
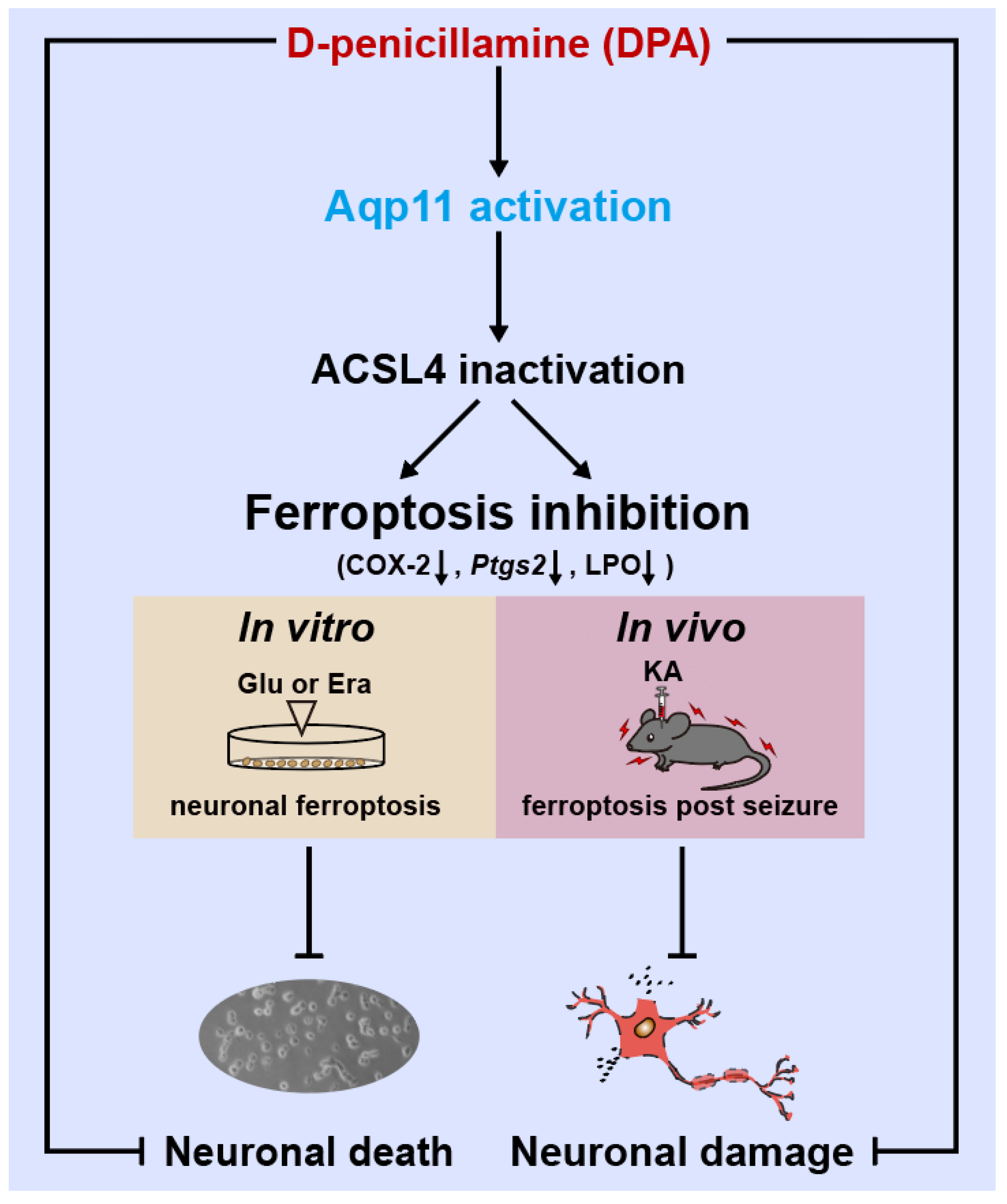
D-Penicillamine Reveals the Amelioration of Seizure-Induced Neuronal Injury via Inhibiting Aqp11-Dependent Ferroptosis


Abstract
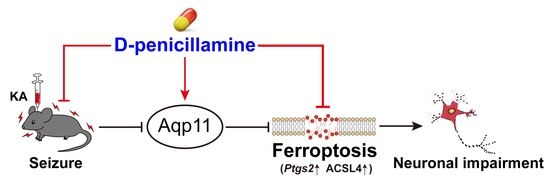
:
1. Introduction
2. Materials and Methods
2.1. Preparation of KA-Induced Seizure Mouse Model and Drug Treatment
2.2. Aqp11 siRNA Transfer in Mice Brain
2.3. Racine Score
2.4. Nissl Staining
2.5. Fluoro-Jade B Assay
2.6. Cell Culture
2.7. RNA-Seq Analysis
2.8. Real-Time Quantitative PCR
2.9. Western Blot Assay
2.10. RNA Interference in HT22 Cell
2.11. Measurement of Lipid Peroxide (LPO) Levels
2.12. Immunofluorescence
2.13. Measurement of Copper Level in Mice Brain
2.14. Statistical Analysis
3. Results
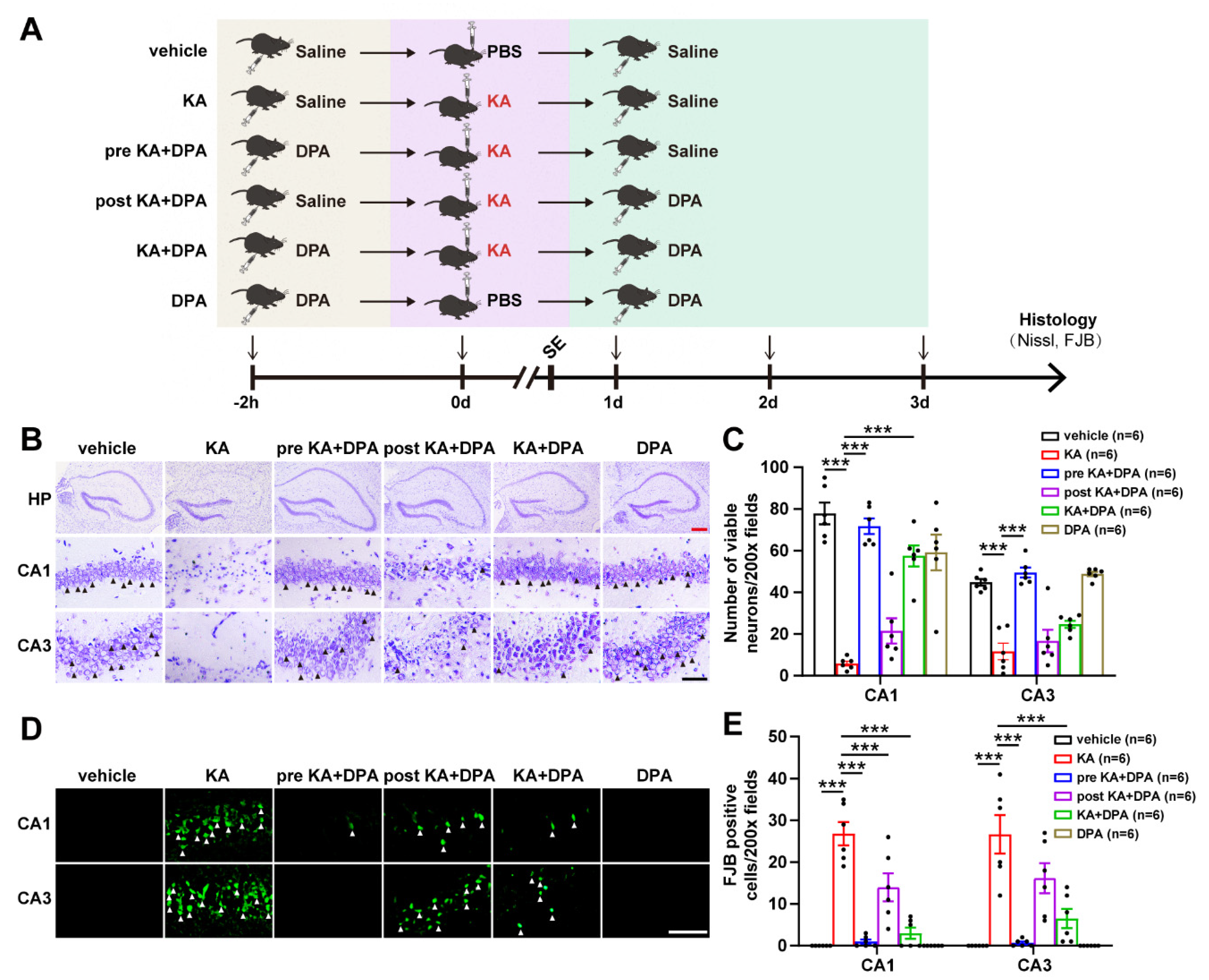
3.1. DPA Ameliorates Seizure-Induced Neuronal Injury in KA-Treated Mouse Model
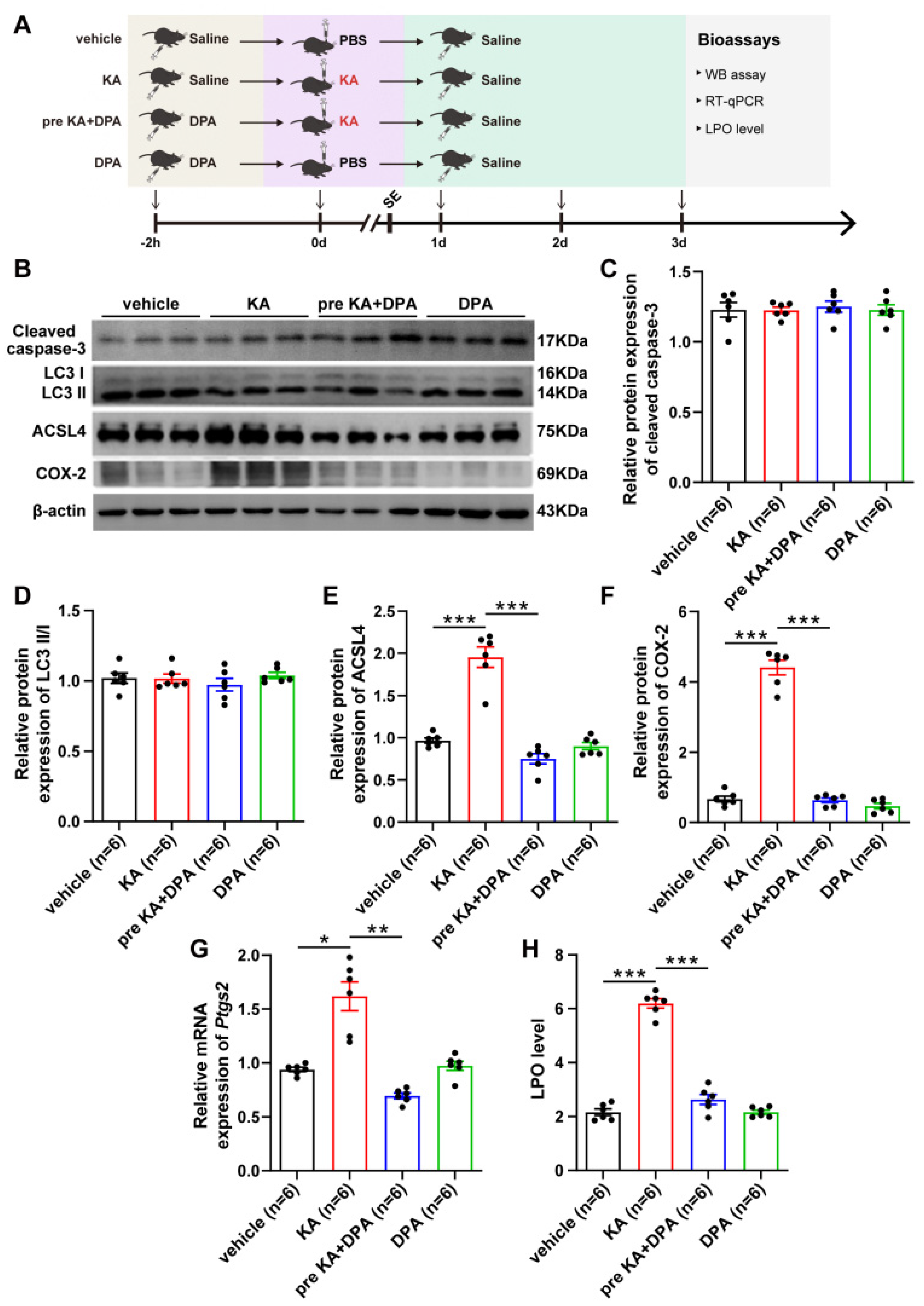
3.2. Ferroptosis Is Involved in the Protection of DPA against Seizure-Related Neuronal Injury
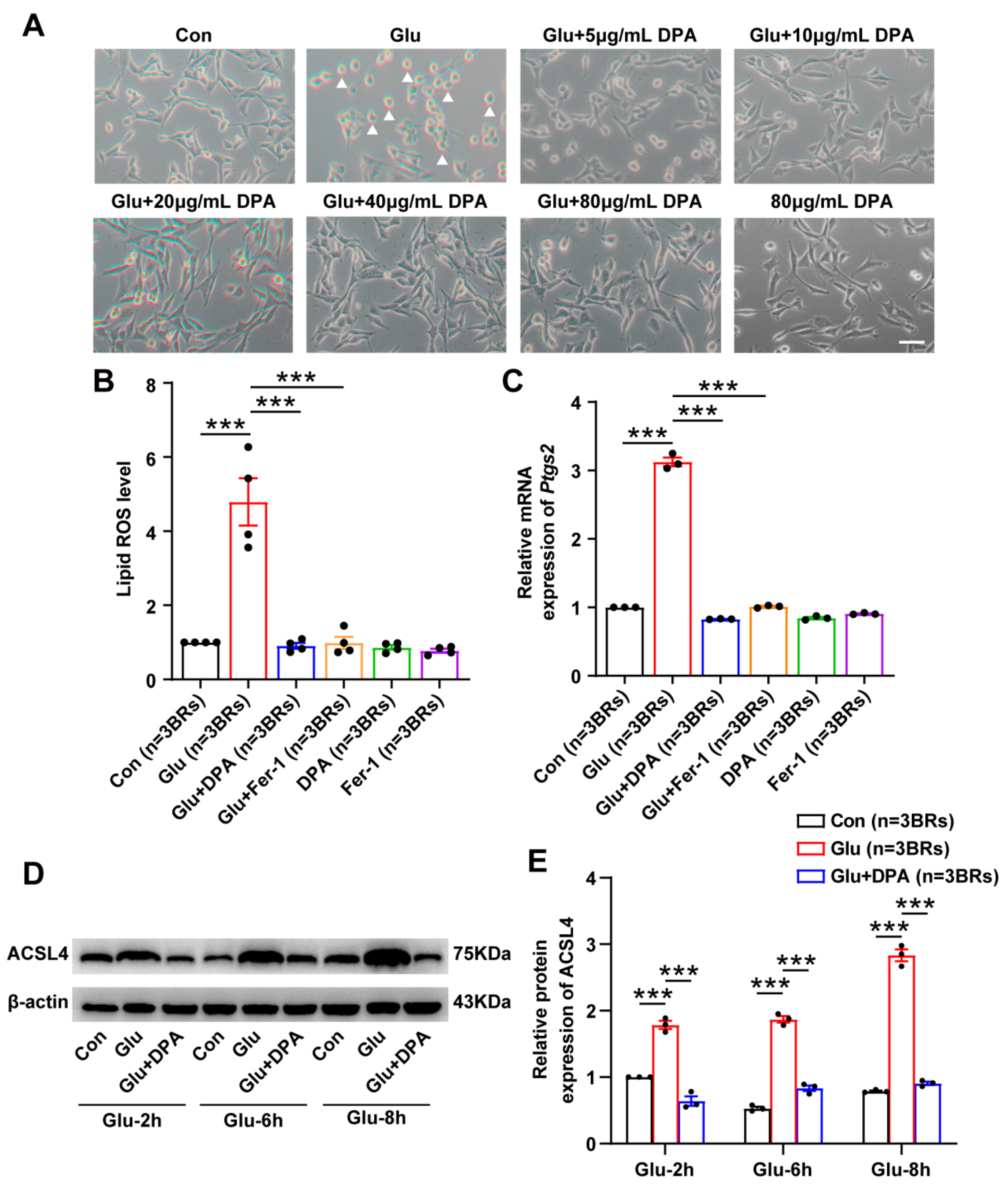
3.3. DPA Inhibits Glutamate-Induced Neuronal Ferroptosis In Vitro
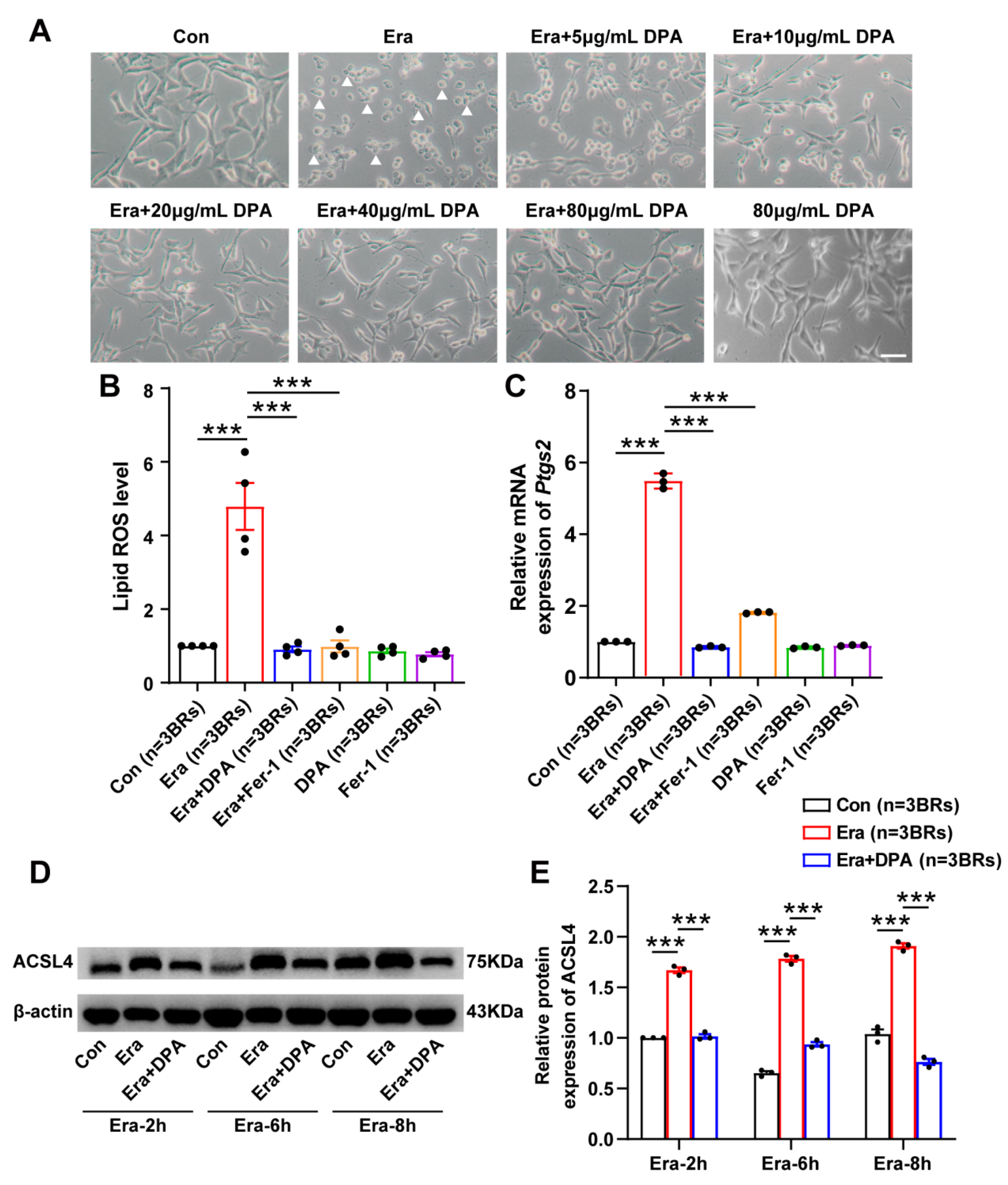
3.4. DPA Inhibits Erastin-Induced Neuronal Ferroptosis In Vitro
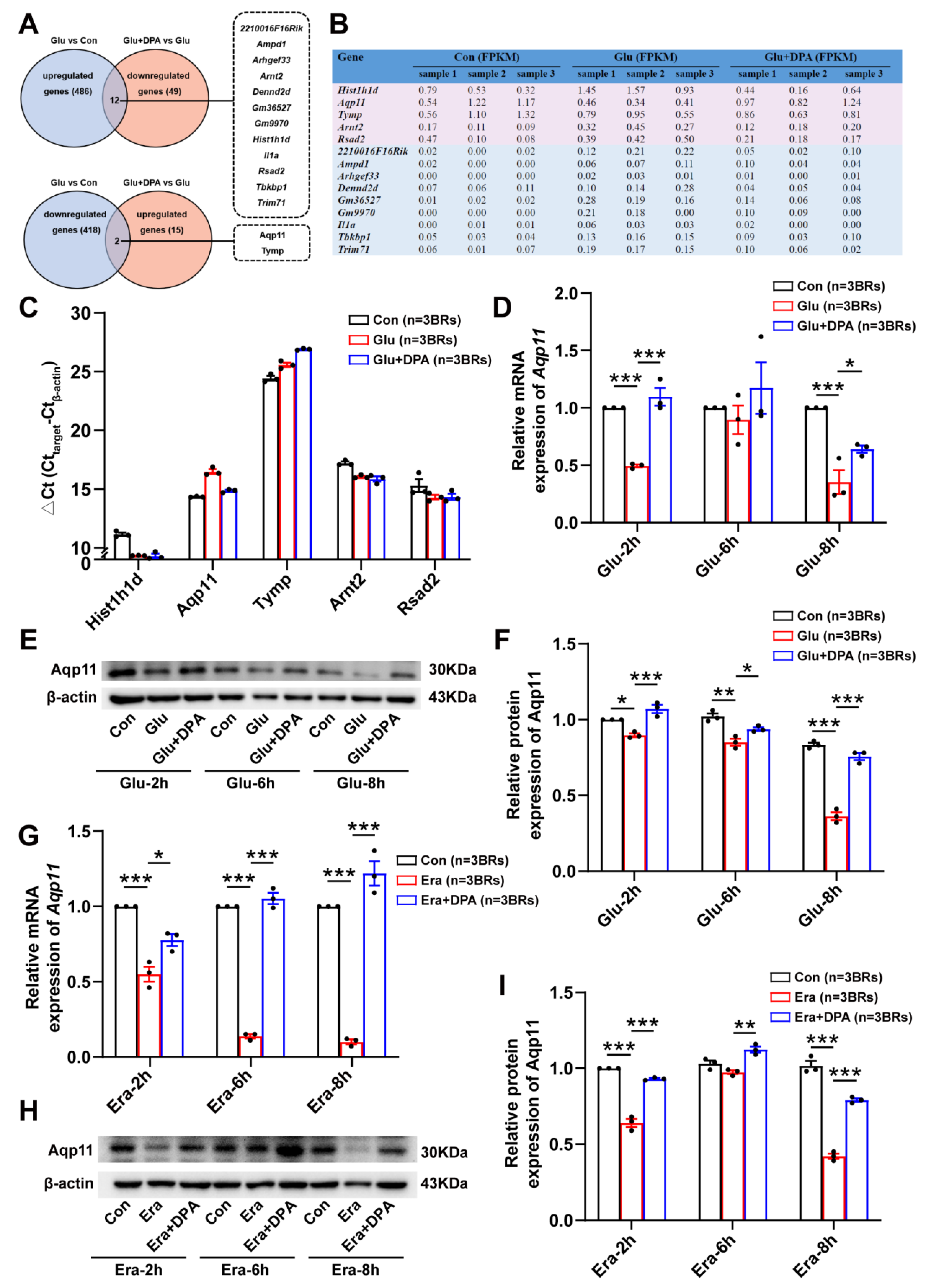
3.5. Aqp11 Is a Key Target Involved in the Protection of DPA against Ferroptosis In Vitro
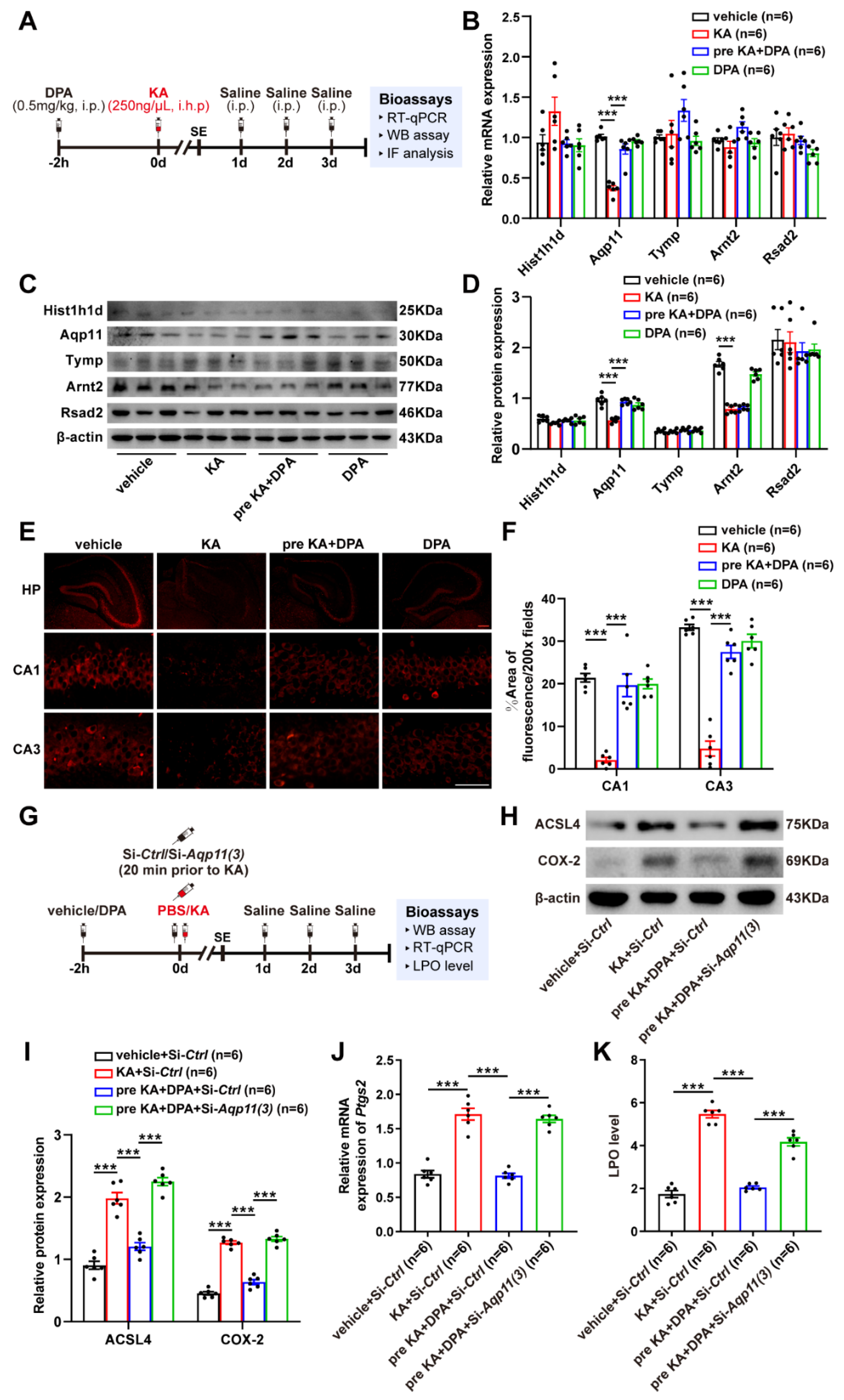
3.6. DPA Increases the Expression and Distribution of Aqp11 in KA-Induced Seizures in Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Ferreira-Atuesta, C.; Döhler, N.; Erdélyi-Canavese, B.; Felbecker, A.; Siebel, P.; Scherrer, N.; Bicciato, G.; Schweizer, J.; Sinka, L.; Imbach, L.L.; et al. Seizures after Ischemic Stroke: A Matched Multicenter Study. Ann. Neurol. 2021, 90, 808–820. [Google Scholar] [CrossRef] [PubMed]
- Mazzucchi, E.; Vollono, C.; Pauletto, G.; Lettieri, C.; Budai, R.; Gigli, G.L.; Sabatino, G.; La Rocca, G.; Skrap, M.; Ius, T. The persistence of seizures after tumor resection negatively affects survival in low-grade glioma patients: A clinical retrospective study. J. Neurol. 2022, 269, 2627–2633. [Google Scholar] [CrossRef] [PubMed]
- Proix, T.; Truccolo, W.; Leguia, M.G.; Tcheng, T.K.; King-Stephens, D.; Rao, V.R.; Baud, M.O. Forecasting seizure risk in adults with focal epilepsy: A development and validation study. Lancet Neurol. 2021, 20, 127–135. [Google Scholar] [CrossRef]
