
Ferroptosis and Its Emerging Role in Pre-Eclampsia


Abstract
:1. Introduction
2. Ferroptosis
2.1. Characteristics of Ferroptosis
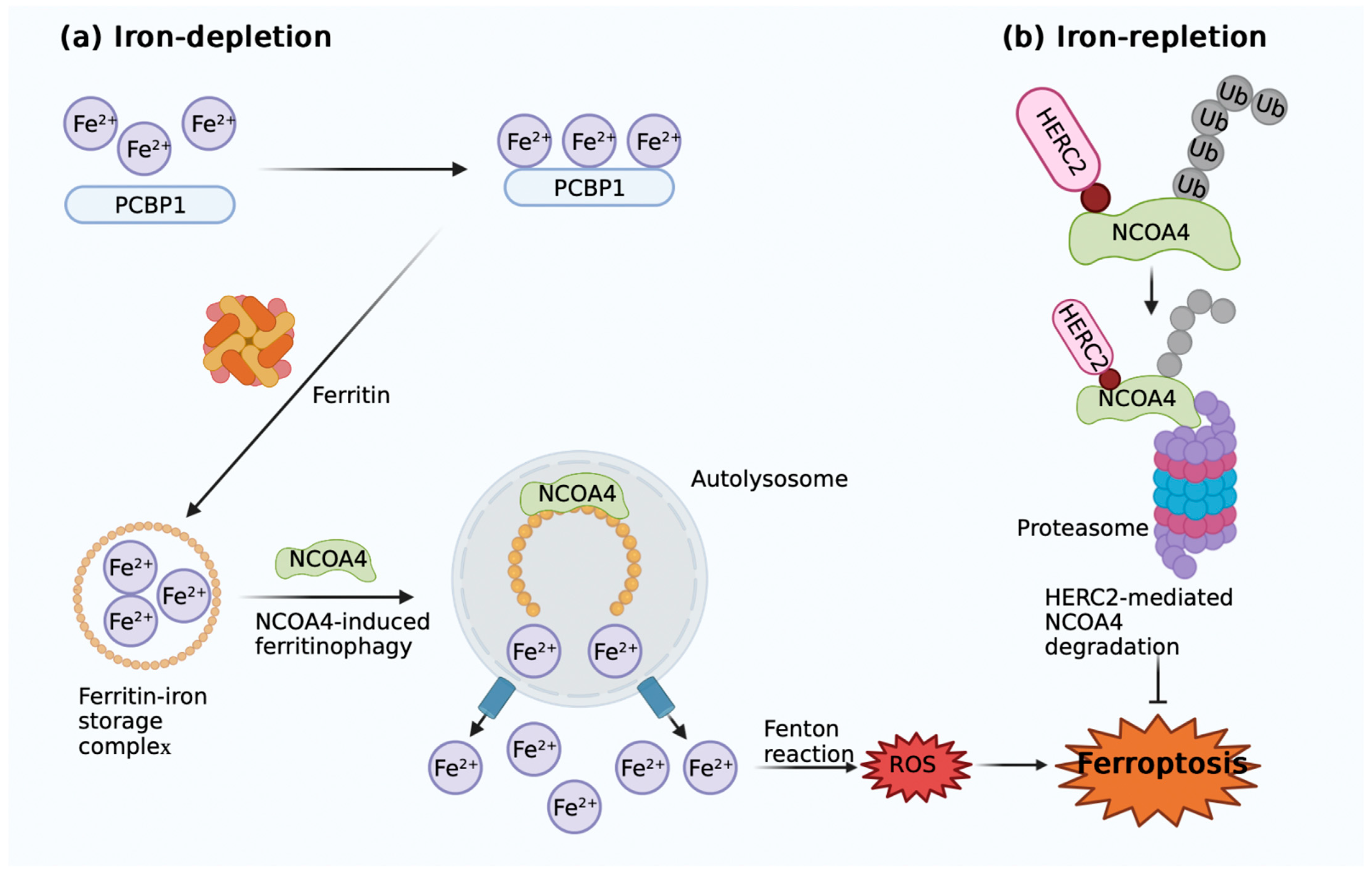
2.2. Iron Metabolism
2.3. Lipid Peroxidation Metabolism
2.4. GSH-Dependent Antioxidant Pathways: System Xc− and GPX4
2.5. GSH-Independent Antioxidant Pathways
3. Ferroptosis and Pre-Eclampsia
3.1. The Role of Iron in PE Pathology
3.2. The Role of Oxidative Stress in PE Pathology
3.3. The Role of Other Ferroptosis Regulators in PE Pathology
4. Ferroptosis and PE Therapy
5. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Kuklina, E.V.; Ayala, C.; Callaghan, W.M. Hypertensive Disorders and Severe Obstetric Morbidity in the United States. Obstet. Gynecol. 2009, 113, 1299–1306. [Google Scholar] [CrossRef] [Green Version]
- Wallis, A.B.; Saftlas, A.F.; Hsia, J.; Atrash, H.K. Secular trends in the rates of preeclampsia, eclampsia, and gestational hypertension, United States, 1987–2004. Am. J. Hypertens 2008, 21, 521–526. [Google Scholar] [CrossRef] [Green Version]
- Ananth, C.V.; Keyes, K.M.; Wapner, R.J. Pre-eclampsia rates in the United States, 1980–2010: Age-period-cohort analysis. BMJ 2013, 347, f6564. [Google Scholar] [CrossRef] [Green Version]
- Abalos, E.; Cuesta, C.; Grosso, A.L.; Chou, D.; Say, L. Global and regional estimates of preeclampsia and eclampsia: A systematic review. Eur. J. Obstet. Gynecol. Reprod. Biol. 2013, 170, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Ghulmiyyah, L.; Sibai, B. Maternal Mortality From Preeclampsia/Eclampsia. Semin. Perinatol. 2012, 36, 56–59. [Google Scholar] [CrossRef] [PubMed]
- Bouter, A.R.; Duvekot, J.J. Evaluation of the clinical impact of the revised ISSHP and ACOG definitions on preeclampsia. Pregnancy Hypertens 2020, 19, 206–211. [Google Scholar] [CrossRef]
- Roberts, J.M.; August, P.A.; Bakris, G.; Barton, J.R.; Bernstein, I.M.; Druzin, M.; Gaiser, R.R.; Granger, J.P.; Jeyabalan, A.; Johnson, D.D.; et al. Hypertension in Pregnancy Report of the American College of Obstetricians and Gynecologists’ Task Force on Hypertension in Pregnancy. Obstet. Gynecol. 2013, 122, 1122–1131. [Google Scholar]
- Robillard, P.Y.; Dekker, G.; Iacobelli, S.; Chaouat, G. An essay of reflection: Why does preeclampsia exist in humans, and why are there such huge geographical differences in epidemiology? J. Reprod. Immunol. 2016, 114, 44–47. [Google Scholar] [CrossRef]
- Lisonkova, S.; Joseph, K.S. Incidence of preeclampsia: Risk factors and outcomes associated with early-versus late-onset disease. Am. J. Obstet. Gynecol. 2013, 209, 544.e1–544.e12. [Google Scholar] [CrossRef]
- Rasmussen, S.; Irgens, L.M.; Espinoza, J. Maternal obesity and excess of fetal growth in pre-eclampsia. BJOG 2014, 121, 1351–1357. [Google Scholar] [CrossRef] [Green Version]
- Alese, M.O.; Moodley, J.; Naicker, T. Preeclampsia and HELLP syndrome, the role of the liver. J. Matern. Fetal Neonatal. Med. 2021, 34, 117–123. [Google Scholar] [CrossRef]
- Walker, I.; Chappell, L.C.; Williamson, C. Abnormal liver function tests in pregnancy. BMJ 2013, 347, f6055. [Google Scholar] [CrossRef] [Green Version]
- Walker, C.K.; Krakowiak, P.; Baker, A.; Hansen, R.L.; Ozonoff, S.; Hertz-Picciotto, I. Preeclampsia, placental insufficiency, and autism spectrum disorder or developmental delay. JAMA Pediatr. 2015, 169, 154–162. [Google Scholar] [CrossRef]
