, 387–388. [Google Scholar] [CrossRef]
- Tung, S.M.; Unal, C.; Ley, A.; Pena, C.; Tunggal, B.; Noegel, A.A.; Krut, O.; Steinert, M.; Eichinger, L. Loss of Dictyostelium ATG9 results in a pleiotropic phenotype affecting growth, development, phagocytosis and clearance and replication of Legionella pneumophila. Cell. Microbiol. 2010, 12, 765–780. [Google Scholar] [CrossRef]
- Liu, Y.; Shoji-Kawata, S.; Sumpter, R.M., Jr.; Wei, Y.; Ginet, V.; Zhang, L.; Posner, B.; Tran, K.A.; Green, D.R.; Xavier, R.J.; et al. Autosis is a Na+,K+-ATPase-regulated form of cell death triggered by autophagy-inducing peptides, starvation, and hypoxia-ischemia. Proc. Natl. Acad. Sci. USA 2013, 110, 20364–20371. [Google Scholar] [CrossRef]
- Gao, W.; Shen, Z.; Shang, L.; Wang, X. Upregulation of human autophagy-initiation kinase ULK1 by tumor suppressor p53 contributes to DNA-damage-induced cell death. Cell Death Differ. 2011, 18, 1598–1607. [Google Scholar] [CrossRef]
- Iershov, A.; Nemazanyy, I.; Alkhoury, C.; Girard, M.; Barth, E.; Cagnard, N.; Montagner, A.; Chretien, D.; Rugarli, E.I.; Guillou, H.; et al. The class 3 PI3K coordinates autophagy and mitochondrial lipid catabolism by controlling nuclear receptor PPARalpha. Nat. Commun. 2019, 10, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Mercer, T.J.; Ohashi, Y.; Boeing, S.; Jefferies, H.B.J.; De Tito, S.; Flynn, H.; Tremel, S.; Zhang, W.; Wirth, M.; Frith, D.; et al. Phosphoproteomic identification of ULK substrates reveals VPS15-dependent ULK/VPS34 interplay in the regulation of autophagy. EMBO J. 2021, 40, e105985. [Google Scholar] [CrossRef]
- Goodall, M.L.; Fitzwalter, B.E.; Zahedi, S.; Wu, M.; Rodriguez, D.; Mulcahy-Levy, J.M.; Green, D.R.; Morgan, M.; Cramer, S.D.; Thorburn, A. The Autophagy Machinery Controls Cell Death Switching between Apoptosis and Necroptosis. Dev. Cell 2016, 37, 337–349. [Google Scholar] [CrossRef] [PubMed]
- Strappazzon, F.; Di Rita, A.; Cianfanelli, V.; D’Orazio, M.; Nazio, F.; Fimia, G.M.; Cecconi, F. Prosurvival AMBRA1 turns into a proapoptotic BH3-like protein during mitochondrial apoptosis. Autophagy 2016, 12, 963–975. [Google Scholar] [CrossRef] [PubMed]
- Nazio, F.; Po, A.; Abballe, L.; Ballabio, C.; Diomedi Camassei, F.; Bordi, M.; Camera, A.; Caruso, S.; Caruana, I.; Pezzullo, M.; et al. Targeting cancer stem cells in medulloblastoma by inhibiting AMBRA1 dual function in autophagy and STAT3 signalling. Acta Neuropathol. 2021, 142, 537–564. [Google Scholar] [CrossRef] [PubMed]
- Cianfanelli, V.; Fuoco, C.; Lorente, M.; Salazar, M.; Quondamatteo, F.; Gherardini, P.F.; De Zio, D.; Nazio, F.; Antonioli, M.; D’Orazio, M.; et al. AMBRA1 links autophagy to cell proliferation and tumorigenesis by promoting c-Myc dephosphorylation and degradation. Nat. Cell Biol. 2015, 17, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Zhao, H.; Martinez, J.; Doggett, T.A.; Kolesnikov, A.V.; Tang, P.H.; Ablonczy, Z.; Chan, C.C.; Zhou, Z.; Green, D.R.; et al. Noncanonical autophagy promotes the visual cycle. Cell 2013, 154, 365–376. [Google Scholar] [CrossRef]
- Li, X.; Yang, K.B.; Chen, W.; Mai, J.; Wu, X.Q.; Sun, T.; Wu, R.Y.; Jiao, L.; Li, D.D.; Ji, J.; et al. CUL3 (cullin 3)-mediated ubiquitination and degradation of BECN1 (beclin 1) inhibit autophagy and promote tumor progression. Autophagy 2021, 17, 4323–4340. [Google Scholar] [CrossRef]
