
Analysis of Thioredoxins and Glutaredoxins in Soybean: Evidence of Translational Regulation under Water Restriction

 ,
,  and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sequence Retrieval and Identification and Initial Characterization of Trx and Grx Family Members
2.2. Plant Growth and Drought Assay
2.3. Polysomal Fraction Purification by Sucrose Cushion Centrifugation
2.3.1. Preparation of Cytoplasmic Lysates
2.3.2. Sucrose Cushion Centrifugation and Polysome Purification
2.4. TOTAL and PAR RNA Fraction Extraction and Transcriptome Sequencing
2.5. Processing of Sequencing Data
3. Results
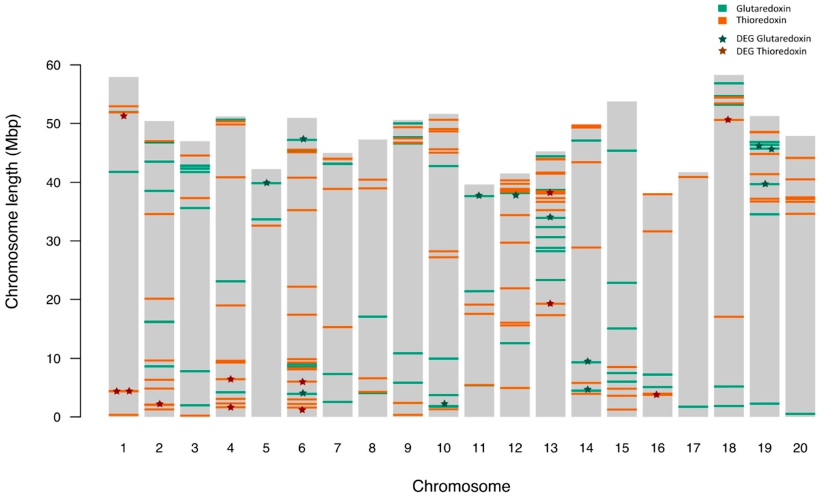
3.1. Trx and Grx Family Member Identification and Initial Characterization in Glycine max
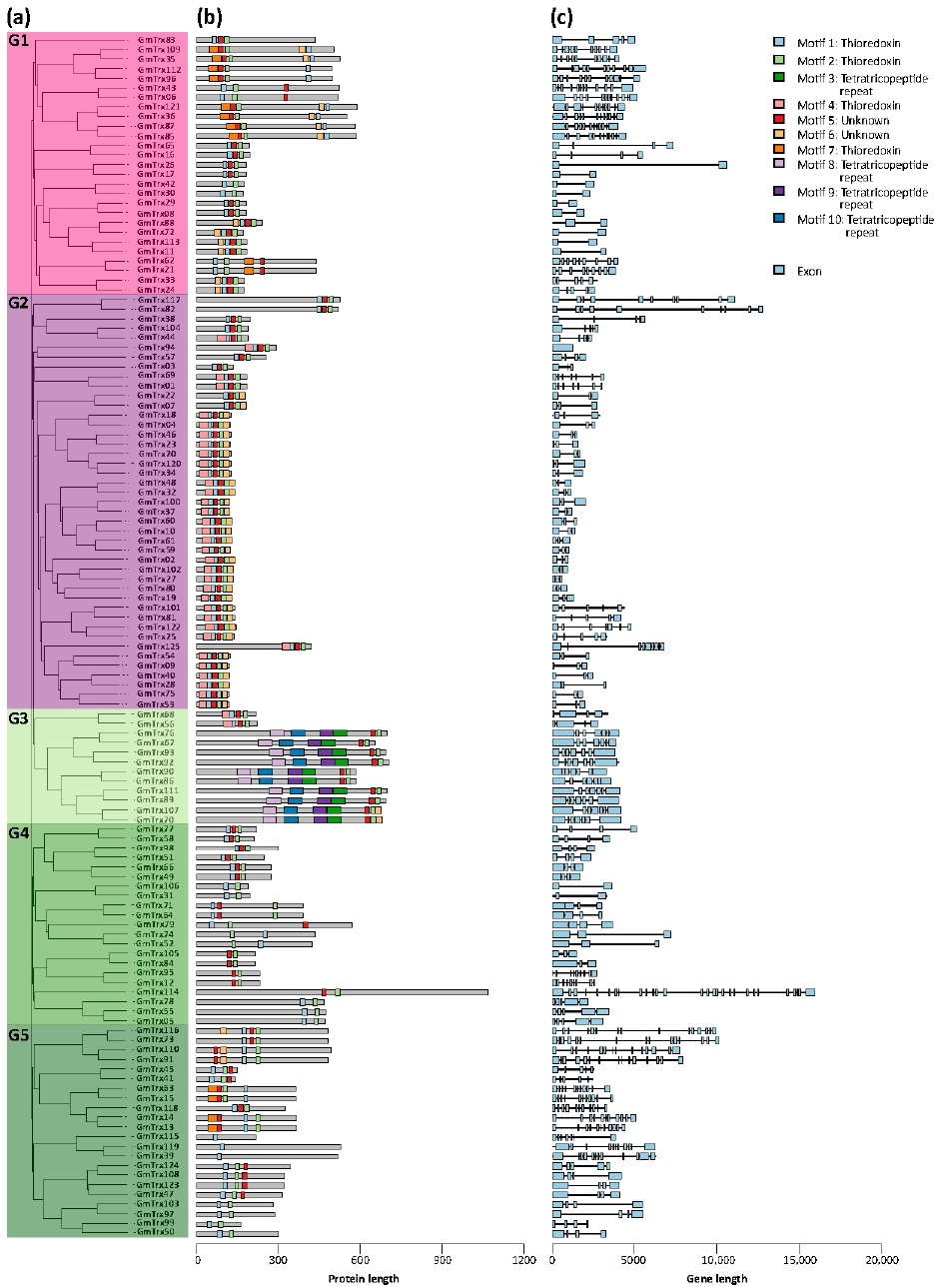
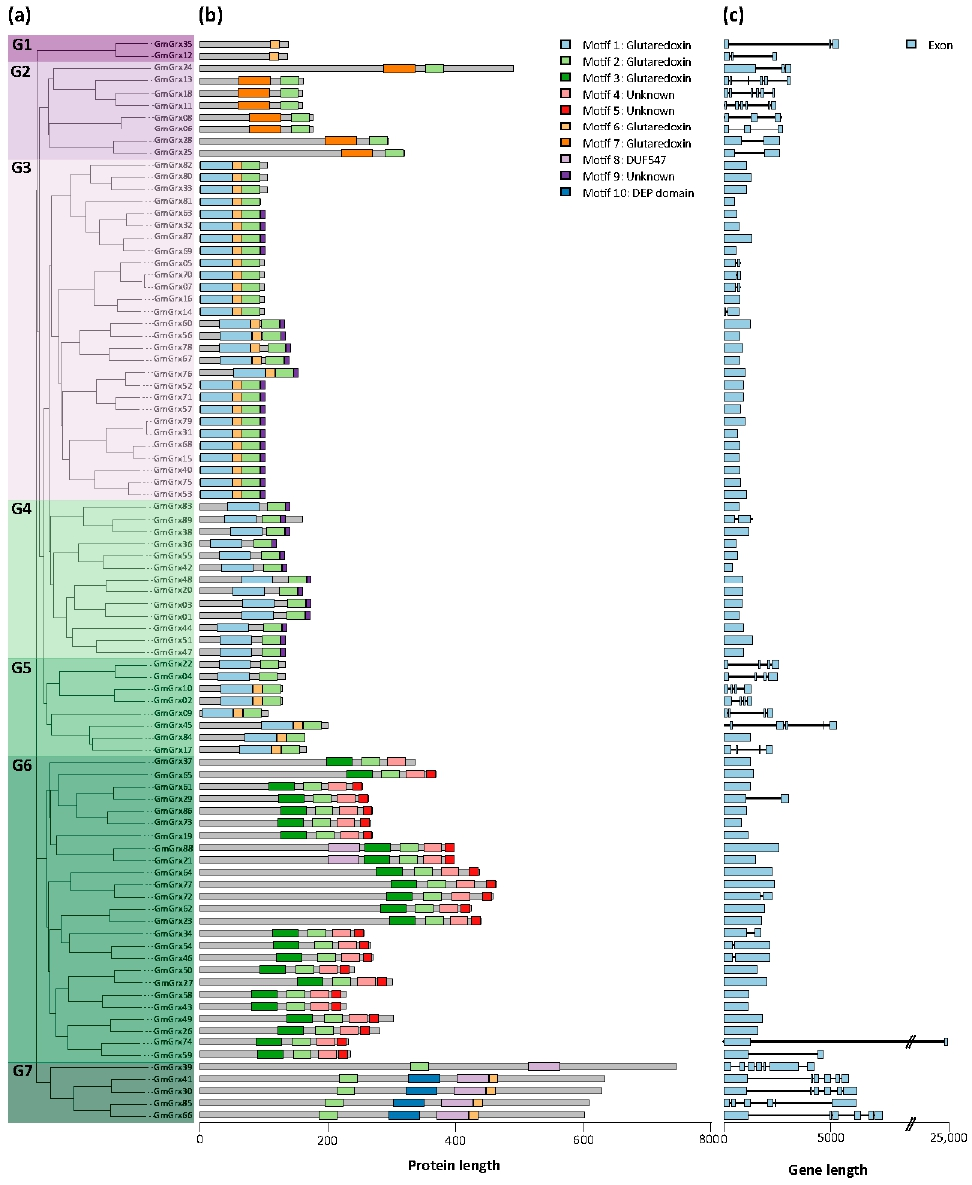
3.2. Phylogenetic, Gene Structure, and Conserved Motif Analyses of GmTrx and GmGrx Gene Family
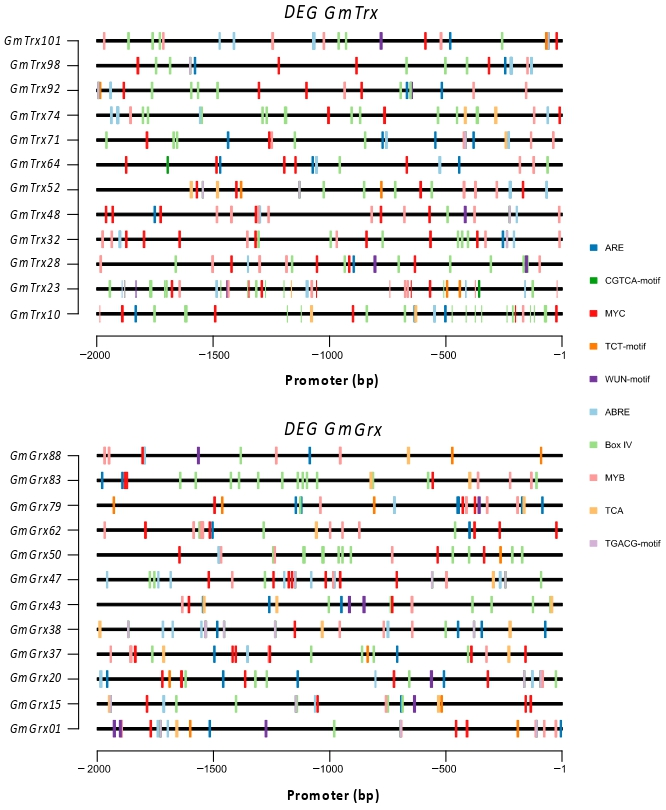
3.3. Cis-Regulatory Elements (CRE) for GmTrx and GmGrx
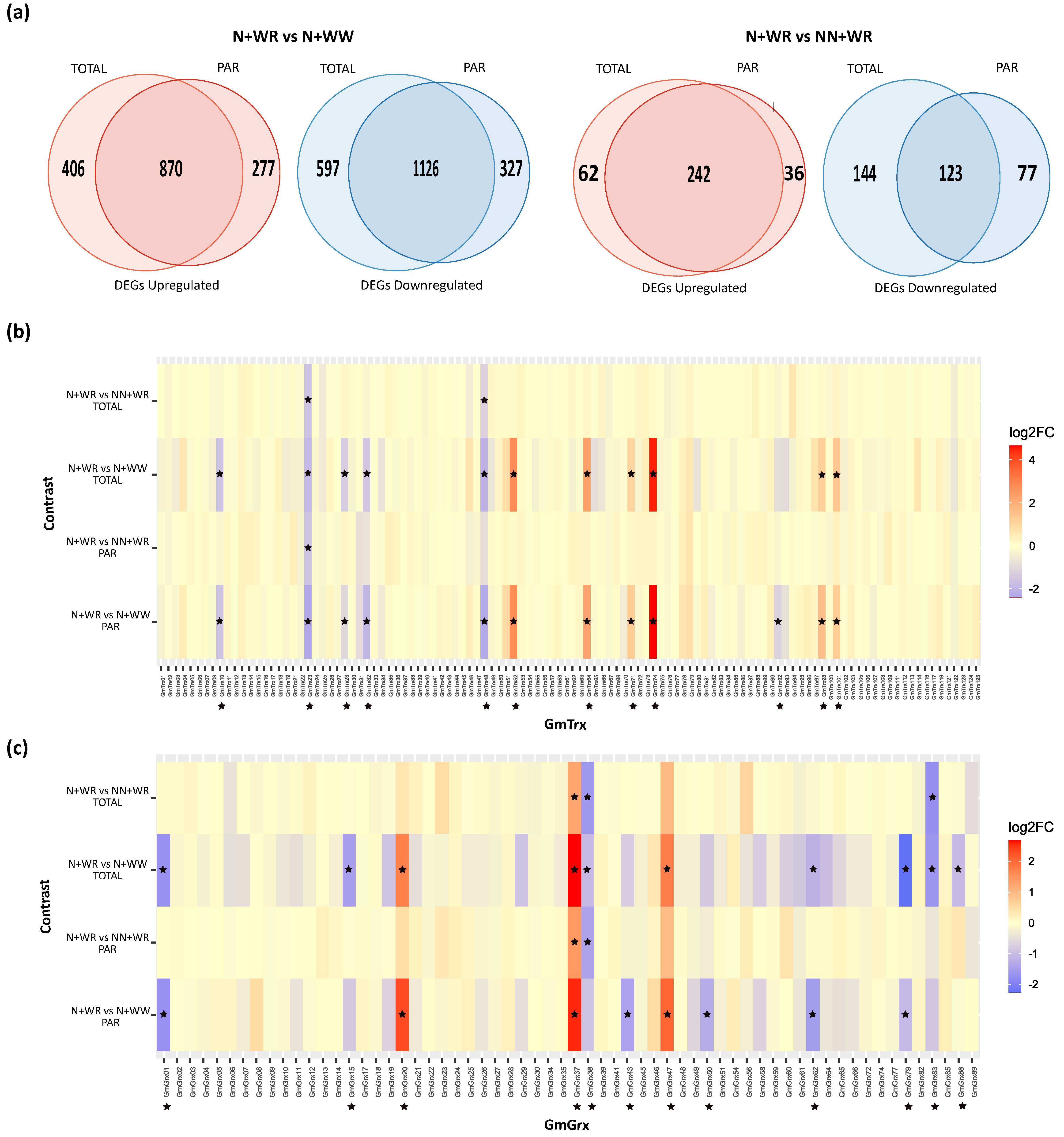
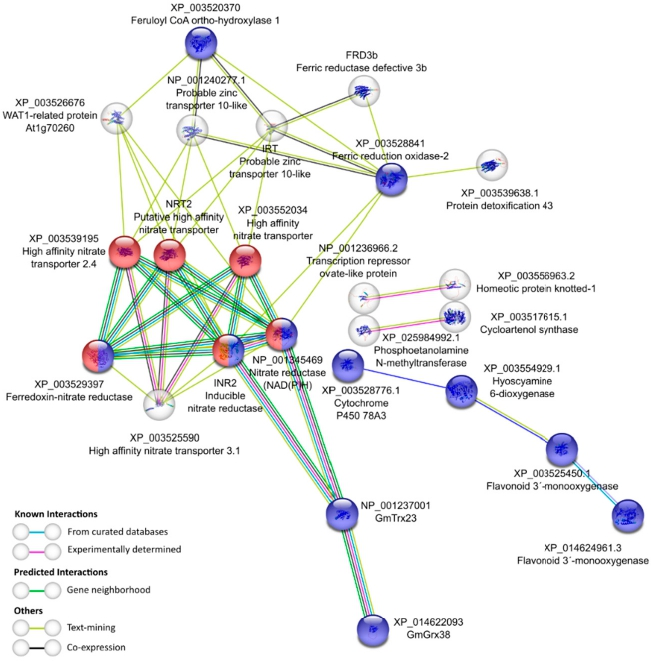
3.4. Identification of the Differentially Expressed GmTrx and GmGrx Genes in Nodulated and Water-Restricted Soybean Plants
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Verma, G.; Srivastava, D.; Tiwari, P.; Chakrabarty, D. ROS Modulation in Crop Plants Under Drought Stress. In Reactive Oxygen, Nitrogen and Sulfur Species in Plants: Production, Metabolism, Signaling and Defense Mechanisms; Hasanuzzaman, M., Fotopoulos, V., Fujita, M., Eds.; John Wiley and Sons, Inc.: Hoboken, NJ, USA, 2019; Volume 1, pp. 311–336. [Google Scholar]