- Shahul, S.; Tung, A.; Minhaj, M.; Nizamuddin, J.; Wenger, J.; Mahmood, E.; Mueller, A.; Shaefi, S.; Scavone, B.; Kociol, R.D.; et al. Racial Disparities in Comorbidities, Complications, and Maternal and Fetal Outcomes in Women With Preeclampsia/eclampsia. Hypertens. Pregnancy 2015, 34, 506–515. [Google Scholar] [CrossRef] [Green Version]
- Hutcheon, J.A.; Lisonkova, S.; Joseph, K.S. Epidemiology of pre-eclampsia and the other hypertensive disorders of pregnancy. Best Pract. Res. Clin. Obstet. Gynaecol. 2011, 25, 391–403. [Google Scholar] [CrossRef]
- Bartsch, E.; Medcalf, K.E.; Park, A.L.; Ray, J.G.; High Risk of Pre-eclampsia Identification Group. Clinical risk factors for pre-eclampsia determined in early pregnancy: Systematic review and meta-analysis of large cohort studies. BMJ 2016, 353, i1753. [Google Scholar] [CrossRef] [Green Version]
- Phipps, E.A.; Thadhani, R.; Benzing, T.; Karumanchi, S.A. Pre-eclampsia: Pathogenesis, novel diagnostics and therapies. Nat. Rev. Nephrol. 2019, 15, 275–289. [Google Scholar] [CrossRef]
- Karumanchi, S.A. Angiogenic Factors in Preeclampsia From Diagnosis to Therapy. Hypertension 2016, 67, 1072–1079. [Google Scholar] [CrossRef]
- Cnattingius, S.; Reilly, M.; Pawitan, Y.; Lichtenstein, P. Maternal and fetal genetic factors account for most of familial aggregation of preeclampsia: A population-based Swedish cohort study. Am. J. Med. Genet. A 2004, 130, 365–371. [Google Scholar] [CrossRef]
- McGinnis, R.; Steinthorsdottir, V.; Williams, N.O.; Thorleifsson, G.; Shooter, S.; Hjartardottir, S.; Bumpstead, S.; Stefansdottir, L.; Hildyard, L.; Sigurdsson, J.K.; et al. Variants in the fetal genome near FLT1 are associated with risk of preeclampsia. Nat. Genet. 2017, 49, 1255. [Google Scholar] [CrossRef]
- Poon, L.C.; Shennan, A.; Hyett, J.A.; Kapur, A.; Hadar, E.; Divakar, H.; McAuliffe, F.; da Silva Costa, F.; von Dadelszen, P.; McIntyre, H.D.; et al. The International Federation of Gynecology and Obstetrics (FIGO) initiative on pre-eclampsia: A pragmatic guide for first-trimester screening and prevention. Int. J. Gynaecol. Obstet. 2019, 145 (Suppl. S1), 1–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snead, C.M.; Strassberg, E.; Overcash, R.; Stark, L.; Paglia, M.J.; Schulkin, J.; Jelin, A. Obstetricians’ knowledge and practices regarding the management of preeclampsia. J. Matern. Fetal Neonatal. Med. 2020, 33, 2970–2975. [Google Scholar] [CrossRef] [PubMed]
- Dhariwal, N.K.; Lynde, G.C. Update in the Management of Patients with Preeclampsia. Anesthesiol. Clin. 2017, 35, 95–106. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, D.R. The Coming Decade of Cell Death Research: Five Riddles. Cell 2019, 177, 1094–1107. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Hou, W.; Song, X.; Yu, Y.; Huang, J.; Sun, X.; Kang, R.; Tang, D. Ferroptosis: Process and function. Cell Death Differ. 2016, 23, 369–379. [Google Scholar] [CrossRef] [Green Version]
- Ng, S.W.; Norwitz, S.G.; Norwitz, E.R. The Impact of Iron Overload and Ferroptosis on Reproductive Disorders in Humans: Implications for Preeclampsia. Int. J. Mol. Sci. 2019, 20, 3283. [Google Scholar] [CrossRef] [Green Version]
- Soares, M.J.; Iqbal, K.; Kozai, K. Hypoxia and Placental Development. Birth Defects Res. 2017, 109, 1309–1329. [Google Scholar] [CrossRef]
- Alotaibi, M.; Arrowsmith, S.; Wray, S. Hypoxia-induced force increase (HIFI) is a novel mechanism underlying the strengthening of labor contractions, produced by hypoxic stresses. Proc. Natl. Acad. Sci. USA 2015, 112, 9763–9768. [Google Scholar] [CrossRef] [Green Version]