- Frangez, Z.; Fernandez-Marrero, Y.; Stojkov, D.; Seyed Jafari, S.M.; Hunger, R.E.; Djonov, V.; Riether, C.; Simon, H.U. BIF-1 inhibits both mitochondrial and glycolytic ATP production: Its downregulation promotes melanoma growth. Oncogene 2020, 39, 4944–4955. [Google Scholar] [CrossRef]
- Takahashi, Y.; Coppola, D.; Matsushita, N.; Cualing, H.D.; Sun, M.; Sato, Y.; Liang, C.; Jung, J.U.; Cheng, J.Q.; Mule, J.J.; et al. Bif-1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesis. Nat. Cell Biol. 2007, 9, 1142–1151. [Google Scholar] [CrossRef]
- Cheng, X.; Sun, Q. RUBCNL/Pacer and RUBCN/Rubicon in regulation of autolysosome formation and lipid metabolism. Autophagy 2019, 15, 1120–1121. [Google Scholar] [CrossRef]
- O’Sullivan, T.E.; Geary, C.D.; Weizman, O.E.; Geiger, T.L.; Rapp, M.; Dorn, G.W., 2nd; Overholtzer, M.; Sun, J.C. Atg5 Is Essential for the Development and Survival of Innate Lymphocytes. Cell Rep. 2016, 15, 1910–1919. [Google Scholar] [CrossRef] [PubMed]
- Murrow, L.; Debnath, J. ATG12-ATG3 connects basal autophagy and late endosome function. Autophagy 2015, 11, 961–962. [Google Scholar] [CrossRef]
- DeSelm, C.J.; Miller, B.C.; Zou, W.; Beatty, W.L.; van Meel, E.; Takahata, Y.; Klumperman, J.; Tooze, S.A.; Teitelbaum, S.L.; Virgin, H.W. Autophagy proteins regulate the secretory component of osteoclastic bone resorption. Dev. Cell 2011, 21, 966–974. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.; Maloney, N.S.; Bruinsma, M.W.; Goel, G.; Duan, E.; Zhang, L.; Shrestha, B.; Diamond, M.S.; Dani, A.; Sosnovtsev, S.V.; et al. Nondegradative role of Atg5-Atg12/ Atg16L1 autophagy protein complex in antiviral activity of interferon gamma. Cell Host Microbe 2012, 11, 397–409. [Google Scholar] [CrossRef]
- Pei, B.; Zhao, M.; Miller, B.C.; Vela, J.L.; Bruinsma, M.W.; Virgin, H.W.; Kronenberg, M. Invariant NKT cells require autophagy to coordinate proliferation and survival signals during differentiation. J. Immunol. 2015, 194, 5872–5884. [Google Scholar] [CrossRef]
- Dupont, N.; Jiang, S.; Pilli, M.; Ornatowski, W.; Bhattacharya, D.; Deretic, V. Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1beta. EMBO J. 2011, 30, 4701–4711. [Google Scholar] [CrossRef]
- Xu, X.; Araki, K.; Li, S.; Han, J.H.; Ye, L.; Tan, W.G.; Konieczny, B.T.; Bruinsma, M.W.; Martinez, J.; Pearce, E.L.; et al. Autophagy is essential for effector CD8(+) T cell survival and memory formation. Nat. Immunol. 2014, 15, 1152–1161. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Kenny, S.J.; Ge, L.; Xu, K.; Schekman, R. Translocation of interleukin-1beta into a vesicle intermediate in autophagy-mediated secretion. Elife 2015, 4, e11205. [Google Scholar] [CrossRef]
- Kimmey, J.M.; Huynh, J.P.; Weiss, L.A.; Park, S.; Kambal, A.; Debnath, J.; Virgin, H.W.; Stallings, C.L. Unique role for ATG5 in neutrophil-mediated immunopathology during M. tuberculosis infection. Nature 2015, 528, 565–569. [Google Scholar] [CrossRef]
- Lee, I.H.; Kawai, Y.; Fergusson, M.M.; Rovira, I.I.; Bishop, A.J.; Motoyama, N.; Cao, L.; Finkel, T. Atg7 modulates p53 activity to regulate cell cycle and survival during metabolic stress. Science 2012, 336, 225–228. [Google Scholar] [CrossRef] [Green Version]