- Juan, C.A.; de la Lastra, J.M.P.; Plou, F.J.; Pérez-Lebeña, E. The chemistry of reactive oxygen species (ROS) revisited: Outlining their role in biological macromolecules (dna, lipids and proteins) and induced pathologies. Int. J. Mol. Sci. 2021, 22, 4642. [Google Scholar] [CrossRef] [PubMed]
- Mittler, R. ROS Are Good. Trends Plant Sci. 2017, 22, 11–19. [Google Scholar] [CrossRef]
- Meyer, Y.; Riondet, C.; Constans, L.; Abdelgawwad, M.R.; Reichheld, J.P.; Vignols, F. Evolution of redoxin genes in the green lineage. Photosynth. Res. 2006, 89, 179–192. [Google Scholar] [CrossRef]
- Meyer, Y.; Belin, C.; Delorme-Hinoux, V.; Reichheld, J.P.; Riondet, C. Thioredoxin and glutaredoxin systems in plants: Molecular mechanisms, crosstalks, and functional significance. Antioxid. Redox Signal. 2012, 17, 1124–1160. [Google Scholar] [CrossRef] [PubMed]
- Lakhssassi, N.; Doblas, V.G.; Rosado, A.; del Valle, A.E.; Posé, D.; Jimenez, A.J.; Castillo, A.G.; Valpuesta, V.; Borsani, O.; Botella, M.A. The arabidopsis TETRATRICOPEPTIDE THIOREDOXIN-LIKE gene family is required for osmotic stress tolerance and male sporogenesis. Plant Physiol. 2012, 158, 1252–1266. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Li, Y.; Huang, X. Genome-Wide Analysis of the Thioredoxin Gene Family in Gossypium hirsutum L. and the Role of the Atypical Thioredoxin Gene GhTRXL3-2 in Flowering. J. Plant Biol. 2021, 64, 461–473. [Google Scholar] [CrossRef]
