
Dicarbonyl-Dependent Modification of LDL as a Key Factor of Endothelial Dysfunction and Atherosclerotic Vascular Wall Damage



{kind=link}
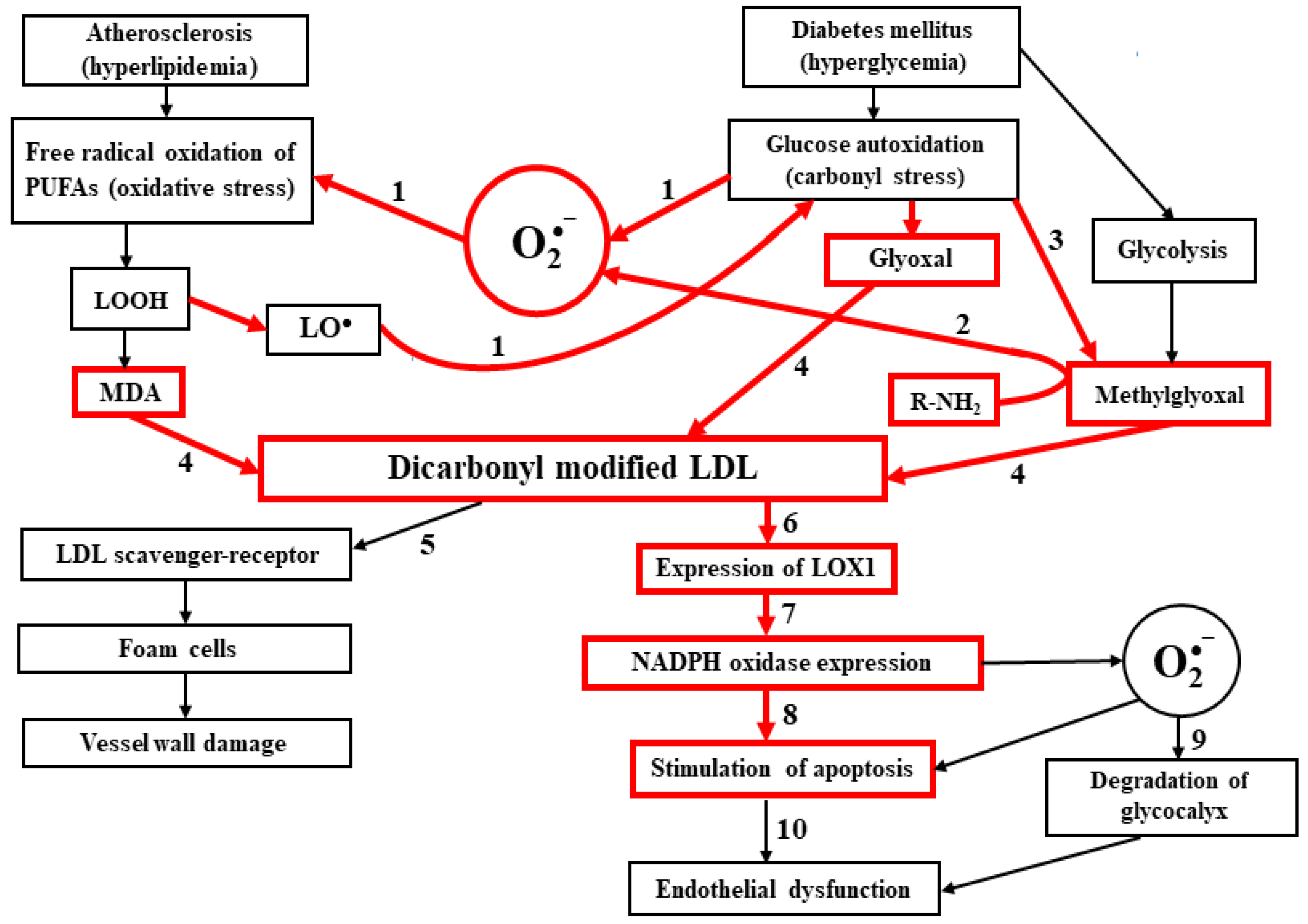
Abstract
:1. Introduction: Free-Radical Oxidation in Atherosclerosis—Background
2. Oxidative Stress in Atherosclerosis and Atherogenic Modification of LDL
3. Carbonyl Stress in Diabetes and the Role of Modified LDL in Vascular Wall Damage
4. Oxidatively Modified LDL, LOX-1 and Endothelial Dysfunction
5. Active Oxygen Species and Glycocalyx Degradation
6. Antioxidants in Cardiology: Pro Et Contra
7. Conclusions: Dicarbonyl-Modification LDL Is a Key Factor of Endothelial Dysfunction and Atherogenesis
Funding
Conflicts of Interest
References
- Harman, D. Aging: A Theory Based on Free Radical and Radiation Chemistry. J. Gerontol. 1956, 11, 298–300. [Google Scholar] [CrossRef] [PubMed]
- Harman, D. Free radical theory of aging: The “free radical” diseases. Age 1984, 7, 111–131. [Google Scholar] [CrossRef]
- Harman, D. The Free Radical Theory of Aging. Antioxid. Redox Signal. 2003, 5, 557–561. [Google Scholar] [CrossRef] [PubMed]
- Woodford, F.; Böttcher, C.; Oette, K.; Ahrens, E. The artifactual nature of lipid peroxides detected in extracts of human aorta. J. Atheroscler. Res. 1965, 5, 311–316. [Google Scholar] [CrossRef]
- Oette, K.; Peterson, M.L.; McAuley, R.L. A highly sensitive method for measurement of lipid hydroperoxides by iodometry and amperometric endpoint. J. Lipid Res. 1963, 4, 212–215. [Google Scholar] [CrossRef]
- Lankin, V.Z.; Tikhaze, A.K. Atherosclerosis as a free radical pathology and antioxidative therapy of this disease. In Free Radicals, NO and Inflammation; IOS Press: Amsterdam, The Netherland, 2003; Volume 344, pp. 218–231. [Google Scholar]
- Kühn, H.; Belkner, J.; Wiesner, R.; Schewe, T.; Lankin, V.Z.; Tikhaze, A.K. Structure elucidation of oxygenated lipids in human atherosclerotic lesions. Eicosanoids 1992, 5, 17–22. [Google Scholar]
- Harland, W.; Gilbert, J.D.; Brooks, C.J. Lipids of human atheroma: VIII. Oxidised derivatives of cholesteryl linoleate. Biochim. Biophys. Acta 1973, 316, 378–385. [Google Scholar] [CrossRef]
- Carpenter, K.L.; Taylor, S.E.; Ballantine, J.A.; Fussell, B.; Halliwell, B.; Mitchinson, M.J. Lipids and oxidised lipids in human atheroma and normal aorta. Biochim. Biophys. Acta 1993, 1167, 121–130. [Google Scholar] [CrossRef]
- Lankin, V.Z.; Vikhert, A.M.; Kosykh, V.A.; Tikhaze, A.K.; Galakhov, I.E.; Orekhov, A.N.; Repin, V.N. Enzymatic detoxication of superoxide anion-radical and lipoperoxides in intima and media of atherosclerotic aorta. Biomed. Biochim. Acta 1984, 43, 797–802. [Google Scholar] [PubMed]
- Lankin, V.; Tikhaze, A. Role of Oxidative Stress in the Genesis of Atherosclerosis and Diabetes Mellitus: A Personal Look Back on 50 Years of Research. Curr. Aging Sci. 2017, 10, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Esterbauer, H.; Gebicki, J.; Puhl, H.; Jürgens, G. The role of lipid peroxidation and antioxidants in oxidative modification of LDL. Free Radic. Biol. Med. 1992, 13, 341–390. [Google Scholar] [CrossRef]
- Steinbrecher, U.P.; Parthasarathy, S.; Leake, D.S.; Witztum, J.L.; Steinberg, D. Modification of low density lipoprotein by endothelial cells involves lipid peroxidation and degradation of low density lipoprotein phospholipids. Proc. Natl. Acad. Sci. USA 1984, 81, 3883–3887. [Google Scholar] [CrossRef]