- Henault, J.; Martinez, J.; Riggs, J.M.; Tian, J.; Mehta, P.; Clarke, L.; Sasai, M.; Latz, E.; Brinkmann, M.M.; Iwasaki, A.; et al. Noncanonical autophagy is required for type I interferon secretion in response to DNA-immune complexes. Immunity 2012, 37, 986–997. [Google Scholar] [CrossRef] [PubMed]
- Selleck, E.M.; Orchard, R.C.; Lassen, K.G.; Beatty, W.L.; Xavier, R.J.; Levine, B.; Virgin, H.W.; Sibley, L.D. A Noncanonical Autophagy Pathway Restricts Toxoplasma gondii Growth in a Strain-Specific Manner in IFN-gamma-Activated Human Cells. mBio 2015, 6, e01157-15. [Google Scholar] [CrossRef] [PubMed]
- Sanjuan, M.A.; Dillon, C.P.; Tait, S.W.; Moshiach, S.; Dorsey, F.; Connell, S.; Komatsu, M.; Tanaka, K.; Cleveland, J.L.; Withoff, S.; et al. Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature 2007, 450, 1253–1257. [Google Scholar] [CrossRef]
- Haller, M.; Hock, A.K.; Giampazolias, E.; Oberst, A.; Green, D.R.; Debnath, J.; Ryan, K.M.; Vousden, K.H.; Tait, S.W. Ubiquitination and proteasomal degradation of ATG12 regulates its proapoptotic activity. Autophagy 2014, 10, 2269–2278. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Chitiprolu, M.; Roncevic, L.; Javalet, C.; Hemming, F.J.; Trung, M.T.; Meng, L.; Latreille, E.; Tanese de Souza, C.; McCulloch, D.; et al. Atg5 Disassociates the V1V0-ATPase to Promote Exosome Production and Tumor Metastasis Independent of Canonical Macroautophagy. Dev. Cell 2017, 43, 716–730.e7. [Google Scholar] [CrossRef] [PubMed]
- Sorbara, M.T.; Ellison, L.K.; Ramjeet, M.; Travassos, L.H.; Jones, N.L.; Girardin, S.E.; Philpott, D.J. The protein ATG16L1 suppresses inflammatory cytokines induced by the intracellular sensors Nod1 and Nod2 in an autophagy-independent manner. Immunity 2013, 39, 858–873. [Google Scholar] [CrossRef]
- Alirezaei, M.; Flynn, C.T.; Wood, M.R.; Harkins, S.; Whitton, J.L. Coxsackievirus can exploit LC3 in both autophagy-dependent and -independent manners in vivo. Autophagy 2015, 11, 1389–1407. [Google Scholar] [CrossRef]
- Al-Younes, H.M.; Al-Zeer, M.A.; Khalil, H.; Gussmann, J.; Karlas, A.; Machuy, N.; Brinkmann, V.; Braun, P.R.; Meyer, T.F. Autophagy-independent function of MAP-LC3 during intracellular propagation of Chlamydia trachomatis. Autophagy 2011, 7, 814–828. [Google Scholar] [CrossRef]
- Wong, J.; Zhang, J.; Si, X.; Gao, G.; Mao, I.; McManus, B.M.; Luo, H. Autophagosome supports coxsackievirus B3 replication in host cells. J. Virol. 2008, 82, 9143–9153. [Google Scholar] [CrossRef]
- Joo, J.H.; Wang, B.; Frankel, E.; Ge, L.; Xu, L.; Iyengar, R.; Li-Harms, X.; Wright, C.; Shaw, T.I.; Lindsten, T.; et al. The Noncanonical Role of ULK/ATG1 in ER-to-Golgi Trafficking Is Essential for Cellular Homeostasis. Mol. Cell 2016, 62, 491–506. [Google Scholar] [CrossRef] [Green Version]
- Liang, C.; Lee, J.S.; Inn, K.S.; Gack, M.U.; Li, Q.; Roberts, E.A.; Vergne, I.; Deretic, V.; Feng, P.; Akazawa, C.; et al. Beclin1-binding UVRAG targets the class C Vps complex to coordinate autophagosome maturation and endocytic trafficking. Nat. Cell Biol. 2008, 10, 776–787. [Google Scholar] [CrossRef] [PubMed]
- Tan, E.H.N.; Tang, B.L. Rab7a and Mitophagosome Formation. Cells 2019, 8, 224. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Kroemer, G. Biological Functions of Autophagy Genes: A Disease Perspective. Cell 2019, 176, 11–42. [Google Scholar] [CrossRef] [PubMed]