- Geigenberger, P.; Thormählen, I.; Daloso, D.M.; Fernie, A.R. The Unprecedented Versatility of the Plant Thioredoxin System. Trends Plant Sci. 2017, 22, 249–262. [Google Scholar] [CrossRef]
- Chibani, K.; Pucker, B.; Dietz, K.J.; Cavanagh, A. Genome-wide analysis and transcriptional regulation of the typical and atypical thioredoxins in Arabidopsis thaliana. FEBS Lett. 2021, 595, 2715–2730. [Google Scholar] [CrossRef]
- Hou, L.-Y.; Lehmann, M.; Geigenberger, P. Thioredoxin h2 and o1 show different subcellular localizations and redox-active functions, and are extrachloroplastics factors influencing photosynthetic performance in fluctation light. Antioxidants 2021, 10, 705. [Google Scholar] [CrossRef]
- Ribeiro, C.W.; Baldacci-Cresp, F.; Pierre, O.; Larousse, M.; Benyamina, S.; Lambert, A.; Hopkins, J.; Castella, C.; Cazareth, J.; Alloing, G.; et al. Regulation of Differentiation of Nitrogen-Fixing Bacteria by Microsymbiont Targeting of Plant Thioredoxin s1. Curr. Biol. 2017, 27, 250–256. [Google Scholar] [CrossRef]
- Alkhalfioui, F.; Renard, M.; Frendo, P.; Keichinger, C.; Meyer, Y.; Gelhaye, E.; Hirasawa, M.; Knaff, D.B.; Ritzenthaler, C.; Montrichard, F. A novel type of thioredoxin dedicated to symbiosis in legumes. Plant Physiol. 2008, 148, 424–435. [Google Scholar] [CrossRef] [PubMed]
- Meyer, Y.; Riondet, C.; Constans, L.; Abdelgawwad, M.R.; Reichheld, J.P.; Vignols, F. Evolution of redoxin genes in the green lineage. Photosynth. Res. 2008, 96, 113–114. [Google Scholar] [CrossRef]
- Rouhier, N.; Couturier, J.; Jacquot, J.P. Genome-wide analysis of plant glutaredoxin systems. J. Exp. Bot. 2006, 57, 1685–1696. [Google Scholar] [CrossRef] [PubMed]