- Goldstein, J.L.; Ho, Y.K.; Basu, S.K.; Brown, M.S. Binding site on macrophages that mediates uptake and degradation of acetylated low density lipoprotein, producing massive cholesterol deposition. Proc. Natl. Acad. Sci. USA 1979, 76, 333–337. [Google Scholar] [CrossRef]
- Epstein, F.H.; Steinberg, D.; Parthasarathy, S.; Carew, T.E.; Khoo, J.C.; Witztum, J.L. Beyond Cholesterol. Modification of low density lipoprotein that increase its atherogenicity. N. Engl. J. Med. 1989, 320, 915–924. [Google Scholar] [CrossRef]
- Steibrecher, U.P.; Lougheed, M.; Kwan, W.-C.; Dirks, M. Recognition of oxidized low density lipoprotein by the scavenger receptor of macrophages results from derivatization of apoprotein B by products fatty acid peroxidation. J. Biol. Chem. 1989, 264, 15216–15223. [Google Scholar] [CrossRef]
- Kita, T.; Ishii, K.; Yokode, M.; Kume, N.; Nagano, Y.; Arai, H.; Kawai, C. The role of oxidized low density lipoprotein in the pathogenesis of atherosclerosis. Eur. Heart J. 1990, 11 (Suppl. E), 122–127. [Google Scholar] [CrossRef]
- Witztum, J.L.; Steinberg, D. Role of oxidized low density lipoprotein in atherogenesis. J. Clin. Investig. 1991, 88, 1785–1792. [Google Scholar] [CrossRef]
- Witztum, J. The oxidation hypothesis of atherosclerosis. Lancet 1994, 344, 793–795. [Google Scholar] [CrossRef]
- Ylä-Herttuala, S. Macrophages and Oxidized Low Density Lipoproteins in the Pathogenesis of Atherosclerosis. Ann. Med. 1991, 23, 561–567. [Google Scholar] [CrossRef]
- Yla-Herttuala, S. Role of lipid and lipoprotein oxidation in the pathogenesis of atherosclerosis. Drugs Today 1994, 30, 507–514. [Google Scholar]
- Steinberg, D. Role of Oxidized LDL and Antioxidants in Atherosclerosis. Adv. Exp. Med. Biol. 1995, 369, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Estévez, M.; Padilla, P.; Carvalho, L.; Martín, L.; Carrapiso, A.; Delgado, J. Malondialdehyde interferes with the formation and detection of primary carbonyls in oxidized proteins. Redox. Biol. 2019, 26, 101277. [Google Scholar] [CrossRef] [PubMed]
- Fogelman, A.M.; Shechter, I.; Seager, J.; Hokom, M.; Child, J.S.; Edwards, P.A. Malondialdehyde alteration of low density lipoproteins leads to the cholesteryl ester accumulation in human monocyte-macrophages. Proc. Natl. Acad. Sci. USA 1980, 77, 2214–2218. [Google Scholar] [CrossRef]
- Lankin, V.Z. Lipid peroxides and atherosclerosis. Hypothesis: The role of cholesterol and free-radical lipid peroxidation in altering cell membrane properties in hypercholesterolemia and atherosclerosis. Kardiologiia 1980, 20, 42–48. [Google Scholar]
- Halliwell, B. Reactive Species and Antioxidants. Redox Biology Is a Fundamental Theme of Aerobic Life. Plant Physiol. 2006, 141, 312–322. [Google Scholar] [CrossRef]
- Lankin, V.Z.; Tikhaze, A.K.; Kumskova, E.M. Macrophages actively accumulate malonyldialdehyde-modified but not enzymatically oxidized low density lipoprotein. Mol. Cell. Biochem. 2012, 365, 93–98. [Google Scholar] [CrossRef]
- Schewe, T.; Rapoport, S.M.; Kühn, H. Enzymology and Physiology of Reticulocyte Lipoxygenase: Comparison with Other Lipoxygenases. Adv. Enzymol. 1986, 58, 191–272. [Google Scholar] [CrossRef] [PubMed]
- Summerhill, V.; Orekhov, A. Pericytes in Atherosclerosis. Adv. Exp. Med. Biol. 2019, 1147, 279–297. [Google Scholar] [CrossRef] [PubMed]
- Lankin, V.; Viigimaa, M.; Tikhaze, A.; Kumskova, E.; Konovalova, G.; Abina, J.; Zemtsovskaya, G.; Kotkina, T.; Yanushevskaya, E.; Vlasik, T. Cholesterol-rich low density lipoproteins are also more oxidized. Mol. Cell. Biochem. 2011, 355, 187–191. [Google Scholar] [CrossRef]