- Sangkhae, V.; Nemeth, E. Placental iron transport: The mechanism and regulatory circuits. Free Radical. Biol. Med. 2019, 133, 254–261. [Google Scholar] [CrossRef]
- Aouache, R.; Biquard, L.; Vaiman, D.; Miralles, F. Oxidative Stress in Preeclampsia and Placental Diseases. Int. J. Mol. Sci. 2018, 19, 1496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, X.; Lin, Y.; Li, J.; Liu, M.; Wang, J.; Li, X.; Liu, J.; Jia, X.; Jing, Z.; Huang, Z.; et al. Evaluation of Glutathione Peroxidase 4 role in Preeclampsia. Sci. Rep. 2016, 6, 33300. [Google Scholar] [CrossRef] [PubMed]
- Beharier, O.; Tyurin, V.A.; Goff, J.P.; Guerrero-Santoro, J.; Kajiwara, K.; Chu, T.J.; Tyurina, Y.Y.; St Croix, C.M.; Wallace, C.T.; Parry, S.; et al. PLA2G6 guards placental trophoblasts against ferroptotic injury. Proc. Natl. Acad. Sci. USA 2020, 117, 27319–27328. [Google Scholar] [CrossRef]
- Jacobson, M.D.; Raff, M.C. Programmed Cell-Death and Bcl-2 Protection in Very-Low Oxygen. Nature 1995, 374, 814–816. [Google Scholar] [CrossRef] [PubMed]
- Gibellini, L.; Moro, L. Programmed Cell Death in Health and Disease. Cells 2021, 10, 1765. [Google Scholar] [CrossRef]
- Dolma, S.; Lessnick, S.L.; Hahn, W.C.; Stockwell, B.R. Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell 2003, 3, 285–296. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.S.; Stockwell, B.R. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem. Biol. 2008, 15, 234–245. [Google Scholar] [CrossRef] [Green Version]
- Yagoda, N.; von Rechenberg, M.; Zaganjor, E.; Bauer, A.J.; Yang, W.S.; Fridman, D.J.; Wolpaw, A.J.; Smukste, I.; Peltier, J.M.; Boniface, J.J.; et al. RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature 2007, 447, 864–868. [Google Scholar] [CrossRef] [Green Version]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar]
- Stockwell, B.R. Ferroptosis: Death by lipid peroxidation. Free Radical. Biol. Med. 2018, 120, S7. [Google Scholar] [CrossRef] [Green Version]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascon, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Cao, F.; Yin, H.L.; Huang, Z.J.; Lin, Z.T.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, present and future. Cell Death Dis. 2020, 11, 88. [Google Scholar] [CrossRef] [PubMed]
- Angeli, J.P.F.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.A.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E.; et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 2014, 16, 1180–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, L.; Kon, N.; Li, T.; Wang, S.J.; Su, T.; Hibshoosh, H.; Baer, R.; Gu, W. Ferroptosis as a p53-mediated activity during tumour suppression. Nature 2015, 520, 57–62. [Google Scholar] [CrossRef] [Green Version]
- Doll, S.; Proneth, B.; Tyurina, Y.Y.; Panzilius, E.; Kobayashi, S.; IngoId, I.; Irmler, M.; Beckers, J.; Aichler, M.; Walch, A.; et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 2017, 13, 91–98. [Google Scholar] [CrossRef]
- Sun, X.F.; Ou, Z.H.; Chen, R.C.; Niu, X.H.; Chen, D.; Kang, R.; Tang, D.L. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology 2016, 63, 173–184. [Google Scholar] [CrossRef]
- Wiernicki, B.; Dubois, H.; Tyurina, Y.Y.; Hassannia, B.; Bayir, H.; Kagan, V.E.; Vandenabeele, P.; Wullaert, A.; Vanden Berghe, T. Excessive phospholipid peroxidation distinguishes ferroptosis from other cell death modes including pyroptosis. Cell Death Dis. 2020, 11, 922. [Google Scholar] [CrossRef] [PubMed]