- Rouhier, N. Plant glutaredoxins: Pivotal players in redox biology and iron-sulphur centre assembly. New Phytol. 2010, 186, 365–372. [Google Scholar] [CrossRef] [PubMed]
- Alloing, G.; Mandon, K.; Boncompagni, E.; Montrichard, F.; Frendo, P. Involvement of Glutaredoxin and Thioredoxin Systems in the Nitrogen-Fixing Symbiosis between Legumes and Rhizobia. Antioxidants 2018, 7, 182. [Google Scholar] [CrossRef] [PubMed]
- King, C.A.; Purcell, L.C.; Bolton, A.; Specht, J.E. A Possible Relationship Between Shoot N Concentration and the Sensitivity of N Fixation to Drought in Soybean. Crop Sci. 2014, 54, 746–756. [Google Scholar] [CrossRef]
- Bailey-Serres, J. Selective translation of cytoplasmic. Trends Plant Sci. 1999, 4, 142–148. [Google Scholar] [CrossRef]
- Wang, T.; Cui, Y.; Jin, J.; Guo, J.; Wang, G.; Yin, X.; He, Q.Y.; Zhang, G. Translating mRNAs strongly correlate to proteins in a multivariate manner and their translation ratios are phenotype specific. Nucleic Acids Res. 2013, 41, 4743–4754. [Google Scholar] [CrossRef]
- Ingolia, N.T. Ribosome profiling: New views of translation, from single codons to genome scale. Nat. Rev. Genet. 2014, 15, 205–213. [Google Scholar] [CrossRef]
- Lukoszek, R.; Feist, P.; Ignatova, Z. Insights into the adaptive response of Arabidopsis thaliana to prolonged thermal stress by ribosomal profiling and RNA-Seq. BMC Plant Biol. 2016, 16, 221. [Google Scholar] [CrossRef]
- The National Center of Biotechnology (NCBI). Available online: https://www.ncbi.nlm.nih.gov/ (accessed on 1 June 2022).