- Khlebus, E.; Kutsenko, V.; Meshkov, A.; Ershova, A.; Kiseleva, A.; Shcherbakova, N.; Zharikova, A.; Drapkina, O.; Shevtsov, A.; Yarovaya, E.; et al. Multiple rare and common variants in APOB gene locus associated with oxidatively modified low-density lipoprotein levels. PLoS ONE 2019, 14, e0217620. [Google Scholar] [CrossRef]
- Tikhaze, A.K.; Kosach, V.Y.; Lankin, V.Z.; Panferova, A.A.; Smirnova, M.D. Indicator Characterizing Carbonyl-Dependent Modification of Erythrocytic Superoxydismutase as a Biochemical Marker of Oxidative Stress in Coronary Heart Disease. Kardiologiia 2020, 60, 47–51. [Google Scholar] [CrossRef] [PubMed]
- Lankin, V.Z.; Shumaev, K.B.; Tikhaze, A.K.; Kurganov, B.I. Influence of dicarbonyls on kinetic characteristics of glutathione peroxidase. Dokl. Biochem. Biophys. 2017, 475, 287–290. [Google Scholar] [CrossRef] [PubMed]
- Nishizawa, T.; Bornfeldt, K.E. Diabetic vascular disease and the potential role of macrophage glucose metabolism. Ann. Med. 2012, 44, 555–563. [Google Scholar] [CrossRef]
- Bornfeldt, K.E. Does Elevated Glucose Promote Atherosclerosis? Pros and Cons. Circ. Res. 2016, 119, 190–193. [Google Scholar] [CrossRef]
- Poznyak, A.; Grechko, A.V.; Poggio, P.; Myasoedova, V.A.; Alfieri, V.; Orekhov, A.N. The Diabetes Mellitus–Atherosclerosis Connection: The Role of Lipid and Glucose Metabolism and Chronic Inflammation. Int. J. Mol. Sci. 2020, 21, 1835. [Google Scholar] [CrossRef]
- Oberley, L.W. Free radicals and diabetes. Free Radic. Biol. Med. 1988, 5, 113–124. [Google Scholar] [CrossRef]
- Lankin, V.Z.; Tikhaze, A.K.; Kapel’Ko, V.I.; Shepel’Kova, G.S.; Shumaev, K.B.; Panasenko, O.M.; Konovalova, G.G.; Belenkov, Y.N. Mechanisms of oxidative modification of low density lipoproteins under conditions of oxidative and carbonyl stress. Biochemistry 2007, 72, 1081–1090. [Google Scholar] [CrossRef]
- Thornalley, P.J.; Langborg, A.; Minhas, H.S. Formation of glyoxal, methylglyoxal and 3-deoxyglucosone in the glycation of proteins by glucose. Biochem. J. 1999, 344, 109–116. [Google Scholar] [CrossRef]
- Wang, X.-J.; Ma, S.-B.; Liu, Z.-F.; Li, H.; Gao, W.-Y. Elevated levels of α-dicarbonyl compounds in the plasma of type II diabetics and their relevance with diabetic nephropathy. J. Chromatogr. B Analyt. Technol. Biomed. Life. Sci. 2019, 1106–1107, 19–25. [Google Scholar] [CrossRef]
- Spiteller, G. The relation of lipid peroxidation processes with atherogenesis: A new theory on atherogenesis. Mol. Nutr. Food Res. 2005, 49, 999–1013. [Google Scholar] [CrossRef]
- Spiteller, G. Peroxyl Radicals Are Essential Reagents in the Oxidation Steps of the Maillard Reaction Leading to Generation of Advanced Glycation End Products. Ann. N. Y. Acad. Sci. 2008, 1126, 128–133. [Google Scholar] [CrossRef] [PubMed]
- Lankin, V.; Shadyro, O.; Shumaev, K.; Tikhaze, A.; Sladkova, A. Non-Enzymatic Methylglyoxal Formation From glucose Metabolites and Generation of Superoxide Anion Radical during Methylglyoxal-Dependent Cross-Links Reaction. J. Antioxid. Act. 2019, 1, 33–45. [Google Scholar] [CrossRef]
- Lankin, V.; Konovalova, G.; Tikhaze, A.; Shumaev, K.; Kumskova, E.; Viigimaa, M. The initiation of free radical peroxidation of low-density lipoproteins by glucose and its metabolite methylglyoxal: A common molecular mechanism of vascular wall injure in atherosclerosis and diabetes. Mol. Cell. Biochem. 2014, 395, 241–252. [Google Scholar] [CrossRef]