- Drummen, G.P.; van Liebergen, L.C.; Op den Kamp, J.A.; Post, J.A. C11-BODIPY(581/591), an oxidation-sensitive fluorescent lipid peroxidation probe: (micro)spectroscopic characterization and validation of methodology. Free Radic. Biol. Med. 2002, 33, 473–490. [Google Scholar] [CrossRef]
- Ayala, A.; Munoz, M.F.; Arguelles, S. Lipid peroxidation: Production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid. Med. Cell Longev. 2014, 2014, 360438. [Google Scholar] [CrossRef]
- Xian, Z.Y.; Hu, B.; Wang, T.; Cai, J.L.; Zeng, J.Y.; Zou, Q.; Zhu, P.X. CircABCB10 silencing inhibits the cell ferroptosis and apoptosis by regulating the miR-326/CCL5 axis in rectal cancer. Neoplasma 2020, 67, 1063–1073. [Google Scholar] [CrossRef]
- Mou, Y.; Wang, J.; Wu, J.; He, D.; Zhang, C.; Duan, C.; Li, B. Ferroptosis, a new form of cell death: Opportunities and challenges in cancer. J. Hematol. Oncol. 2019, 12, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doll, S.; Conrad, M. Iron and Ferroptosis: A Still Ill-Defined Liaison. Iubmb Life 2017, 69, 423–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Y.; Xie, Y.; Cao, L.; Yang, L.; Yang, M.; Lotze, M.T.; Zeh, H.J.; Kang, R.; Tang, D. The ferroptosis inducer erastin enhances sensitivity of acute myeloid leukemia cells to chemotherapeutic agents. Mol. Cell Oncol. 2015, 2, e1054549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, R.; Shchepinov, M.S.; Pratt, D.A. Resolving the Role of Lipoxygenases in the Initiation and Execution of Ferroptosis. ACS Cent. Sci. 2018, 4, 387–396. [Google Scholar] [CrossRef]
- Ward, D.M.; Kaplan, J. Ferroportin-mediated iron transport: Expression and regulation. Biochim. Biophys. Acta 2012, 1823, 1426–1433. [Google Scholar] [CrossRef] [Green Version]
- Donovan, A.; Lima, C.A.; Pinkus, J.L.; Pinkus, G.S.; Zon, L.I.; Robine, S.; Andrews, N.C. The iron exporter ferroportin/Slc40a1 is essential for iron homeostasis. Cell Metab. 2005, 1, 191–200. [Google Scholar] [CrossRef] [Green Version]
- Krishnamurthy, P.; Xie, T.; Schuetz, J.D. The role of transporters in cellular heme and porphyrin homeostasis. Pharmacol. Therapeut. 2007, 114, 345–358. [Google Scholar] [CrossRef]
- Keel, S.B.; Doty, R.T.; Yang, Z.; Quigley, J.G.; Chen, J.; Knoblaugh, S.; Kingsley, P.D.; De Domenico, I.; Vaughn, M.B.; Kaplan, J.; et al. A heme export protein is required for red blood cell differentiation and iron homeostasis. Science 2008, 319, 825–828. [Google Scholar] [CrossRef] [Green Version]
- Shaw, G.C.; Cope, J.J.; Li, L.T.; Corson, K.; Hersey, C.; Ackermann, G.E.; Gwynn, B.; Lambert, A.J.; Wingert, R.A.; Traver, D.; et al. Mitoferrin is essential for erythroid iron assimilation. Nature 2006, 440, 96–100. [Google Scholar] [CrossRef]
- Hunter, G.A.; Ferreira, G.C. 5-Aminolevulinate Synthase: Catalysis of the First Step of Heme Biosynthesis. Cell Mol. Biol. 2009, 55, 102–110. [Google Scholar]
- Yoshida, T.; Biro, P.; Cohen, T.; Muller, R.M.; Shibahara, S. Human heme oxygenase cDNA and induction of its mRNA by hemin. Eur. J. Biochem. 1988, 171, 457–461. [Google Scholar] [CrossRef] [PubMed]
- Chifman, J.; Laubenbacher, R.; Torti, S.V. A systems biology approach to iron metabolism. Adv. Exp. Med. Biol. 2014, 844, 201–225. [Google Scholar] [PubMed] [Green Version]