- Hei, C.; Liu, P.; Yang, X.; Niu, J.; Li, P.A. Inhibition of mTOR signaling Confers Protection against Cerebral Ischemic Injury in Acute Hyperglycemic Rats. Int. J. Biol. Sci. 2017, 13, 878–887. [Google Scholar] [CrossRef]
- Vercelli, A.; Biggi, S.; Sclip, A.; Repetto, I.E.; Cimini, S.; Falleroni, F.; Tomasi, S.; Monti, R.; Tonna, N.; Morelli, F.; et al. Exploring the role of MKK7 in excitotoxicity and cerebral ischemia: A novel pharmacological strategy against brain injury. Cell Death Dis. 2015, 6, e1854. [Google Scholar] [CrossRef]
- Wang, Y.; Zhen, Y.; Wu, X.; Jiang, Q.; Li, X.; Chen, Z.; Zhang, G.; Dong, L. Vitexin protects brain against ischemia/reperfusion injury via modulating mitogen-activated protein kinase and apoptosis signaling in mice. Phytomedicine 2015, 22, 379–384. [Google Scholar] [CrossRef]
- Han, D.; Scott, E.L.; Dong, Y.; Raz, L.; Wang, R.; Zhang, Q. Attenuation of mitochondrial and nuclear p38alpha signaling: A novel mechanism of estrogen neuroprotection in cerebral ischemia. Mol. Cell. Endocrinol. 2015, 400, 21–31. [Google Scholar] [CrossRef]
- Lv, B.; Li, F.; Han, J.; Fang, J.; Xu, L.; Sun, C.; Hua, T.; Zhang, Z.; Feng, Z.; Jiang, X. Hif-1alpha Overexpression Improves Transplanted Bone Mesenchymal Stem Cells Survival in Rat MCAO Stroke Model. Front. Mol. Neurosci. 2017, 10, 80. [Google Scholar] [CrossRef]
- Li, X.; Zhang, D.; Bai, Y.; Xiao, J.; Jiao, H.; He, R. Ginaton improves neurological function in ischemic stroke rats via inducing autophagy and maintaining mitochondrial homeostasis. Neuropsychiatr. Dis. Treat. 2019, 15, 1813–1822. [Google Scholar] [CrossRef] [PubMed]
- Yan, B.C.; Wang, J.; Rui, Y.; Cao, J.; Xu, P.; Jiang, D.; Zhu, X.; Won, M.H.; Bo, P.; Su, P. Neuroprotective Effects of Gabapentin Against Cerebral Ischemia Reperfusion-Induced Neuronal Autophagic Injury via Regulation of the PI3K/Akt/mTOR Signaling Pathways. J. Neuropathol. Exp. Neurol. 2019, 78, 157–171. [Google Scholar] [CrossRef] [Green Version]
- Tao, J.; Shen, C.; Sun, Y.; Chen, W.; Yan, G. Neuroprotective effects of pinocembrin on ischemia/reperfusion-induced brain injury by inhibiting autophagy. Biomed. Pharmacother. 2018, 106, 1003–1010. [Google Scholar] [CrossRef] [PubMed]
- Shi, Q.; Zhang, Q.; Peng, Y.; Zhang, X.; Wang, Y.; Shi, L. A natural diarylheptanoid protects cortical neurons against oxygen-glucose deprivation-induced autophagy and apoptosis. J. Pharm. Pharmacol. 2019, 71, 1110–1118. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Zhao, T.Z.; Zou, Y.J.; Zhang, J.H.; Feng, H. Hypoxia Induces autophagic cell death through hypoxia-inducible factor 1alpha in microglia. PLoS ONE 2014, 9, e96509. [Google Scholar] [CrossRef]
- Chen, C.M.; Wu, C.T.; Yang, T.H.; Chang, Y.A.; Sheu, M.L.; Liu, S.H. Green Tea Catechin Prevents Hypoxia/Reperfusion-Evoked Oxidative Stress-Regulated Autophagy-Activated Apoptosis and Cell Death in Microglial Cells. J. Agric. Food Chem. 2016, 64, 4078–4085. [Google Scholar] [CrossRef] [PubMed]
- Mo, Y.; Sun, Y.Y.; Liu, K.Y. Autophagy and inflammation in ischemic stroke. Neural Regen. Res. 2020, 15, 1388–1396. [Google Scholar] [CrossRef]