- Jones, P.; Binns, D.; Chang, H.-Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F. BLAST Algorithm. eLS 2014, 1–5. [Google Scholar] [CrossRef]
- Charif, D.; Lobry, J.R. Seqin R 1.0-2: A Contributed Package to the R Project for Statistical Computing Devoted to Biological Sequences Retrieval and Analysis. In Structural Approaches to Sequence Evolution; Bastolla, U., Porto, M., Roman, H.E., Vendruscolo, M., Eds.; Springer: Berlin/Heidelberg, Germany, 2007; ISBN 978-3-540-35305-8. [Google Scholar]
- Chou, K.C.; Shen, H. Bin Plant-mPLoc: A top-down strategy to augment the power for predicting plant protein subcellular localization. PLoS ONE 2010, 5, e11335. [Google Scholar] [CrossRef] [PubMed]
- Lescot, M.; Déhais, P.; Thijs, G.; Marchal, K.; Moreau, Y.; Van de Peer, Y.; Rouzé, P.; Rombauts, S. PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 2002, 30, 325–327. [Google Scholar] [CrossRef] [PubMed]
- Bailey, T.L.; Johnson, J.; Grant, C.E.; Noble, W.S. The MEME Suite. Nucleic Acids Res. 2015, 43, 39–49. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Core Team: Vienna, Austria, 2021. [Google Scholar]
- Kolde, R. Pheatmap: Pretty Heatmaps. 2019. Available online: https://cran.r-project.org/web/packages/pheatmap/index.html (accessed on 1 June 2022).
- The Arabidopsis Information Resource (TAIR). Available online: www.arabidopsis.org/aboutarabidopsis.html (accessed on 1 June 2022).
- Bodenhofer, U.; Bonatesta, E.; Horejs-Kainrath, C.; Hochreiter, S. msa: An R package for multiple sequence alignment. Bioinformatics 2015, 31, 3997–3999. [Google Scholar] [CrossRef]
- Schliep, K.P. phangorn: Phylogenetic analysis in R. Bioinformatics 2011, 27, 592–593. [Google Scholar] [CrossRef]
- Vincent, J. A Manual for the Practical Study of Root-Nodule Bacteria; IBP Handbo; Blackwell Scientific Publications: Oxford, UK, 1970. [Google Scholar]
- Broughton, W.J.; Dilworth, M.J. Control of Leghaemoglobin Synthesis in Snake Beans. Biochem. J. 1971, 125, 1075–1080. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef]
- Soneson, C.; Love, M.I.; Robinson, M.D. Differential analyses for RNA-seq: Transcript-level estimates improve gene-level inferences. F1000Research 2016, 4, 1521. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016; ISBN 978-3-319-24277-4. [Google Scholar]
- Jensen, L.J.; Kuhn, M.; Stark, M.; Chaffron, S.; Creevey, C.; Muller, J.; Doerks, T.; Julien, P.; Roth, A.; Simonovic, M.; et al. STRING 8—A global view on proteins and their functional interactions in 630 organisms. Nucleic Acids Res. 2009, 37, 412–416. [Google Scholar] [CrossRef] [PubMed]
- Nuruzzaman, M.; Sharoni, A.M.; Satoh, K.; Al-Shammari, T.; Shimizu, T.; Sasaya, T.; Omura, T.; Kikuchi, S. The thioredoxin gene family in rice: Genome-wide identification and expression profiling under different biotic and abiotic treatments. Biochem. Biophys. Res. Commun. 2012, 423, 417–423. [Google Scholar] [CrossRef]
- Elasad, M.; Wei, H.; Wang, H.; Su, J.; Ondati, E.; Yu, S. Genome- Wide Analysis and Characterization of the TRX Gene Family in Upland Cotton. Trop. Plant Biol. 2018, 11, 119–130. [Google Scholar] [CrossRef]
- Zhang, J.; Zhao, T.; Yan, F.; Wang, L.; Tang, Y.; Wang, Y.; Zhang, C. Genome-Wide Identification and Expression Analysis of Thioredoxin (Trx) Genes in Seed Development of Vitis vinifera. J. Plant Growth Regul. 2021. [Google Scholar] [CrossRef]
- Avashthi, H.; Pathak, R.K.; Gaur, V.S.; Singh, S.; Gupta, V.K.; Ramteke, P.W.; Kumar, A. Comparative analysis of ROS-scavenging gene families in finger millet, rice, sorghum, and foxtail millet revealed potential targets for antioxidant activity and drought tolerance improvement. Netw. Model. Anal. Health Inform. Bioinform. 2020, 9, 33. [Google Scholar] [CrossRef]
- Wang, C.; Gao, G.; Cao, S.; Xie, Q.; Qi, H. Isolation and functional validation of the CmLOX08 promoter associated with signalling molecule and abiotic stress responses in oriental melon, Cucumis melo var. makuwa Makino. BMC Plant Biol. 2019, 19, 75. [Google Scholar] [CrossRef]