- Feng, H.; Schorpp, K.; Jin, J.; Yozwiak, C.E.; Hoffstrom, B.G.; Decker, A.M.; Rajbhandari, P.; Stokes, M.E.; Bender, H.G.; Csuka, J.M.; et al. Transferrin Receptor Is a Specific Ferroptosis Marker. Cell Rep. 2020, 30, 3411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trowbridge, I.S.; Omary, M.B. Human Cell-Surface Glycoprotein Related to Cell-Proliferation Is the Receptor for Transferrin. Proc. Natl. Acad. Sci.-Biol. 1981, 78, 3039–3043. [Google Scholar] [CrossRef] [Green Version]
- Hentze, M.W.; Kuhn, L.C. Molecular control of vertebrate iron metabolism: mRNA-based regulatory circuits operated by iron, nitric oxide, and oxidative stress. Proc. Natl. Acad. Sci. USA 1996, 93, 8175–8182. [Google Scholar] [CrossRef] [Green Version]
- Feder, J.N.; Gnirke, A.; Thomas, W.; Tsuchihashi, Z.; Ruddy, D.A.; Basava, A.; Dormishian, F.; Domingo, R.; Ellis, M.C.; Fullan, A.; et al. A novel MHC class I-like gene is mutated in patients with hereditary haemochromatosis. Nat. Genet. 1996, 13, 399–408. [Google Scholar] [CrossRef]
- Gao, J.W.; Chen, J.X.; De Domenico, I.; Koeller, D.M.; Harding, C.O.; Fleming, R.E.; Koeberl, D.D.; Enns, C.A. Hepatocyte-targeted HFE and TFR2 control hepcidin expression in mice. Blood 2010, 115, 3374–3381. [Google Scholar] [CrossRef] [Green Version]
- Abboud, S.; Haile, D.J. A novel mammalian iron-regulated protein involved in intracellular iron metabolism. J. Biol. Chem. 2000, 275, 19906–19912. [Google Scholar] [CrossRef] [Green Version]
- McKie, A.T.; Marciani, P.; Rolfs, A.; Brennan, K.; Wehr, K.; Barrow, D.; Miret, S.; Bomford, A.; Peters, T.J.; Farzaneh, F.; et al. A novel duodenal iron-regulated transporter, IREG1, implicated in the basolateral transfer of iron to the circulation. Mol. Cell 2000, 5, 299–309. [Google Scholar] [CrossRef]
- Mancias, J.D.; Pontano Vaites, L.; Nissim, S.; Biancur, D.E.; Kim, A.J.; Wang, X.; Liu, Y.; Goessling, W.; Kimmelman, A.C.; Harper, J.W. Ferritinophagy via NCOA4 is required for erythropoiesis and is regulated by iron dependent HERC2-mediated proteolysis. Elife 2015, 4, e10308. [Google Scholar] [CrossRef]
- Mancias, J.D.; Wang, X.; Gygi, S.P.; Harper, J.W.; Kimmelman, A.C. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 2014, 509, 105–109. [Google Scholar] [CrossRef]
- Lee, J.Y.; Kim, W.K.; Bae, K.H.; Lee, S.C.; Lee, E.W. Lipid Metabolism and Ferroptosis. Biology 2021, 10, 184. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; Kim, K.J.; Gaschler, M.M.; Patel, M.; Shchepinov, M.S.; Stockwell, B.R. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc. Natl. Acad. Sci. USA 2016, 113, E4966–E4975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kagan, V.E.; Mao, G.; Qu, F.; Angeli, J.P.; Doll, S.; Croix, C.S.; Dar, H.H.; Liu, B.; Tyurin, V.A.; Ritov, V.B.; et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat. Chem. Biol. 2017, 13, 81–90. [Google Scholar] [CrossRef] [Green Version]
- Gaschler, M.M.; Stockwell, B.R. Lipid peroxidation in cell death. Biochem. Biophys. Res. Commun. 2017, 482, 419–425. [Google Scholar] [CrossRef]
- Dixon, S.J.; Winter, G.E.; Musavi, L.S.; Lee, E.D.; Snijder, B.; Rebsamen, M.; Superti-Furga, G.; Stockwell, B.R. Human Haploid Cell Genetics Reveals Roles for Lipid Metabolism Genes in Nonapoptotic Cell Death. ACS Chem. Biol. 2015, 10, 1604–1609. [Google Scholar] [CrossRef] [PubMed]
- Shintoku, R.; Takigawa, Y.; Yamada, K.; Kubota, C.; Yoshimoto, Y.; Takeuchi, T.; Koshiishi, I.; Torii, S. Lipoxygenase-mediated generation of lipid peroxides enhances ferroptosis induced by erastin and RSL3. Cancer Sci. 2017, 108, 2187–2194. [Google Scholar] [CrossRef]