- Lankin, V.Z.; Tikhaze, A.K.; Konovalova, G.G.; Odinokova, A.O.; Doroshchuk, A.N.; Chazova, E.I. Oxidative and carbonyl stress as factors of protein modification and DNA destruction in diabetes mellitus. Ther. Arch. 2018, 90, 46–50. [Google Scholar] [CrossRef] [PubMed]
- Graille, M.; Wild, P.; Sauvain, J.-J.; Hemmendinger, M.; Canu, I.G.; Hopf, N.B. Urinary 8-OHdG as a Biomarker for Oxidative Stress: A Systematic Literature Review and Meta-Analysis. Int. J. Mol. Sci. 2020, 21, 3743. [Google Scholar] [CrossRef]
- Lankin, V.Z.; Konovalova, G.G.; Tikhaze, A.K.; Shumaev, K.B.; Kumskova, E.M.B.; Grechnikova, M.A.; Viigimaa, M. Aldehyde inhibition of antioxidant enzymes in the blood of diabetic patients. J. Diabetes 2016, 8, 398–404. [Google Scholar] [CrossRef] [PubMed]
- Knott, H.M.; Brown, B.E.; Davies, M.J.; Dean, R.T. Glycation and glycoxidation of low-density lipoproteins by glucose and low-molecular mass aldehydes. Formation of modified and oxidized particles. Eur. J. Biol. Chem. 2003, 270, 3572–3582. [Google Scholar] [CrossRef] [PubMed]
- Pirillo, A.; Norata, G.D.; Catapano, A.L. LOX-1, OxLDL, and Atherosclerosis. Mediat. Inflamm. 2013, 2013, 152786. [Google Scholar] [CrossRef] [PubMed]
- Lubrano, V.; Balzan, S. LOX-1 and ROS, inseparable factors in the process of endothelial damage. Free Radic. Res. 2014, 48, 841–848. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Orekhov, A.N.; Bobryshev, Y.V. LOX-1-Mediated Effects on Vascular Cells in Atherosclerosis. Cell. Physiol. Biochem. 2016, 38, 1851–1859. [Google Scholar] [CrossRef]
- Kattoor, A.J.; Kanuri, S.H.; Mehta, J.L. Role of Ox-LDL and LOX-1 in Atherogenesis. Curr. Med. Chem. 2019, 26, 1693–1700. [Google Scholar] [CrossRef] [PubMed]
- Galle, J.; Schneider, R.; Heinloth, A.; Wanner, C.; Galle, P.R.; Conzelmann, E.; Dimmeler, S.; Heermeier, K. Lp(a) and LDL induce apoptosis in human endothelial cells and in rabbit aorta: Role of oxidative stress. Kidney Int. 1999, 55, 1450–1461. [Google Scholar] [CrossRef]
- Lankin, V.Z.; Sharapov, M.G.; Goncharov, R.G.; Antonova, O.A.; Tikhaze, A.K.; Konovalova, G.G. Expression of LOX-1 and NADPH Oxidase in Endotheliocytes by Dicarbonyl-Modified LDL. Moscow, Russia. 2022; manuscript in preparation. [Google Scholar]
- Sharapov, M.G.; Goncharov, R.G.; Gordeeva, A.E.; Novoselov, V.I.; Antonova, O.A.; Tikhaze, A.K.; Lankin, V.Z. Enzymatic antioxidant system of endotheliocytes. Dokl. Biochem. Biophys. 2016, 471, 410–412. [Google Scholar] [CrossRef]
- Lankin, V.Z.; Sharapov, M.G.; Goncharov, R.G.; Tikhaze, A.K.; Novoselov, V.I. Natural Dicarbonyls Inhibit Peroxidase Activity of Peroxiredoxins. Dokl. Biochem. Biophys. 2019, 485, 132–134. [Google Scholar] [CrossRef] [PubMed]
- Weinbaum, S.; Tarbell, J.M.; Damiano, E.R. The Structure and Function of the Endothelial Glycocalyx Layer. Annu. Rev. Biomed. Eng. 2007, 9, 121–167. [Google Scholar] [CrossRef] [PubMed]
- Reitsma, S.; Slaaf, D.W.; Vink, H.; van Zandvoort, M.A.M.J.; Oude Egbrink, M.G. The endothelial glycocalyx: Composition, functions, and visualization. Pflug. Arch. 2007, 454, 345–359. [Google Scholar] [CrossRef] [PubMed]
- Noble, M.; Drake-Holland, A.; Vink, H. Hypothesis: Arterial glycocalyx dysfunction is the first step in the atherothrombotic process. QJM 2008, 101, 513–518. [Google Scholar] [CrossRef]
- Becker, B.F.; Jacob, M.; Leipert, S.; Salmon, A.H.J.; Chappell, D. Degradation of the endothelial glycocalyx in clinical settings: Searching for the sheddases. Br. J. Clin. Pharmacol. 2015, 80, 389–402. [Google Scholar] [CrossRef]