- Bai, B.; Lu, N.; Li, Y.; Guo, S.; Yin, H.; He, Y.; Sun, W.; Li, W.; Xie, X. OsBBX14 promotes photomorphogenesis in rice by activating OsHY5L1 expression under blue light conditions. Plant Sci. 2019, 284, 192–202. [Google Scholar] [CrossRef]
- Wang, J.; Ling, L.; Cai, H.; Guo, C. Gene-wide identification and expression analysis of the PMEI family genes in soybean (Glycine max). 3 Biotech 2020, 10, 335. [Google Scholar] [CrossRef]
- Zheng, Z.; Yang, X.; Fu, Y.; Zhu, L.; Wei, H.; Lin, X. Overexpression of PvPin1, a bamboo homolog of PIN1-type parvulin 1, delays flowering time in transgenic Arabidopsis and rice. Front. Plant Sci. 2017, 8, 1526. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.Y.; Hamayun, M.; Lee, S.-K.; Lee, I.-J. Methyl jasmonate alleviated salinity stress in soybean. J. Crop Sci. Biotechnol. 2009, 12, 63–68. [Google Scholar] [CrossRef]
- Liao, P.; Huang, J.; Tong, P.; Nie, W.; Yan, X.; Feng, Y.; Peng, H.; Peng, X.; Li, S. Characterization and expression analysis of inositolphosphorylceramide synthase family genes in rice (Oryza sativa L.). Genes Genom. 2017, 39, 485–492. [Google Scholar] [CrossRef]
- Guan, S.; Xu, Q.; Ma, D.; Zhang, W.; Xu, Z.; Zhao, M.; Guo, Z. Transcriptomics profiling in response to cold stress in cultivated rice and weedy rice. Gene 2019, 685, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.J.; Yin, L.J.; Ma, J.; Zheng, J.C.; Wang, Y.X.; Lan, J.H.; Fu, J.D.; Chen, M.; Xu, Z.S.; Ma, Y.Z. The Roles of GmERF135 in Improving Salt Tolerance and Decreasing ABA Sensitivity in Soybean. Front. Plant Sci. 2019, 10, 940. [Google Scholar] [CrossRef] [PubMed]
- Gebauer, F.; Hentze, M.W. Molecular mechanisms of translational control. Nat. Rev. Mol. Cell Biol. 2004, 5, 827–835. [Google Scholar] [CrossRef]
- Hershey, J.W.B.; Sonenberg, N.; Mathews, M.B. Principles of Translational Control: An Overview. Cold Spring Harb. Perspect. Biol. 2012, 4, a011528. [Google Scholar] [CrossRef]
- Wang, L.; Wessler, S.R. Role of mRNA Secondary Structure in Translational Repression of the Maize Transcriptional Activator Lc. Plant Physiol. 2001, 125, 1380–1387. [Google Scholar] [CrossRef]
- Hummel, M.; Rahmani, F.; Smeekens, S.; Hanson, J. Sucrose-mediated translational control. Ann. Bot. 2009, 104, 1–7. [Google Scholar] [CrossRef]
- Sablok, G.; Powell, J.J.; Kazan, K. Emerging Roles and Landscape of Translating mRNAs in Plants. Front. Plant Sci. 2017, 8, 1443. [Google Scholar] [CrossRef]
- Merchante, C.; Stepanova, A.N.; Alonso, J.M. Translation regulation in plants: An interesting past, an exciting present and a promising future. Plant J. 2017, 90, 628–653. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, R.; Girke, T.; Bray, E.A.; Bailey-Serres, J. Differential mRNA translation contributes to gene regulation under non-stress and dehydration stress conditions in Arabidopsis thaliana. Plant J. 2004, 38, 823–839. [Google Scholar] [CrossRef] [PubMed]
- Mustroph, A.; Zanetti, M.E.; Jang, C.J.H.; Holtan, H.E.; Repetti, P.P.; Galbraith, D.W.; Girke, T.; Bailey-Serres, J. Profiling translatomes of discrete cell populations resolves altered cellular priorities during hypoxia in Arabidopsis. Proc. Natl. Acad. Sci. USA 2009, 106, 18843–18848. [Google Scholar] [CrossRef]
- Juntawong, P.; Girke, T.; Bazin, J.; Bailey-Serres, J. Translational dynamics revealed by genome-wide profiling of ribosome footprints in Arabidopsis. Proc. Natl. Acad. Sci. USA 2014, 111, E203–E212. [Google Scholar] [CrossRef] [PubMed]
- Lei, L.; Shi, J.; Chen, J.; Zhang, M.; Sun, S.; Xie, S.; Li, X.; Zeng, B.; Peng, L. Ribosome profiling reveals dynamic translational landscape in maize seedlings under drought stress. Plant J. 2015, 84, 1206–1218. [Google Scholar] [CrossRef] [PubMed]
- Borsani, O.; Díaz, P.; Monza, J. Proline is involved in water stress responses of Lotus corniculatus nitrogen fixing and nitrate fed plants. J. Plant Physiol. 1999, 155, 269–273. [Google Scholar] [CrossRef]
- Lodeiro, A.R.; González, P.; Hernández, A.; Balagué, L.J.; Favelukes, G. Comparison of drought tolerance in nitrogen-fixing and inorganic nitrogen-grown common beans. Plant Sci. 2000, 154, 31–41. [Google Scholar] [CrossRef]