- Khanna, S.; Roy, S.; Ryu, H.; Bahadduri, P.; Swaan, P.W.; Ratan, R.R.; Sen, C.K. Molecular basis of vitamin E action: Tocotrienol modulates 12-lipoxygenase, a key mediator of glutamate-induced neurodegeneration. J. Biol. Chem. 2003, 278, 43508–43515. [Google Scholar] [CrossRef] [Green Version]
- Zilka, O.; Shah, R.; Li, B.; Friedmann Angeli, J.P.; Griesser, M.; Conrad, M.; Pratt, D.A. On the Mechanism of Cytoprotection by Ferrostatin-1 and Liproxstatin-1 and the Role of Lipid Peroxidation in Ferroptotic Cell Death. ACS Cent. Sci. 2017, 3, 232–243. [Google Scholar] [CrossRef]
- Zhao, J.M.; O’Donnell, V.B.; Balzar, S.; Croix, C.M.S.; Trudeau, J.B.; Wenzel, S.E. 15-Lipoxygenase 1 interacts with phosphatidylethanolamine-binding protein to regulate MAPK signaling in human airway epithelial cells. Proc. Natl. Acad. Sci. USA 2011, 108, 14246–14251. [Google Scholar] [CrossRef] [Green Version]
- Wenzel, S.E.; Tyurina, Y.Y.; Zhao, J.M.; Croix, C.M.S.; Dar, H.H.; Mao, G.W.; Tyurin, V.A.; Anthonymuthu, T.S.; Kapralov, A.A.; Amoscato, A.A.; et al. PEBP1 Wardens Ferroptosis by Enabling Lipoxygenase Generation of Lipid Death Signals. Cell 2017, 171, 628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ursini, F.; Maiorino, M.; Gregolin, C. The Selenoenzyme Phospholipid Hydroperoxide Glutathione-Peroxidase. Biochim. Biophys. Acta 1985, 839, 62–70. [Google Scholar] [CrossRef]
- Seiler, A.; Schneider, M.; Forster, H.; Roth, S.; Wirth, E.K.; Culmsee, C.; Plesnila, N.; Kremmer, E.; Radmark, O.; Wurst, W.; et al. Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and AIF-Mediated cell death. Cell Metab. 2008, 8, 237–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bersuker, K.; Hendricks, J.M.; Li, Z.P.; Magtanong, L.; Ford, B.; Tang, P.H.; Roberts, M.A.; Tong, B.Q.; Maimone, T.J.; Zoncu, R.; et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 2019, 575, 688. [Google Scholar] [CrossRef]
- Doll, S.; Freitas, F.P.; Shah, R.; Aldrovandi, M.; da Silva, M.C.; Ingold, I.; Grocin, A.G.; da Silva, T.N.X.; Panzilius, E.; Scheel, C.H.; et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 2019, 575, 693. [Google Scholar] [CrossRef]
- Liu, M.R.; Zhu, W.T.; Pei, D.S. System Xc(-): A key regulatory target of ferroptosis in cancer. Investig. New Drugs 2021, 39, 1123–1131. [Google Scholar] [CrossRef]
- Liu, J.; Xia, X.; Huang, P. xCT: A Critical Molecule That Links Cancer Metabolism to Redox Signaling. Mol. Ther. 2020, 28, 2358–2366. [Google Scholar] [CrossRef]
- Liu, D.S.; Duong, C.P.; Haupt, S.; Montgomery, K.G.; House, C.M.; Azar, W.J.; Pearson, H.B.; Fisher, O.M.; Read, M.; Guerra, G.R.; et al. Inhibiting the system xC(-)/glutathione axis selectively targets cancers with mutant-p53 accumulation. Nat. Commun. 2017, 8, 14844. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.L.; Yang, M.C.; Deng, J.; Li, P.; Su, W.J.; Jiang, R. Upregulation and activation of p53 by erastin-induced reactive oxygen species contribute to cytotoxic and cytostatic effects in A549 lung cancer cells. Oncol. Rep. 2018, 40, 2363–2370. [Google Scholar] [CrossRef]
- Guan, J.; Lo, M.; Dockery, P.; Mahon, S.; Karp, C.M.; Buckley, A.R.; Lam, S.; Gout, P.W.; Wang, Y.Z. The xc- cystine/glutamate antiporter as a potential therapeutic target for small-cell lung cancer: Use of sulfasalazine. Cancer Chem. Pharmacol. 2009, 64, 463–472. [Google Scholar] [CrossRef]