- Pillinger, N.L.; Kam, P.C.A. Endothelial Glycocalyx: Basic Science and Clinical Implications. Anaesth. Intensive Care 2017, 45, 295–307. [Google Scholar] [CrossRef]
- Nieuwdorp, M.; van Haeften, T.W.; Gouverneur, M.C.; Mooij, H.L.; van Lieshout, M.H.; Levi, M.; Meijers, J.C.; Holleman, F.; Hoekstra, J.B.; Vink, H.; et al. Loss of Endothelial Glycocalyx during Acute Hyperglycemia Coincides with Endothelial Dysfunction and Coagulation Activation In Vivo. Diabetes 2006, 55, 480–486. [Google Scholar] [CrossRef] [PubMed]
- Curry, F.E.; Adamson, R.H. Endothelial Glycocalyx: Permeability Barrier and Mechanosensor. Ann. Biomed. Eng. 2012, 40, 828–839. [Google Scholar] [CrossRef] [PubMed]
- Mulivor, A.W.; Lipowsky, H.H. Role of glycocalyx in leukocyte-endothelial cell adhesion. Am. J. Physiol.-Heart Circ. Physiol. 2002, 283, H1282–H1291. [Google Scholar] [CrossRef] [PubMed]
- Egbrink, M.G.A.O.; Heijnen, V.V.T.; Megens, R.T.A.; Engels, W.; Vink, H.; Slaaf, D.W.; van Zandvoort, M.A.M.J.; Reitsma, S. Endothelial glycocalyx thickness and platelet-vessel wall interactions during atherogenesis. Thromb. Haemost. 2011, 106, 939–946. [Google Scholar] [CrossRef] [PubMed]
- Alphonsus, C.S.; Rodseth, R. The endothelial glycocalyx: A review of the vascular barrier. Anaesthesia 2014, 69, 777–784. [Google Scholar] [CrossRef] [PubMed]
- Henrich, M.; Gruss, M.; Weigand, M.A. Sepsis-Induced Degradation of Endothelial Glycocalix. Sci. World J. 2010, 10, 917–923. [Google Scholar] [CrossRef] [PubMed]
- Melkumyants, A.M.; Balashov, S.A.; Khayutin, V.M. Control of arterial lumen by shear stress on endothelium. NIPS 1995, 10, 204–210. [Google Scholar] [CrossRef]
- Gouverneur, M.; Berg, B.; Nieuwdorp, M.; Stroes, E.; Vink, H. Vasculoprotective properties of the endothelial glycocalyx: Effects of fluid shear stress. J. Intern. Med. 2006, 259, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Berg, B.M.V.D.; Spaan, J.A.E.; Vink, H. Impaired glycocalyx barrier properties contribute to enhanced intimal low-density lipoprotein accumulation at the carotid artery bifurcation in mice. Pflüg. Arch.-Eur. J. Physiol. 2009, 457, 1199–1206. [Google Scholar] [CrossRef] [PubMed]
- Dogné, S.; Flamion, B. Endothelial Glycocalyx Impairment in Disease: Focus on hyaluronan shedding. Am. J. Pathol. 2020, 190, 768–780. [Google Scholar] [CrossRef] [PubMed]
- Rehm, M.; Bruegger, D.; Christ, F.; Conzen, P.; Thiel, M.; Jacob, M.; Chappell, D.; Stoeckelhuber, M.; Welsch, U.; Reichart, B.; et al. Shedding of the Endothelial Glycocalyx in Patients Undergoing Major Vascular Surgery with Global and Regional Ischemia. Circulation 2007, 116, 1896–1906. [Google Scholar] [CrossRef] [PubMed]
- Chappell, D.; Jacob, M.; Hofmann-Kiefer, K.; Rehm, M.; Welsch, U.; Conzen, P.; Becker, B.F. Antithrombin reduces shedding of the endothelial glycocalyx following ischaemia/reperfusion. Cardiovasc. Res. 2009, 83, 388–396. [Google Scholar] [CrossRef] [PubMed]
- Rubio-Gayosso, I.; Platts, S.H.; Duling, B.R. Reactive oxygen species mediate modification of glycocalyx during ischemia-reperfusion injury. Am. J. Physiol.-Heart Circ. Physiol. 2006, 290, H2247–H2256. [Google Scholar] [CrossRef]
- Moseley, R.; Waddington, R.; Embery, G. Degradation of glycosaminoglycans by reactive oxygen species derived from stimulated polymorphonuclear leukocytes. Biochim. Biophys. Acta 1997, 1362, 221–231. [Google Scholar] [CrossRef]