- Staudinger, C.; Mehmeti-Tershani, V.; Gil-Quintana, E.; Gonzalez, E.M.; Hofhansl, F.; Bachmann, G.; Wienkoop, S. Evidence for a rhizobia-induced drought stress response strategy in Medicago truncatula. J. Proteom. 2016, 136, 202–213. [Google Scholar] [CrossRef]
- Liu, Y.; Guo, Z.; Shi, H. Rhizobium Symbiosis Leads to Increased Drought Tolerance in Chinese Milk Vetch (Astragalus sinicus L.). Agronomy 2022, 12, 725. [Google Scholar] [CrossRef]
- Traubenik, S.; Reynoso, M.A.; Hobecker, K.; Lancia, M.; Hummel, M.; Rosen, B.; Town, C.; Bailey-Serres, J.; Blanco, F.; Zanetti, M.E. Reprogramming of root cells during nitrogen-fixing symbiosis involves dynamic polysome association of coding and noncoding RNAs. Plant Cell 2020, 32, 352–373. [Google Scholar] [CrossRef]
- Verma, D.P.; Fortin, M.; Stanley, J.; Mauro, V.; Purohit, S.; Morrison, N. Nodulins and nodulin genes of Glycine max. Plant Mol. Biol. 1986, 7, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Gamm, M.; Peviani, A.; Honsel, A.; Snel, B.; Smeekens, S.; Hanson, J. Increased sucrose levels mediate selective mRNA translation in Arabidopsis. BMC Plant Biol. 2014, 14, 306. [Google Scholar] [CrossRef] [PubMed]
- Branco-Price, C.; Kaiser, K.A.; Jang, C.J.H.; Larive, C.K.; Bailey-Serres, J. Selective mRNA translation coordinates energetic and metabolic adjustments to cellular oxygen deprivation and reoxygenation in Arabidopsis thaliana. Plant J. 2008, 56, 743–755. [Google Scholar] [CrossRef] [PubMed]
- Palusa, S.; Reddy, A.S. Differential Recruitment of Splice Variants from SR Pre-mRNAs to Polysomes during Development and in Response to Stresses. Plant Cell Physiol. 2015, 56, 421–427. [Google Scholar] [CrossRef]
- Hu, Y.; Wu, Q.; Sprague, S.; Park, J.; Oh, M.; Rajashekar, C.; Koiwa, H.; Nakata, P.; Cheng, N.; Hirschi, K.; et al. Tomato expressing Arabidopsis glutaredoxin gene AtGRXS17 confers tolerance to chilling stress via modulating cold responsive components. Hortic. Res. 2015, 2, 15051. [Google Scholar] [CrossRef]
- Wu, Q.; Yang, J.; Cheng, N.; Hirschi, K.D.; White, F.F.; Park, S. Glutaredoxins in plant development, abiotic stress response, and iron homeostasis: From model organisms to crops. Environ. Exp. Bot. 2017, 139, 91–98. [Google Scholar] [CrossRef]
- Vanacker, H.; Guichard, M.; Bohrer, A.S.; Issakidis-Bourguet, E. Redox regulation of monodehydroascorbate reductase by thioredoxin y in plastids revealed in the context of water stress. Antioxidants 2018, 7, 183. [Google Scholar] [CrossRef]
- Cuadrado-Pedetti, M.B.; Rauschert, I.; Sainz, M.M.; Amorim-Silva, V.; Botella, M.A.; Borsani, O.; Sotelo-Silveira, M. The arabidopsis tetratricopeptide thioredoxin-like 1 gene is involved in anisotropic root growth during osmotic stress adaptation. Genes 2021, 12, 236. [Google Scholar] [CrossRef]
- Kneeshaw, S.; Keyani, R.; Delorme-Hinoux, V.; Imrie, L.; Loake, G.J.; Le Bihan, T.; Reichheld, J.P.; Spoel, S.H. Nucleoredoxin guards against oxidative stress by protecting antioxidant enzymes. Proc. Natl. Acad. Sci. USA 2017, 114, 8414–8419. [Google Scholar] [CrossRef]
- Le, D.T.; Nishiyama, R.; Watanabe, Y.; Tanaka, M.; Seki, M.; Ham, L.H.; Yamaguchi-shinozaki, K.; Shinozaki, K.; Tran, L.P. Differential Gene Expression in Soybean Leaf Tissues at Late Developmental Stages under Drought Stress Revealed by Genome-Wide Transcriptome Analysis. PLoS ONE 2012, 7, e49522. [Google Scholar] [CrossRef]
- Li, N.; Muthreich, M.; Huang, L.J.; Thurow, C.; Sun, T.; Zhang, Y.; Gatz, C. TGACG-BINDING FACTORs (TGAs) and TGA-interacting CC-type glutaredoxins modulate hyponastic growth in Arabidopsis thaliana. New Phytol. 2019, 221, 1906–1918. [Google Scholar] [CrossRef] [PubMed]
- Aerts, N.; Pereira Mendes, M.; Van Wees, S.C.M. Multiple levels of crosstalk in hormone networks regulating plant defense. Plant J. 2021, 105, 489–504. [Google Scholar] [CrossRef] [PubMed]