- Yu, H.C.; Yang, C.C.; Jian, L.; Guo, S.P.; Chen, R.; Li, K.; Qu, F.L.; Tao, K.; Fu, Y.; Luo, F.; et al. Sulfasalazine-induced ferroptosis in breast cancer cells is reduced by the inhibitory effect of estrogen receptor on the transferrin receptor. Oncol. Rep. 2019, 42, 826–838. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Patel, D.N.; Welsch, M.; Skouta, R.; Lee, E.D.; Hayano, M.; Thomas, A.G.; Gleason, C.E.; Tatonetti, N.P.; Slusher, B.S.; et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife 2014, 3, e02523. [Google Scholar] [CrossRef] [PubMed]
- Houessinon, A.; Gicquel, A.; Bochereau, F.; Louandre, C.; Nyga, R.; Godin, C.; Degonville, J.; Fournier, E.; Saidak, Z.; Drullion, C.; et al. Alpha-fetoprotein is a biomarker of unfolded protein response and altered proteostasis in hepatocellular carcinoma cells exposed to sorafenib. Cancer Lett. 2016, 370, 242–249. [Google Scholar] [CrossRef]
- Ursini, F.; Maiorino, M.; Valente, M.; Ferri, L.; Gregolin, C. Purification from Pig-Liver of a Protein Which Protects Liposomes and Biomembranes from Peroxidative Degradation and Exhibits Glutathione-Peroxidase Activity on Phosphatidylcholine Hydroperoxides. Biochim. Biophys. Acta 1982, 710, 197–211. [Google Scholar] [CrossRef]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of Ferroptotic Cancer Cell Death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef] [Green Version]
- Seibt, T.M.; Proneth, B.; Conrad, M. Role of GPX4 in ferroptosis and its pharmacological implication. Free Radic. Biol. Med. 2019, 133, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.J.; Yang, F.; Che, J.T.; Han, Y.; Wang, Y.K.; Chen, N.; Bak, D.W.; Lai, S.C.; Xie, X.; Weerapana, E.; et al. Selenium-Encoded Isotopic Signature Targeted Profiling. ACS Cent. Sci. 2018, 4, 960–970. [Google Scholar] [CrossRef] [PubMed]
- Abrams, R.P.; Carroll, W.L.; Woerpel, K.A. Five-Membered Ring Peroxide Selectively Initiates Ferroptosis in Cancer Cells. ACS Chem. Biol. 2016, 11, 1305–1312. [Google Scholar] [CrossRef] [Green Version]
- Gaschler, M.M.; Andia, A.A.; Liu, H.R.; Csuka, J.M.; Hurlocker, B.; Vaiana, C.A.; Heindel, D.W.; Zuckerman, D.S.; Bos, P.H.; Reznik, E.; et al. FINO2 initiates ferroptosis through GPX4 inactivation and iron oxidation. Nat. Chem. Biol. 2018, 14, 507. [Google Scholar] [CrossRef]
- Shimada, K.; Hayano, M.; Pagano, N.C.; Stockwell, B.R. Cell-Line Selectivity Improves the Predictive Power of Pharmacogenomic Analyses and Helps Identify NADPH as Biomarker for Ferroptosis Sensitivity. Cell Chem. Biol. 2016, 23, 225–235. [Google Scholar] [CrossRef] [Green Version]
- Stockwell, B.R.; Jiang, X.; Gu, W. Emerging Mechanisms and Disease Relevance of Ferroptosis. Trends Cell. Biol. 2020, 30, 478–490. [Google Scholar] [CrossRef] [PubMed]
- Shimada, K.; Skouta, R.; Kaplan, A.; Yang, W.S.; Hayano, M.; Dixon, S.J.; Brown, L.M.; Valenzuela, C.A.; Wolpaw, A.J.; Stockwell, B.R. Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nat. Chem. Biol. 2016, 12, 497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, C.; Liu, X.G.; Zhang, Y.L.; Lei, G.; Yan, Y.L.; Lee, H.M.; Koppula, P.; Wu, S.Q.; Zhuang, L.; Fang, B.L.; et al. DHODH-mediated ferroptosis defence is a targetable vulnerability in cancer. Nature 2021, 593, 586, Erratum in Nature 2021, 596, E13. [Google Scholar] [CrossRef] [PubMed]
- Kraft, V.A.N.; Bezjian, C.T.; Pfeiffer, S.; Ringelstetter, L.; Muller, C.; Zandkarimi, F.; Merl-Pham, J.; Bao, X.W.; Anastasov, N.; Kossl, J.; et al. GTP Cyclohydrolase 1/Tetrahydrobiopterin Counteract Ferroptosis through Lipid Remodeling. ACS Central Sci. 2020, 6, 41–53. [Google Scholar] [CrossRef] [Green Version]