- Vink, H.; Constantinescu, A.A.; Spaan, J.A.E. Oxidized Lipoproteins Degrade the Endothelial Surface Layer: Implications for platelet-endothelial cell adhesion. Circulation 2000, 101, 1500–1502. [Google Scholar] [CrossRef]
- Constantinescu, A.A.; Vink, H.; Spaan, J.A.E. Elevated capillary tube hematocrit reflects degradation of endothelial cell glycocalyx by oxidized LDL. Am. J. Physiol.-Heart Circ. Physiol. 2001, 280, H1051–H1057. [Google Scholar] [CrossRef]
- Ermishkin, V.V.; Lukoshkova, E.V.; Melkumyants, A.M. Malonyldialdehyde- and Methylglyoxal-Induced Suppression of Endothelium-Mediated Dilation of Rat Iliac Artery in Response to Elevation of Blood Flow. J. Evol. Biochem. Physiol. 2021, 57, 792–802. [Google Scholar] [CrossRef]
- Jialal, I.; Traber, M.; Devaraj, S. Is there a vitamin E paradox? Curr. Opin. Lipidol. 2001, 12, 49–53. [Google Scholar] [CrossRef]
- Kuller, L.H. A Time to Stop Prescribing Antioxidant Vitamins to Prevent and Treat Heart Disease? Arter. Thromb. Vasc. Biol. 2001, 21, 1253. [Google Scholar] [CrossRef]
- Steinberg, D. Is there a potential therapeutic role for vitamin E or other antioxidants in atherosclerosis? Curr. Opin. Lipidol. 2000, 11, 603–607. [Google Scholar] [CrossRef] [PubMed]
- Witztum, J.L.; Steinberg, D. The oxidative modification hypothesis of atherosclerosis: Does it hold for humans? Trends Cardiovasc. Med. 2001, 11, 93–102. [Google Scholar] [CrossRef]
- Steinberg, D.; Witztum, J.L. Is the Oxidative Modification Hypothesis Relevant to Human Atherosclerosis? Do the antioxidant trials conducted to date refute the hypothesis? Circulation 2002, 105, 2107–2111. [Google Scholar] [CrossRef] [PubMed]
- Losonczy, K.G.; Harris, T.B.; Havlik, R.J. Vitamin E and vitamin C supplement use and risk of all-cause and coronary heart disease mortality in older persons: The Established Populations for Epidemiologic Studies of the Elderly. Am. J. Clin. Nutr. 1996, 64, 190–196. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, D. Antioxidant Vitamins and Coronary Heart Disease. N. Engl. J. Med. 1993, 328, 1487–1489. [Google Scholar] [CrossRef]
- Steinberg, D. Clinical trials of antioxidants in atherosclerosis: Are we doing the right thing? Lancet 1995, 346, 36–38. [Google Scholar] [CrossRef]
- Hodis, H.N.; Mack, W.J.; La Bree, L.; Cashin-Hemphill, L.; Sevanian, A.; Johnson, R.; Azen, S.P. Serial Coronary Angiographic Evidence That Antioxidant Vitamin Intake Reduces Progression of Coronary Artery Atherosclerosis. JAMA 1995, 273, 1849–1854. [Google Scholar] [CrossRef]
- Rimm, E.B.; Stampfer, M.J.; Ascherio, A.; Giovannucci, E.; Colditz, G.A.; Willett, W.C. Vitamin E Consumption and the Risk of Coronary Heart Disease in Men. N. Engl. J. Med. 1993, 328, 1450–1456. [Google Scholar] [CrossRef]
- Stampfer, M.J.; Hennekens, C.H.; Manson, J.E.; Colditz, G.A.; Rosner, B.; Willett, W.C. Vitamin E Consumption and the Risk of Coronary Disease in Women. N. Engl. J. Med. 1993, 328, 1444–1449. [Google Scholar] [CrossRef]
- Stephens, N.; Parsons, A.; Brown, M.; Schofield, P.; Kelly, F.; Cheeseman, K.; Mitchinson, M. Randomised controlled trial of vitamin E in patients with coronary disease: Cambridge Heart Antioxidant Study (CHAOS). Lancet 1996, 347, 781–786. [Google Scholar] [CrossRef]
- Boaz, M.; Smetana, S.; Weinstein, T.; Matas, Z.; Gafter, U.; Iaina, A.; Knecht, A.; Weissgarten, Y.; Brunner, D.; Fainaru, M.; et al. Secondary prevention with antioxidants of cardiovascular disease in endstage renal disease (SPACE): Randomised placebo-controlled trial. Lancet 2000, 356, 1213–1218. [Google Scholar] [CrossRef]