- Devinsky, O.; Vezzani, A.; O’Brien, T.J.; Jette, N.; Scheffer, I.E.; de Curtis, M.; Perucca, P. Epilepsy. Nat. Rev. Dis. Primers 2018, 4, 18024. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, Z. An update for epilepsy research and antiepileptic drug development: Toward precise circuit therapy. Pharmacol. Ther. 2019, 201, 77–93. [Google Scholar] [CrossRef] [PubMed]
- Löscher, W.; Potschka, H.; Sisodiya, S.M.; Vezzani, A. Drug Resistance in Epilepsy: Clinical Impact, Potential Mechanisms, and New Innovative Treatment Options. Pharmacol. Rev. 2020, 72, 606–638. [Google Scholar] [CrossRef]
- Mao, X.Y.; Zhou, H.H.; Jin, W.L. Redox-Related Neuronal Death and Crosstalk as Drug Targets: Focus on Epilepsy. Front. Neurosci. 2019, 13, 512. [Google Scholar] [CrossRef]
- Lee, D.A.; Lee, J.; Kim, H.C.; Park, K.M.; Kim, S.E. Hippocampal injury in patients with status epilepticus: Quantitative analysis of hippocampal volume and structural co-variance network. Seizure 2022, 95, 84–89. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Mao, X.Y.; Zhou, H.H.; Jin, W.L. Ferroptosis Induction in Pentylenetetrazole Kindling and Pilocarpine-Induced Epileptic Seizures in Mice. Front. Neurosci. 2019, 13, 721. [Google Scholar] [CrossRef]
- Li, Q.; Li, Q.Q.; Jia, J.N.; Sun, Q.Y.; Zhou, H.H.; Jin, W.L.; Mao, X.Y. Baicalein Exerts Neuroprotective Effects in FeCl3-Induced Posttraumatic Epileptic Seizures via Suppressing Ferroptosis. Front. Pharmacol. 2019, 10, 638. [Google Scholar] [CrossRef] [PubMed]
- Jia, J.N.; Yin, X.X.; Li, Q.; Guan, Q.W.; Yang, N.; Chen, K.N.; Zhou, H.H.; Mao, X.Y. Neuroprotective Effects of the Anti-cancer Drug Lapatinib against Epileptic Seizures via Suppressing Glutathione Peroxidase 4-Dependent Ferroptosis. Front. Pharmacol. 2020, 11, 601572. [Google Scholar] [CrossRef]
- Chen, K.N.; Guan, Q.W.; Yin, X.X.; Wang, Z.J.; Zhou, H.H.; Mao, X.Y. Ferrostatin-1 obviates seizures and associated cognitive deficits in ferric chloride-induced posttraumatic epilepsy via suppressing ferroptosis. Free Radic. Biol. Med. 2022, 179, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Stremmel, W.; Meyerrose, K.W.; Niederau, C.; Hefter, H.; Kreuzpaintner, G.; Strohmeyer, G. Wilson disease: Clinical presentation, treatment, and survival. Ann. Intern. Med. 1991, 115, 720–726. [Google Scholar] [CrossRef] [PubMed]
- Wiesner, R.H.; Grambsch, P.M.; Lindor, K.D.; Ludwig, J.; Dickson, E.R. Clinical and statistical analyses of new and evolving therapies for primary biliary cirrhosis. Hepatology 1988, 8, 668–676. [Google Scholar] [CrossRef]