- Kasprowska, D.; Machnik, G.; Kost, A.; Gabryel, B. Time-Dependent Changes in Apoptosis Upon Autophagy Inhibition in Astrocytes Exposed to Oxygen and Glucose Deprivation. Cell. Mol. Neurobiol. 2017, 37, 223–234. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, X.; Wei, Q.; Leng, S.; Li, C.; Han, B.; Bai, Y.; Zhang, H.; Yao, H. Activation of Sigma-1 Receptor Enhanced Pericyte Survival via the Interplay Between Apoptosis and Autophagy: Implications for Blood-Brain Barrier Integrity in Stroke. Transl. Stroke Res. 2020, 11, 267–287. [Google Scholar] [CrossRef]
- Li, H.; Gao, A.; Feng, D.; Wang, Y.; Zhang, L.; Cui, Y.; Li, B.; Wang, Z.; Chen, G. Evaluation of the protective potential of brain microvascular endothelial cell autophagy on blood-brain barrier integrity during experimental cerebral ischemia-reperfusion injury. Transl. Stroke Res. 2014, 5, 618–626. [Google Scholar] [CrossRef]
- Wang, S.; Han, X.; Mao, Z.; Xin, Y.; Maharjan, S.; Zhang, B. MALAT1 lncRNA Induces Autophagy and Protects Brain Microvascular Endothelial Cells Against Oxygen-Glucose Deprivation by Binding to miR-200c-3p and Upregulating SIRT1 Expression. Neuroscience 2019, 397, 116–126. [Google Scholar] [CrossRef]
- Almeida, M.R.; Silva, A.R.; Elias, I.; Fernandes, C.; Machado, R.; Galego, O.; Santo, G.C. SQSTM1 gene as a potential genetic modifier of CADASIL phenotype. J. Neurol. 2021, 268, 1453–1460. [Google Scholar] [CrossRef]
- Hanemaaijer, E.S.; Panahi, M.; Swaddiwudhipong, N.; Tikka, S.; Winblad, B.; Viitanen, M.; Piras, A.; Behbahani, H. Autophagy-lysosomal defect in human CADASIL vascular smooth muscle cells. Eur. J. Cell Biol. 2018, 97, 557–567. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Huang, Y.; Cai, W.; Chen, X.; Men, X.; Lu, T.; Wu, A.; Lu, Z. Age-related cerebral small vessel disease and inflammaging. Cell Death Dis. 2020, 11, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Guan, Z.F.; Zhou, X.L.; Zhang, X.M.; Zhang, Y.; Wang, Y.M.; Guo, Q.L.; Ji, G.; Wu, G.F.; Wang, N.N.; Yang, H.; et al. Beclin-1- mediated autophagy may be involved in the elderly cognitive and affective disorders in streptozotocin-induced diabetic mice. Transl. Neurodegener. 2016, 5, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.X.; Zhang, B.; Xia, R.; Jia, Q.Y. Inflammation, apoptosis and autophagy as critical players in vascular dementia. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 9601–9614. [Google Scholar] [CrossRef] [PubMed]
- Berlit, P.; Keyvani, K.; Kramer, M.; Weber, R. Zerebrale Amyloidangiopathie und Demenz. Nervenarzt 2015, 86, 1248–1254. [Google Scholar] [CrossRef]
- Inami, Y.; Waguri, S.; Sakamoto, A.; Kouno, T.; Nakada, K.; Hino, O.; Watanabe, S.; Ando, J.; Iwadate, M.; Yamamoto, M.; et al. Persistent activation of Nrf2 through p62 in hepatocellular carcinoma cells. J. Cell Biol. 2011, 193, 275–284. [Google Scholar] [CrossRef]
- Ni, H.M.; Boggess, N.; McGill, M.R.; Lebofsky, M.; Borude, P.; Apte, U.; Jaeschke, H.; Ding, W.X. Liver-specific loss of Atg5 causes persistent activation of Nrf2 and protects against acetaminophen-induced liver injury. Toxicol. Sci. 2012, 127, 438–450. [Google Scholar] [CrossRef]
- Ni, H.M.; Woolbright, B.L.; Williams, J.; Copple, B.; Cui, W.; Luyendyk, J.P.; Jaeschke, H.; Ding, W.X. Nrf2 promotes the development of fibrosis and tumorigenesis in mice with defective hepatic autophagy. J. Hepatol. 2014, 61, 617–625. [Google Scholar] [CrossRef]