- Alpha-Tocopherol, Beta Carotene Cancer Prevention Study Group. The Effect of Vitamin E and Beta Carotene on the Incidence of Lung Cancer and Other Cancers in Male Smokers. N. Engl. J. Med. 1994, 330, 1029–1035. [Google Scholar] [CrossRef]
- Gruppo Italiano per lo Studio della Sopravvivenza nell’Infarto Miocardico. Dietary supplementation with n-3 polyunsaturated fatty acids and vitamin E after myocardial infarction: Results of the GISSI-Prevenzione trial. Lancet 1999, 354, 447–455. [Google Scholar] [CrossRef]
- Yusuf, S.; Dagenais, G.; Pogue, J.; Bosch, J.; Sleight, P. Vitamin E Supplementation and Cardiovascular Events in High-Risk Patients. The Heart Outcomes Prevention Evaluation Study Investigators. N. Engl. J. Med. 2000, 342, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Heart Protection Study Collaborative Group. MRC/BHF Heart Protection Study of antioxidant vitamin supplementation in 20,536 high-risk individuals: A randomised placebo-controlled trial. Lancet 2002, 360, 23–33. [Google Scholar] [CrossRef]
- Traber, M.; Burton, G.; Ingold, K.; Kayden, H. RRR- and SRR-alpha-tocopherols are secreted without discrimination in human chylomicrons, but RRR-alpha-tocopherol is preferentially secreted in very low density lipoproteins. J. Lipid Res. 1990, 31, 675–685. [Google Scholar] [CrossRef]
- Bowry, V.; Ingold, K.U.; Stocker, R. Vitamin E in human low-density lipoprotein. When and how this antioxidant becomes a pro-oxidant. Biochem. J. 1992, 288 Pt 2, 341–344. [Google Scholar] [CrossRef] [PubMed]
- Stocker, R.; Bowry, V.W.; Frei, B. Ubiquinol-10 protects human low density lipoprotein more efficiently against lipid peroxidation than does α-tocopherol. Proc. Natl. Acad. Sci. USA 1991, 88, 1646–1650. [Google Scholar] [CrossRef] [PubMed]
- Mohr, D.; Bowry, V.W.; Stocker, R. Dietary supplementation with coenzyme Q10 results in increased levels of ubiquinol-10 within circulating lipoproteins and increased resistance of human low-density lipoprotein to the initiation of lipid peroxidation. Biochim. Biophys. Acta 1992, 1126, 247–254. [Google Scholar] [CrossRef]
- Ahmadvand, H.; Mabuchi, H.; Nohara, A.; Kobayahi, J.; Kawashiri, M.-A. Effects of coenzyme Q(10) on LDL oxidation in vitro. Acta Med. Iran. 2013, 51, 12–18. [Google Scholar]
- Lankin, V.Z.; Tikhaze, A.K.; Kukharchuk, V.V.; Konovalova, G.G.; Pisarenko, O.I.; Kaminnyi, A.I.; Shumaev, K.B.; Belenkov, Y.N. Antioxidants decreases the intensification of low density lipoprotein free radical peroxidation during therapy with statins. Mol. Cell. Biochem. 2003, 249, 129–140. [Google Scholar] [CrossRef]
- Stocker, R. Natural antioxidants and atherosclerosis. Asia Pac. J. Clin. Nutr. 1993, 2 (Suppl. 1), 15–20. [Google Scholar]
- Frei, B.; Kim, M.C.; Ames, B.N. Ubiquinol-10 is an effective lipid-soluble antioxidant at physiological concentrations. Proc. Natl. Acad. Sci. USA 1990, 87, 4879–4883. [Google Scholar] [CrossRef] [PubMed]
- Beyer, R.E. The role of ascorbate in antioxidant protection of biomembranes: Interaction with vitamin E and coenzyme Q. J. Bioenerg. Biomembr. 1994, 26, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Packer, J.E.; Slater, T.F.; Willson, R.L. Direct observation of a free radical interaction between vitamin E and vitamin C. Nature 1979, 278, 737–738. [Google Scholar] [CrossRef] [PubMed]
- Niki, E.; Saito, T.; Kawakami, A.; Kamiya, Y. Inhibition of oxidation of methyl linoleate in solution by vitamin E and vitamin C. J. Biol. Chem. 1984, 259, 4177–4182. [Google Scholar] [CrossRef]