- Gaujoux-Viala, C.; Smolen, J.S.; Landewé, R.; Dougados, M.; Kvien, T.K.; Mola, E.M.; Scholte-Voshaar, M.; van Riel, P.; Gossec, L. Current evidence for the management of rheumatoid arthritis with synthetic disease-modifying antirheumatic drugs: A systematic literature review informing the EULAR recommendations for the management of rheumatoid arthritis. Ann. Rheum. Dis. 2010, 69, 1004–1009. [Google Scholar] [CrossRef] [PubMed]
- Ala, A.; Walker, A.P.; Ashkan, K.; Dooley, J.S.; Schilsky, M.L. Wilson’s disease. Lancet 2007, 369, 397–408. [Google Scholar] [CrossRef]
- Walshe, J.M. Penicillamine, a new oral therapy for Wilson’s disease. Am. J. Med. 1956, 21, 487–495. [Google Scholar] [CrossRef]
- Ge, E.J.; Bush, A.I.; Casini, A.; Cobine, P.A.; Cross, J.R.; DeNicola, G.M.; Dou, Q.P.; Franz, K.J.; Gohil, V.M.; Gupta, S.; et al. Connecting copper and cancer: From transition metal signalling to metalloplasia. Nat. Rev. Cancer 2022, 22, 102–113. [Google Scholar] [CrossRef]
- Das, A.; Ash, D.; Fouda, A.Y.; Sudhahar, V.; Kim, Y.M.; Hou, Y.; Hudson, F.Z.; Stansfield, B.K.; Caldwell, R.B.; McMenamin, M.; et al. Cysteine oxidation of copper transporter CTR1 drives VEGFR2 signalling and angiogenesis. Nat. Cell Biol. 2022, 24, 35–50. [Google Scholar] [CrossRef]
- Wondrak, G.T.; Roberts, M.J.; Jacobson, M.K.; Jacobson, E.L. Photosensitized growth inhibition of cultured human skin cells: Mechanism and suppression of oxidative stress from solar irradiation of glycated proteins. J. Investig. Dermatol. 2002, 119, 489–498. [Google Scholar] [CrossRef] [PubMed]
- Park, M.W.; Cha, H.W.; Kim, J.; Kim, J.H.; Yang, H.; Yoon, S.; Boonpraman, N.; Yi, S.S.; Yoo, I.D.; Moon, J.S. NOX4 promotes ferroptosis of astrocytes by oxidative stress-induced lipid peroxidation via the impairment of mitochondrial metabolism in Alzheimer’s diseases. Redox Biol. 2021, 41, 101947. [Google Scholar] [CrossRef] [PubMed]
- Alley, M.C.; Killam, E.K.; Fisher, G.L. The influence of D-penicillamine treatment upon seizure activity and trace metal status in the Senegalese baboon, Papio papio. J. Pharmacol. Exp. Ther. 1981, 217, 138–146. [Google Scholar] [PubMed]
- Rahimi, N.; Sadeghzadeh, M.; Javadi-Paydar, M.; Heidary, M.R.; Jazaeri, F.; Dehpour, A.R. Effects of D-penicillamine on pentylenetetrazole-induced seizures in mice: Involvement of nitric oxide/NMDA pathways. Epilepsy Behav. 2014, 39, 42–47. [Google Scholar] [CrossRef]