- Lam, H.C.; Cloonan, S.M.; Bhashyam, A.R.; Haspel, J.A.; Singh, A.; Sathirapongsasuti, J.F.; Cervo, M.; Yao, H.; Chung, A.L.; Mizumura, K.; et al. Histone deacetylase 6-mediated selective autophagy regulates COPD-associated cilia dysfunction. J. Clin. Investig. 2020, 130, 6189. [Google Scholar] [CrossRef]
- Tanida, I. Autophagosome formation and molecular mechanism of autophagy. Antioxid. Redox Signal. 2011, 14, 2201–2214. [Google Scholar] [CrossRef]
- Komatsu, M.; Kurokawa, H.; Waguri, S.; Taguchi, K.; Kobayashi, A.; Ichimura, Y.; Sou, Y.S.; Ueno, I.; Sakamoto, A.; Tong, K.I.; et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 2010, 12, 213–223. [Google Scholar] [CrossRef]
- Komatsu, M.; Waguri, S.; Koike, M.; Sou, Y.S.; Ueno, T.; Hara, T.; Mizushima, N.; Iwata, J.; Ezaki, J.; Murata, S.; et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell 2007, 131, 1149–1163. [Google Scholar] [CrossRef] [PubMed]
- Lau, A.; Wang, X.J.; Zhao, F.; Villeneuve, N.F.; Wu, T.; Jiang, T.; Sun, Z.; White, E.; Zhang, D.D. A noncanonical mechanism of Nrf2 activation by autophagy deficiency: Direct interaction between Keap1 and p62. Mol. Cell Biol. 2010, 30, 3275–3285. [Google Scholar] [CrossRef] [PubMed]
- Copple, I.M.; Lister, A.; Obeng, A.D.; Kitteringham, N.R.; Jenkins, R.E.; Layfield, R.; Foster, B.J.; Goldring, C.E.; Park, B.K. Physical and functional interaction of sequestosome 1 with Keap1 regulates the Keap1-Nrf2 cell defense pathway. J. Biol. Chem. 2010, 285, 16782–16788. [Google Scholar] [CrossRef] [PubMed]
- Bae, S.H.; Sung, S.H.; Oh, S.Y.; Lim, J.M.; Lee, S.K.; Park, Y.N.; Lee, H.E.; Kang, D.; Rhee, S.G. Sestrins activate Nrf2 by promoting p62-dependent autophagic degradation of Keap1 and prevent oxidative liver damage. Cell Metab. 2013, 17, 73–84. [Google Scholar] [CrossRef]
- Baird, L.; Lleres, D.; Swift, S.; Dinkova-Kostova, A.T. Regulatory flexibility in the Nrf2-mediated stress response is conferred by conformational cycling of the Keap1-Nrf2 protein complex. Proc. Natl. Acad. Sci. USA 2013, 110, 15259–15264. [Google Scholar] [CrossRef]
- Jain, A.; Lamark, T.; Sjottem, E.; Larsen, K.B.; Awuh, J.A.; Overvatn, A.; McMahon, M.; Hayes, J.D.; Johansen, T. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J. Biol. Chem. 2010, 285, 22576–22591. [Google Scholar] [CrossRef]
- Liao, W.; Wang, Z.; Fu, Z.; Ma, H.; Jiang, M.; Xu, A.; Zhang, W. p62/SQSTM1 protects against cisplatin-induced oxidative stress in kidneys by mediating the cross talk between autophagy and the Keap1-Nrf2 signalling pathway. Free Radic. Res. 2019, 53, 800–814. [Google Scholar] [CrossRef]
- Kong, L.; Deng, J.; Zhou, X.; Cai, B.; Zhang, B.; Chen, X.; Chen, Z.; Wang, W. Sitagliptin activates the p62-Keap1-Nrf2 signalling pathway to alleviate oxidative stress and excessive autophagy in severe acute pancreatitis-related acute lung injury. Cell Death Dis. 2021, 12, 1–11. [Google Scholar] [CrossRef]
- Yang, S.; Li, F.; Lu, S.; Ren, L.; Bian, S.; Liu, M.; Zhao, D.; Wang, S.; Wang, J. Ginseng root extract attenuates inflammation by inhibiting the MAPK/NF-kappaB signaling pathway and activating autophagy and p62-Nrf2-Keap1 signaling in vitro and in vivo. J. Ethnopharmacol. 2022, 283, 114739. [Google Scholar] [CrossRef]