- Tikhaze, A.K.; Konovalova, G.G.; Lankin, V.Z.; Kaminnyi, A.I.; Kaminnaja, V.I.; Ruuge, E.K.; Kukharchuk, V.V. Effect of Ubiquinone Q10 and Antioxidant Vitamins on Free Radical Oxidation of Phospholipids in Biological Membranes of Rat Liver. Bull. Exp. Biol. Med. 2005, 140, 181–183. [Google Scholar] [CrossRef]
- Kagan, V.E.; Freisleben, H.-J.; Tsuchiya, M.; Forte, T.; Packer, L. Generation of Probucol Radicals and Their Reduction by Ascorbate and Dihydrolipoic Acid in Human Low Density Lipoproteins. Free Radic. Res. Commun. 1991, 15, 265–276. [Google Scholar] [CrossRef]
- Shumaev, K.B.; Ruuge, E.K.; Dmitrovsky, A.A.; Bykhovsky, V.Y.; Kukharchuk, V.V. Effect of lipid peroxidation products and antioxidants on the formation of probucol radical in low density lipoproteins. Biochemistry 1997, 62, 657–660. [Google Scholar]
- Tikhaze, A.K.; Lankin, V.Z.; Konovalova, G.G.; Shumaev, K.B.; Kaminnyi, A.I.; Kozachenko, A.I.; Gurevich, S.M.; Nagler, L.G.; Zaitseva, T.M.; Kukharchuk, V.V. Antioxidant probucol as an effective scavenger of lipid radicals in low density lipoproteins in vivo and in vitro. Bull. Exp. Biol. Med. 1999, 128, 818–821. [Google Scholar] [CrossRef]
- Simons, L.A.; Von Konigsmark, M.; Balasubramaniam, S. What dose of vitamin E is required to reduce susceptibility of LDL to oxidation? Aust. N. Z. J. Med. 1996, 26, 496–503. [Google Scholar] [CrossRef]
- Lankin, V.Z.; Tikhaze, A.K.; Konovalova, G.G.; Kozachenko, A.I. Concentration inversion of the antioxidant and pro-oxidant effects of beta-carotene in tissues in vivo. Bull. Eksp. Biol. Med. 1999, 128, 314–316. [Google Scholar]
- Ruggiero-Lopez, D.; Lecomte, M.; Moinet, G.; Patereau, G.; Lagarde, M.; Wiernsperger, N. Reaction of metformin with dicarbonyl compounds. possible implication in the inhibition of advanced glycation end product formation. Biochem. Pharmacol. 1999, 58, 1765–1773. [Google Scholar] [CrossRef]
- Beisswenger, P.; Ruggiero-Lopez, D. Metformin inhibition of glycation processes. Diabetes Metab. 2003, 29, 6S95–6S103. [Google Scholar] [CrossRef]
- Wang, G.; Wang, Y.; Yang, Q.; Xu, C.; Zheng, Y.; Wang, L.; Wu, J.; Zeng, M.; Luo, M. Metformin prevents methylglyoxal-induced apoptosis by suppressing oxidative stress in vitro and in vivo. Cell Death Dis. 2022, 13, 29. [Google Scholar] [CrossRef]
- Boldyrev, A.A.; Aldini, G.; Derave, W. Physiology and Pathophysiology of Carnosine. Physiol. Rev. 2013, 93, 1803–1845. [Google Scholar] [CrossRef]
- Reddy, V.P.; Garrett, M.R.; Perry, G.; Smith, M.A. Carnosine: A Versatile Antioxidant and Antiglycating Agent. Sci. Aging Knowl. Environ. 2005, 2005, pe12. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lankin, V.Z.; Tikhaze, A.K.; Melkumyants, A.M. Dicarbonyl-Dependent Modification of LDL as a Key Factor of Endothelial Dysfunction and Atherosclerotic Vascular Wall Damage. Antioxidants 2022, 11, 1565. https://doi.org/10.3390/antiox11081565
Lankin VZ, Tikhaze AK, Melkumyants AM. Dicarbonyl-Dependent Modification of LDL as a Key Factor of Endothelial Dysfunction and Atherosclerotic Vascular Wall Damage. Antioxidants. 2022; 11(8):1565. https://doi.org/10.3390/antiox11081565
Chicago/Turabian StyleLankin, Vadim Z., Alla K. Tikhaze, and Arthur M. Melkumyants. 2022. "Dicarbonyl-Dependent Modification of LDL as a Key Factor of Endothelial Dysfunction and Atherosclerotic Vascular Wall Damage" Antioxidants 11, no. 8: 1565. https://doi.org/10.3390/antiox11081565