- Mao, X.; Wang, X.; Jin, M.; Li, Q.; Jia, J.; Li, M.; Zhou, H.; Liu, Z.; Jin, W.; Zhao, Y.; et al. Critical involvement of lysyl oxidase in seizure-induced neuronal damage through ERK-Alox5-dependent ferroptosis and its therapeutic implications. Acta Pharm. Sin. B 2022. [Google Scholar] [CrossRef]
- Nair, A.B.; Jacob, S. A simple practice guide for dose conversion between animals and human. J. Basic Clin. Pharm. 2016, 7, 27–31. [Google Scholar] [CrossRef]
- Jiang, T.; Yu, J.T.; Zhu, X.C.; Tan, M.S.; Gu, L.Z.; Zhang, Y.D.; Tan, L. Triggering receptor expressed on myeloid cells 2 knockdown exacerbates aging-related neuroinflammation and cognitive deficiency in senescence-accelerated mouse prone 8 mice. Neurobiol. Aging 2014, 35, 1243–1251. [Google Scholar] [CrossRef]
- Tan, M.S.; Tan, L.; Jiang, T.; Zhu, X.C.; Wang, H.F.; Jia, C.D.; Yu, J.T. Amyloid-β induces NLRP1-dependent neuronal pyroptosis in models of Alzheimer’s disease. Cell Death Dis. 2014, 5, e1382. [Google Scholar] [CrossRef]
- Racine, R.; Okujava, V.; Chipashvili, S. Modification of seizure activity by electrical stimulation. 3. Mechanisms. Electroencephalogr. Clin. Neurophysiol. 1972, 32, 295–299. [Google Scholar] [CrossRef]
- He, M.; Liu, J.; Cheng, S.; Xing, Y.; Suo, W.Z. Differentiation renders susceptibility to excitotoxicity in HT22 neurons. Neural Regen. Res. 2013, 8, 1297–1306. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome. Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Huang, Z.; Duan, J.; Nice, E.C.; Lin, J.; Huang, C. Elesclomol induces copper-dependent ferroptosis in colorectal cancer cells via degradation of ATP7A. Mol. Oncol. 2021, 15, 3527–3544. [Google Scholar] [CrossRef] [PubMed]
- van de Stadt, R.J.; Muijsers, A.O.; Henrichs, A.M.; van der Korst, J.K. D-penicillamine: Biochemical, metabolic and pharmacological aspects. Scand. J. Rheumatol. 1979, 8 (Suppl. S28), 13–20. [Google Scholar] [CrossRef] [PubMed]
- Gupte, A.; Mumper, R.J. Copper chelation by D-penicillamine generates reactive oxygen species that are cytotoxic to human leukemia and breast cancer cells. Free Radic. Biol. Med. 2007, 43, 1271–1278. [Google Scholar] [CrossRef]
- Dhaher, R.; Gruenbaum, S.E.; Sandhu, M.R.S.; Ottestad-Hansen, S.; Tu, N.; Wang, Y.; Lee, T.W.; Deshpande, K.; Spencer, D.D.; Danbolt, N.C.; et al. Network-Related Changes in Neurotransmitters and Seizure Propagation During Rodent Epileptogenesis. Neurology 2021, 96, e2261–e2271. [Google Scholar] [CrossRef]
- Zhang, H.L.; Hu, B.X.; Li, Z.L.; Du, T.; Shan, J.L.; Ye, Z.P.; Peng, X.D.; Li, X.; Huang, Y.; Zhu, X.Y.; et al. PKCβII phosphorylates ACSL4 to amplify lipid peroxidation to induce ferroptosis. Nat. Cell Biol. 2022, 24, 88–98. [Google Scholar] [CrossRef]