- Chen, P.; Yang, J.; Wu, N.; Han, B.; Kastelic, J.P.; Gao, J. Streptococcus lutetiensis Induces Autophagy via Oxidative Stress in Bovine Mammary Epithelial Cells. Oxidative Med. Cell. Longev. 2022, 2022, 2549772. [Google Scholar] [CrossRef] [PubMed]
- Jo, C.; Gundemir, S.; Pritchard, S.; Jin, Y.N.; Rahman, I.; Johnson, G.V. Nrf2 reduces levels of phosphorylated tau protein by inducing autophagy adaptor protein NDP52. Nat. Commun. 2014, 5, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Chan, P.H. Role of oxidants in ischemic brain damage. Stroke 1996, 27, 1124–1129. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Chen, J.; Sun, S.; Zhao, J.; Dong, X.; Wang, J. Effects of Estradiol on Autophagy and Nrf-2/ARE Signals after Cerebral Ischemia. Cell. Physiol. Biochem. 2017, 41, 2027–2036. [Google Scholar] [CrossRef] [PubMed]
- Amin, N.; Du, X.; Chen, S.; Ren, Q.; Hussien, A.B.; Botchway, B.O.A.; Hu, Z.; Fang, M. Therapeutic impact of thymoquninone to alleviate ischemic brain injury via Nrf2/HO-1 pathway. Expert Opin. Ther. Targets 2021, 25, 597–612. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Zhou, X.; Li, Y.; Ma, S.; Pan, L.; Zhang, X.; Zheng, W.; Wu, Z.; Wang, K.; Ahsan, A.; et al. TIGAR alleviates oxidative stress in brain with extended ischemia via a pentose phosphate pathway-independent manner. Redox Biol. 2022, 53, 102323. [Google Scholar] [CrossRef] [PubMed]
- Qi, S.; Zhang, X.; Fu, Z.; Pi, A.; Shi, F.; Fan, Y.; Zhang, J.; Xiao, T.; Shang, D.; Lin, M.; et al. (+/-)-5-bromo-2-(5-fluoro-1-hydroxyamyl) Benzoate Protects Against Oxidative Stress Injury in PC12 Cells Exposed to H2O2 Through Activation of Nrf2 Pathway. Front. Pharmacol. 2022, 13, 943111. [Google Scholar] [CrossRef] [PubMed]
- Cai, S.C.; Yi, C.A.; Hu, X.S.; Tang, G.Y.; Yi, L.M.; Li, X.P. Isoquercitrin Upregulates Aldolase C Through Nrf2 to Ameliorate OGD/R-Induced Damage in SH-SY5Y Cells. Neurotox. Res. 2021, 39, 1959–1969. [Google Scholar] [CrossRef]
- Kaviarasi, S.; Yuba, E.; Harada, A.; Krishnan, U.M. Emerging paradigms in nanotechnology for imaging and treatment of cerebral ischemia. J. Control. Release 2019, 300, 22–45. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Autophagy Process | Protein | Autophagic Role | Non-Autophagic Role(s) | References |
|---|---|---|---|---|
| initiation | FIP200 | component of ULK complex (possibly scaffolding function) | pathogen control | [63,74] |
| ULK1 | serine/threonine kinase; initiates autophagy by phosphorylating components of the autophagy machinery | cytokine secretion, ER-to-GA anterograde transport, vesicular trafficking | [63,75,76,77] | |
| ULK2 | initiates autophagy by phosphorylating components of the autophagy machinery | ER-to-GA anterograde transport, vesicular trafficking | [76,77] | |
| UVRAG | a binding partner and promoter of the BECN1/Beclin 1-associated lipid kinase PIK3C3/Vps34, autophagosome biogenesis | cell proliferation, centrosome functions, cytokinesis, DNA repair, endocytosis, GA-to-ER retrograde transport, LAP, melanogenesis, anti-inflammatory, tumor suppression | [78,79,80,81,82,83,84,85,86] | |
| ATG9 | delivery of membrane material to the phagophore | ADCD, phagocytosis | [63,78,87] | |
| ATG14 | PI3KC3–C1 targeting the PAS and expanding phagophore | ADCD | [63,88] | |
| nucleation | ATG13 | adaptor mediating the interaction between ULK1 and FIP200, enhances ULK1 kinase activity | ADCD, pathogen control | [63,74,89] |