- Zander, M.; Chen, S.; Imkampe, J.; Thurow, C.; Gatz, C. Repression of the arabidopsis thaliana jasmonic acid/ethylene-induced defense pathway by tga-interacting glutaredoxins depends on their c-terminal alwl motif. Mol. Plant 2012, 5, 831–840. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ProtID_Trx | GmTrx | Class | Chromosome # | Putative Localization | Length (aa) | Condition DEG | Status | Regulation Level |
|---|---|---|---|---|---|---|---|---|
| NP_001237762.1 | GmTrx32 | h II | 1 | Cytoplasm. | 138 | N+WR vs. N+WW | Downregulated | TOTAL+PAR |
| NP_001236052.1 | GmTrx10 | h II | 1 | Cytoplasm. | 126 | N+WR vs. N+WW | Downregulated | TOTAL+PAR |
| NP_001237535.2 | GmTrx28 | h I | 1 | Cytoplasm. | 120 | N+WR vs. N+WW | Downregulated | TOTAL+PAR |
| NP_001240862.1 | GmTrx48 | h II | 2 | Cytoplasm. | 138 | N+WR vs. N+WW | Downregulated | TOTAL+PAR |
| N+WR vs. NN+WR | TOTAL | |||||||
| XP_003522672.1 | GmTrx64 | Nucleoredoxin | 4 | Chloroplast. Cytoplasm. Nucleus. | 389 | N+WR vs. N+WW | Upregulated | TOTAL+PAR |
| NP_001276271.2 | GmTrx52 | Nucleoredoxin | 4 | Cytoplasm. | 423 | N+WR vs. N+WW | Upregulated | TOTAL+PAR |
| XP_003527521.1 | GmTrx74 | Nucleoredoxin | 6 | Cytoplasm. Nucleus. | 434 | N+WR vs. N+WW | Upregulated | TOTAL+PAR |
| XP_003526462.1 | GmTrx71 | Nucleoredoxin | 6 | Nucleus. | 389 | N+WR vs. N+WW | Upregulated | TOTAL+PAR |
| NP_001237001.1 | GmTrx23 | h III | 13 | Chloroplast. Cytoplasm. | 122 | N+WR vs. N+WW | Downregulated | TOTAL+PAR |
| N+WR vs. NN+WR | ||||||||
| XP_003543765.1 | GmTrx92 | TTL | 13 | Nucleus. | 703 | N+WR vs. N+WW | Downregulated | PAR |
| XP_003548763.1 | GmTrx98 | Lilium | 16 | Chloroplast. | 299 | N+WR vs. N+WW | Upregulated | TOTAL+PAR |
| XP_003552324.1 | GmTrx101 | h III | 18 | Cytoplasm. | 139 | N+WR vs. N+WW | Upregulated | TOTAL+PAR |
| ProtID_Grx | GmGrx | Class | Chromosome # | Putative Localization | Length (aa) | Condition DEG | Status | Regulation Level |
|---|---|---|---|---|---|---|---|---|
| XP_003525338.1 | GmGrx37 | 4CxxC | 5. | Chloroplast. | 337. | N+WR vs. N+WW | Upregulated | TOTAL+PAR |
| N+WR vs. NN+WR | ||||||||
| XP_003526012.1 | GmGrx38 | CCxS | 6 | Chloroplast. | 140 | N+WR vs. N+WW | Downregulated | TOTAL |
| N+WR vs. NN+WR | TOTAL+PAR | |||||||
| NP_001235171.1 | GmGrx01 | CCxS | 6 | Chloroplast. | 172 | N+WR vs. N+WW | Downregulated | TOTAL+PAR |
| NP_001238068.1 | GmGrx15 | CCxS | 10 | Chloroplast. | 102 | N+WR vs. N+WW | Downregulated | TOTAL |
| XP_003537494.1 | GmGrx43 | 4CxxC | 11 | Chloroplast. Nucleus. | 229 | N+WR vs. N+WW | Downregulated | PAR |
| NP_001240908.1 | GmGrx20 | CCxS | 12 | Chloroplast. | 160 | N+WR vs. N+WW | Upregulated | TOTAL+PAR |
| XP_003543019.1 | GmGrx47 | CCxS | 13 | Chloroplast. | 133 | N+WR vs. N+WW | Upregulated | TOTAL+PAR |
| XP_003545225.1 | GmGrx50 | 4CxxC | 14 | Chloroplast. | 242 | N+WR vs. N+WW | Downregulated | PAR |
| XP_014622093.1 | GmGrx83 | CCxS | 14 | Chloroplast. | 141 | N+WR vs. N+WW | Downregulated | TOTAL |
| N+WR vs. NN+WR | ||||||||
| XP_003554133.1 | GmGrx62 | 4CxxC | 19 | Chloroplast. Nucleus. | 424 | N+WR vs. N+WW | Downregulated | TOTAL+PAR |
| XP_040868600.1 | GmGrx88 | 4CxxC | 19 | Chloroplast. Nucleus. | 398 | N+WR vs. N+WW | Downregulated | TOTAL |
| XP_006604701.1 | GmGrx79 | CCxS | 19 | Chloroplast. | 102 | N+WR vs. N+WW | Downregulated | TOTAL+PAR |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sainz, M.M.; Filippi, C.V.; Eastman, G.; Sotelo-Silveira, J.; Borsani, O.; Sotelo-Silveira, M. Analysis of Thioredoxins and Glutaredoxins in Soybean: Evidence of Translational Regulation under Water Restriction. Antioxidants 2022, 11, 1622. https://doi.org/10.3390/antiox11081622
Sainz MM, Filippi CV, Eastman G, Sotelo-Silveira J, Borsani O, Sotelo-Silveira M. Analysis of Thioredoxins and Glutaredoxins in Soybean: Evidence of Translational Regulation under Water Restriction. Antioxidants. 2022; 11(8):1622. https://doi.org/10.3390/antiox11081622
Chicago/Turabian StyleSainz, María Martha, Carla Valeria Filippi, Guillermo Eastman, José Sotelo-Silveira, Omar Borsani, and Mariana Sotelo-Silveira. 2022. "Analysis of Thioredoxins and Glutaredoxins in Soybean: Evidence of Translational Regulation under Water Restriction" Antioxidants 11, no. 8: 1622. https://doi.org/10.3390/antiox11081622