- Doll, S.; Proneth, B.; Tyurina, Y.Y.; Panzilius, E.; Kobayashi, S.; Ingold, I.; Irmler, M.; Beckers, J.; Aichler, M.; Walch, A.; et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 2017, 13, 91–98. [Google Scholar] [CrossRef]
- Jiang, L.; Kon, N.; Li, T.; Wang, S.J.; Su, T.; Hibshoosh, H.; Baer, R.; Gu, W. Ferroptosis as a p53-mediated activity during tumour suppression. Nature 2015, 520, 57–62. [Google Scholar] [CrossRef]
- McClements, B.M.; Callender, M.E. D-penicillamine therapy in patients with HBsAg-negative chronic active hepatitis and major prednisolone-induced adverse effects. J. Hepatol. 1990, 11, 322–325. [Google Scholar] [CrossRef]
- Pisetsky, D.S.; St Clair, E.W. Progress in the treatment of rheumatoid arthritis. JAMA 2001, 286, 2787–2790. [Google Scholar] [CrossRef] [PubMed]
- Steen, V.D.; Medsger, T.A., Jr.; Rodnan, G.P. D-Penicillamine therapy in progressive systemic sclerosis (scleroderma): A retrospective analysis. Ann. Intern. Med. 1982, 97, 652–659. [Google Scholar] [CrossRef] [PubMed]
- Matloff, D.S.; Alpert, E.; Resnick, R.H.; Kaplan, M.M. A prospective trial of D-penicillamine in primary biliary cirrhosis. N. Engl. J. Med. 1982, 306, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.; Scheuer, P.J.; Samourian, S.; McGee, J.O. A controlled trial of D-penicillamine therapy in primary biliary cirrhosis. Lancet. 1977, 1, 831–834. [Google Scholar] [CrossRef]
- Yokoyama, K.; Araki, S.; Abe, H. Distribution of nerve conduction velocities in acute thallium poisoning. Muscle Nerve 1990, 13, 117–120. [Google Scholar] [CrossRef]
- Thompson, C.; Dent, J.; Saxby, P. Effects of thallium poisoning on intellectual function. Br. J. Psychiatry 1988, 153, 396–399. [Google Scholar] [CrossRef]
- Squitti, R.; Rossini, P.M.; Cassetta, E.; Moffa, F.; Pasqualetti, P.; Cortesi, M.; Colloca, A.; Rossi, L.; Finazzi-Agró, A. d-penicillamine reduces serum oxidative stress in Alzheimer’s disease patients. Eur. J. Clin. Investig. 2002, 32, 51–59. [Google Scholar] [CrossRef]
- Cui, Z.; Lockman, P.R.; Atwood, C.S.; Hsu, C.H.; Gupte, A.; Allen, D.D.; Mumper, R.J. Novel D-penicillamine carrying nanoparticles for metal chelation therapy in Alzheimer’s and other CNS diseases. Eur. J. Pharm. Biopharm. 2005, 59, 263–272. [Google Scholar] [CrossRef]
- Olney, J.W.; de Gubareff, T.; Labruyere, J. Seizure-related brain damage induced by cholinergic agents. Nature 1983, 301, 520–522. [Google Scholar] [CrossRef]