| VPS15 | ULK-dependent phosphorylation | cytokinesis, endocytosis, mitochondrial metabolism | [82,90,91] | |
| VPS34 | catalytic component of PI3KC3–C1, generates PI3P in the phagophore, and stabilizes the ULK complex | ADCD, cytokinesis, endocytosis, GA-to-ER retrograde transport, LAP, vesicular trafficking | [63,75,82,84,92] | |
| AMBRA1 | downstream target of mTOR | ADCD, tumor suppression, cell proliferation | [93,94,95] | |
| BECN1 | autophagosomes formation, extension, and maturation | ADCD, LAP, centrosome functions, cytokinesis, endocytosis, vision cycle, tumor progression | [79,82,84,88,96,97] | |
| BIF-1 | colocalization with Atg5 and LC3, autophagosome formation | cytokinesis, endocytosis, tumor progress or suppression | [82,98,99] | |
| RUBCN | the class III PtdIns3K and HOPS complexes engagement | LAP, vision cycle | [84,96,100] | |
| elongation | ATG3 | E2-like enzyme, conjugation of activated ATG8s to membranal PE | cell proliferation, exosome secretion, LAP | [63,84,101,102] |
| ATG4B | cysteine protease that processes pro-ATG8s, deconjugation of lipidated LC3 and ATG8s | granule exocytosis, LAP | [63,84,103] | |
| ATG5 | E3-like complex that couples ATG8s to PE | ADCD, cell proliferation, exosome secretion, granule exocytosis, immunological memory, LAP, non-canonical protein secretion, pathogen control, vision cycle | [63,84,96,103,104,105,106,107,108,109] | |
| ATG7 | E1-like enzyme, activation of ATG8, conjugation of ATG12 to ATG5 | ADCD, cell proliferation, cytokine secretion, exosome secretion, granule exocytosis, immunological memory, LAP, pathogen control, PRR signaling | [63,84,92,103,106,107,110,111,112,113] | |
| ATG12 | E3-like complex that couples ATG8s to PE | ADCD, exosome secretion, LAP, pathogen control | [63,84,102,104,114] | |
| ATG16L1 | E3-like complex that couples ATG8s to PE | exosome secretion, LAP, phagocytosis, pathogen control, PRR signaling | [63,84,104,113,115,116] | |
| cargo selection | LC3 | interaction with autophagy receptors, phagophore expansion, and sealing | bacterial replication, cytokine secretion, granule exocytosis, LAP, pathogen control, vision cycle, viral replication, viral release | [63,84,96,106,111,113,117,118,119] |
| NDP52 | autophagy receptor | pathogen control | [63,112] | |
| p62 | autophagy receptor | ADCD, pathogen control | [63,92,112] | |
| fusion | RAB7A | correct targeting of ATG9a-bearing vesicles | endocytosis, exosome secretion, granule exocytosis, non-canonical protein secretion | [103,113,120,121,122] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, Y.; Luo, Y.; Zheng, Y. Nrf2 Pathway and Autophagy Crosstalk: New Insights into Therapeutic Strategies for Ischemic Cerebral Vascular Diseases. Antioxidants 2022, 11, 1747. https://doi.org/10.3390/antiox11091747
Hu Y, Luo Y, Zheng Y. Nrf2 Pathway and Autophagy Crosstalk: New Insights into Therapeutic Strategies for Ischemic Cerebral Vascular Diseases. Antioxidants. 2022; 11(9):1747. https://doi.org/10.3390/antiox11091747
Chicago/Turabian StyleHu, Yue, Yumin Luo, and Yangmin Zheng. 2022. "Nrf2 Pathway and Autophagy Crosstalk: New Insights into Therapeutic Strategies for Ischemic Cerebral Vascular Diseases" Antioxidants 11, no. 9: 1747. https://doi.org/10.3390/antiox11091747