- Ben-Ari, Y. Limbic seizure and brain damage produced by kainic acid: Mechanisms and relevance to human temporal lobe epilepsy. Neuroscience 1985, 14, 375–403. [Google Scholar] [CrossRef]
- Holmes, G.L. Seizure-induced neuronal injury: Animal data. Neurology 2002, 59, S3–S6. [Google Scholar] [CrossRef] [PubMed]
- Ciuffi, M.; Gentilini, G.; Franchi-Micheli, S.; Zilletti, L. D-penicillamine affects lipid peroxidation and iron content in the rat brain cortex. Neurochem Res. 1992, 17, 1241–1246. [Google Scholar] [CrossRef] [PubMed]
- Verkman, A.S.; Anderson, M.O.; Papadopoulos, M.C. Aquaporins: Important but elusive drug targets. Nat. Rev. Drug Discov. 2014, 13, 259–277. [Google Scholar] [CrossRef]
- Koike, S.; Tanaka, Y.; Matsuzaki, T.; Morishita, Y.; Ishibashi, K. Aquaporin-11 (AQP11) Expression in the Mouse Brain. Int. J. Mol. Sci. 2016, 17, 861. [Google Scholar] [CrossRef]
- Yeung, C.H.; Cooper, T.G. Aquaporin AQP11 in the testis: Molecular identity and association with the processing of residual cytoplasm of elongated spermatids. Reproduction 2010, 139, 209–216. [Google Scholar] [CrossRef]
- Morishita, Y.; Matsuzaki, T.; Hara-chikuma, M.; Andoo, A.; Shimono, M.; Matsuki, A.; Kobayashi, K.; Ikeda, M.; Yamamoto, T.; Verkman, A.; et al. Disruption of aquaporin-11 produces polycystic kidneys following vacuolization of the proximal tubule. Mol. Cell Biol. 2005, 25, 7770–7779. [Google Scholar] [CrossRef]
- Gorelick, D.A.; Praetorius, J.; Tsunenari, T.; Nielsen, S.; Agre, P. Aquaporin-11: A channel protein lacking apparent transport function expressed in brain. BMC Biochem. 2006, 7, 14. [Google Scholar] [CrossRef]
- Xi, T.; Jin, F.; Zhu, Y.; Wang, J.; Tang, L.; Wang, Y.; Liebeskind, D.S.; Scalzo, F.; He, Z. miR-27a-3p protects against blood-brain barrier disruption and brain injury after intracerebral hemorrhage by targeting endothelial aquaporin-11. J. Biol. Chem. 2018, 293, 20041–20050. [Google Scholar] [CrossRef]
- Hoshino, Y.; Sonoda, H.; Nishimura, R.; Mori, K.; Ishibashi, K.; Ikeda, M. Involvement of the NADPH oxidase 2 pathway in renal oxidative stress in Aqp11-/- mice. Biochem. Biophys. Rep. 2019, 17, 169–176. [Google Scholar] [CrossRef]
- Weiss, K.H.; Thurik, F.; Gotthardt, D.N.; Schäfer, M.; Teufel, U.; Wiegand, F.; Merle, U.; Ferenci-Foerster, D.; Maieron, A.; Stauber, R.; et al. Efficacy and safety of oral chelators in treatment of patients with Wilson disease. Clin. Gastroenterol. Hepatol. 2013, 11, e1021–e1022. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.; Klingenberg, S.L.; Gluud, C. Systematic review and meta-analysis: D-Penicillamine vs. placebo/no intervention in patients with primary biliary cirrhosis—Cochrane Hepato-Biliary Group. Aliment. Pharmacol. Ther. 2006, 24, 1535–1544. [Google Scholar] [CrossRef]
- Boehler, A.; Vogt, P.; Speich, R.; Weder, W.; Russi, E.W. Bronchiolitis obliterans in a patient with localized scleroderma treated with D-penicillamine. Eur. Respir. J. 1996, 9, 1317–1319. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chemical | Source | Identifier |
|---|---|---|
| Kainic acid | Sigma | K0250, more than 99% purity |
| D-penicillamine | Sigma | P4875, 98–101% purity |
| Glutamate | Sigma | G8415, 98.5–100.5% purity |
| Erastin | Selleck | S7242 |
| Ferrostatin-1 | Selleck | S7243 |
| Dimethyl sulfoxide | Sigma | D2650, more than 99.7% purity |
| Dulbecco’s modified Eagle’s medium | Gibco | C11995500BT |
| Hank’s Balanced Salt Solution | Gibco | 14065056BT |
| Fetal bovine serum | Gibco | 10099–141 |
| Penicillin-Streptomycin | Gibco | 15140122 |
| Lipid peroxidase kit | Jiancheng Biotechnology | A106 |
| Copper Colorimetric Assay Kit | Elabscience | E-BC-K300-M |
| BODIPYTM 581/591 C11 | Invitrogen | D3861 |
| Illumina TruSeq RNA Sample Prep Kit | Illumina | FC-122-1001 |
| TRIzol | Invitrogen | 15596026 |
| PrimeScriptTM RT reagent Kit with gDNA Eraser | TAKARA | RR047A |
| cDNA synthesis kit | TAKARA | RR820A |
| Diethypyrocarbonate-treated Water | Beyotime Biotechnology | R0021 |
| Lysis buffer | Beyotime Biotechnology | P0013 |
| ECL prime Western blotting det | GE | RPN2232 |
| Triton X-100 solution | Beyotime Biotechnology | ST797 |
| Phosphate-buffered saline | Solarbio | P1010 |
| Normal donkey serum | Solarbio | SL050 |
| Fluoro-Jade B | AAT Bioquest | 23061 |
| Nissl staining solution | Beyotime Biotechnology | C0117 |
| LipofectamineTM RNAiMAX | Thermo | 13778075 |
| EntransterTM-in vivo | Engreen Biosystem | 18668-11-1 |
| siRNA Sequences | |||
| Name/Gene ID | Nomenclature in Our Paper | Target Sequences (5′-3′) | |
| Aqp11 (66333) | Si-Aqp11 (1) | CTCTGACACTGATCTACTT | |
| Si-Aqp11 (2) | CAAGTACCATTACGACGAA | ||
| Si-Aqp11 (3) | CTTCCATGGCTGCATAACA | ||
| RT-qPCR primers | |||
| Name/Gene ID | Oligonucleotide | Primer sequences (5′-3′) | Product size (bp) |
| Ptgs2 (19225) | Upper primer | GGGAGTCTGGAACATTGTGAA | 112 |
| Lower primer | GTGCACATTGTAAGTAGGTGGACT | ||
| Hist1h1d (14957) | Upper primer | GTGGAGAAGACACCTGTGAAG | 535 |
| Lower primer | CCTTGGCTGGACTCTTTGCT | ||
| Aqp11 (66333) | Upper primer | TGGGGCTAATGCTGCTGTTC | 300 |
| Lower primer | CACCCATTTCGGGGGACATA | ||
| Tymp (72962) | Upper primer | CGCGGTGATAGATGGAAGAGC | 187 |
| Lower primer | CACACCTCCTGTGGAGTGTT | ||
| Arnt2 (11864) | Upper primer | TTATCACGTTTGTGGACCCCA | 269 |
| Lower primer | GTTGGTGCAGGTGACGTACT | ||
| Rsad2 (58185) | Upper primer | TGCTGGCTGAGAATAGCATTAGG | 112 |
| Lower primer | GCTGAGTGCTGTTCCCATCT | ||
| β-actin (11461) | Upper primer | GTGACGTTGACATCCGTAAAGA | 245 |
| Lower primer | GCCGGACTCATCGTACTCC | ||
| Application | Antibody | Species | Dilution | Cat. No. | Source |
|---|---|---|---|---|---|
| Western blot (Primary antibodies) | Cleaved caspase-3 LC3II/I | mouse rabbit | 1:500 1:1000 | sc-373730 4108S | SantaCruz Cell Signaling Technology |
| ACSL4 | mouse | 1:500 | sc-365230 | SantaCruz | |
| COX-2 | rabbit | 1:3000 | A5787 | Abclonal | |
| Aqp11 | rabbit | 1:2000 | AP5805b | Abgent | |
| Hist1h1d | rabbit | 1:1000 | 12177-1 | Absci | |
| Tymp | rabbit | 1:1000 | 12383-1-AP | Ptglab | |
| Arnt2 | rabbit | 1:1000 | A8060 | Abclonal | |
| Rsad2 | rabbit | 1:1000 | A8271 | Abclonal | |
| β-actin | rabbit | 1:10,000 | AP0060 | Bioworld | |
| Immunofluorescence (Primary antibodies) | Aqp11 | rabbit | 1:500 | AP5805b | Abgent |
| Western blot (Secondary antibodies) | Goat anti-mouse IgG HRP Goat anti-rabbit IgG HRP | goat goat | 1:10,000 1:10,000 | ab97023 A9169 | Abcam Sigma |
| Immunofluorescence (Secondary antibodies) | Alexa Fluor®488 Donkey anti-Rabbit IgG (H + L) | donkey | 1:250 | A-21206 | Thermo |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, N.; Zhang, K.; Guan, Q.-W.; Wang, Z.-J.; Chen, K.-N.; Mao, X.-Y. D-Penicillamine Reveals the Amelioration of Seizure-Induced Neuronal Injury via Inhibiting Aqp11-Dependent Ferroptosis. Antioxidants 2022, 11, 1602. https://doi.org/10.3390/antiox11081602
Yang N, Zhang K, Guan Q-W, Wang Z-J, Chen K-N, Mao X-Y. D-Penicillamine Reveals the Amelioration of Seizure-Induced Neuronal Injury via Inhibiting Aqp11-Dependent Ferroptosis. Antioxidants. 2022; 11(8):1602. https://doi.org/10.3390/antiox11081602
Chicago/Turabian StyleYang, Nan, Kai Zhang, Qi-Wen Guan, Zhao-Jun Wang, Kang-Ni Chen, and Xiao-Yuan Mao. 2022. "D-Penicillamine Reveals the Amelioration of Seizure-Induced Neuronal Injury via Inhibiting Aqp11-Dependent Ferroptosis" Antioxidants 11, no. 8: 1602. https://doi.org/10.3390/antiox11081602