- Fisher, A.L.; Nemeth, E. Iron homeostasis during pregnancy. Am. J. Clin. Nutr. 2017, 106, 1567s–1574s. [Google Scholar] [CrossRef]
- Young, M.F.; Griffin, I.; Pressman, E.; McIntyre, A.W.; Cooper, E.; McNanley, T.; Harris, Z.L.; Westerman, M.; O’Brien, K.O. Maternal hepcidin is associated with placental transfer of iron derived from dietary heme and nonheme sources. J. Nutr. 2012, 142, 33–39. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.X.; Chen, D.; Li, M.X.; Hua, Y. Increased serum iron levels in pregnant women with preeclampsia: A meta-analysis of observational studies. J. Obstet. Gynaecol. 2019, 39, 11–16. [Google Scholar] [CrossRef]
- Shaji Geetha, N.; Bobby, Z.; Dorairajan, G.; Jacob, S.E. Increased hepcidin levels in preeclampsia: A protective mechanism against iron overload mediated oxidative stress? J. Matern. Fetal Neonatal. Med. 2022, 35, 636–641. [Google Scholar] [CrossRef]
- Toldi, G.; Stenczer, B.; Molvarec, A.; Takats, Z.; Beko, G.; Rigo, J.; Vasarhelyi, B. Hepcidin concentrations and iron homeostasis in preeclampsia. Clin. Chem. Lab. Med. 2010, 48, 1423–1426. [Google Scholar] [CrossRef]
- Brunacci, F.; Rocha, V.S.; De Carli, E.; Esposito, B.P.; Ruano, R.; Colli, C. Increased serum iron in preeclamptic women is likely due to low hepcidin levels. Nutr. Res. 2018, 53, 32–39. [Google Scholar] [CrossRef]
- Cardaropoli, S.; Todros, T.; Nuzzo, A.M.; Rolfo, A. Maternal serum levels and placental expression of hepcidin in preeclampsia. Pregnancy Hypertens 2018, 11, 47–53. [Google Scholar] [CrossRef]
- Aires Rodrigues de Freitas, M.; Vieira da Costa, A.; Alves de Medeiros, L.; da Silva Garrote Filho, M.; Lemos Debs Diniz, A.; Penha-Silva, N. Are There Differences in the Anthropometric, Hemodynamic, Hematologic, and Biochemical Profiles between Late- and Early-Onset Preeclampsia? Obstet. Gynecol. Int. 2018, 2018, 9628726. [Google Scholar] [CrossRef] [Green Version]
- Rayman, M.P.; Barlis, J.; Evans, R.W.; Redman, C.W.G.; King, L.J. Abnormal iron parameters in the pregnancy syndrome preeclampsia. Am. J. Obstet. Gynecol. 2002, 187, 412–418. [Google Scholar] [CrossRef] [Green Version]
- Switzer, J.W.; Valerio, D.A.; Martin, D.P.; Valerio, M.G.; Rininger, B.F. Hematologic Changes Associated with Pregnancy and Parturition in Macaca-Mulatta. Lab Anim. Care 1970, 20, 930. [Google Scholar]
- Erlandsson, L.; Masoumi, Z.; Hansson, L.R.; Hansson, S.R. The roles of free iron, heme, haemoglobin, and the scavenger proteins haemopexin and alpha-1-microglobulin in preeclampsia and fetal growth restriction. J. Intern. Med. 2021, 290, 952–968. [Google Scholar] [CrossRef]
- Kajiwara, K.; Beharier, O.; Chng, C.P.; Goff, J.P.; Ouyang, Y.; St Croix, C.M.; Huang, C.; Kagan, V.E.; Hsia, K.J.; Sadovsky, Y. Ferroptosis induces membrane blebbing in placental trophoblasts. J. Cell Sci. 2022, 135, jcs255737. [Google Scholar] [CrossRef]
- Ziaei, S.; Norrozi, M.; Faghihzadeh, S.; Jafarbegloo, E. A randomised placebo-controlled trial to determine the effect of iron supplementation on pregnancy outcome in pregnant women with haemoglobin ≥ 13.2 g/dl. BJOG 2007, 114, 684–688. [Google Scholar] [CrossRef]
- Camaschella, C.; Nai, A.; Silvestri, L. Iron metabolism and iron disorders revisited in the hepcidin era. Haematologica 2020, 105, 260–272. [Google Scholar] [CrossRef] [Green Version]
- Borzychowski, A.M.; Sargent, I.L.; Redman, C.W. Inflammation and pre-eclampsia. Semin. Fetal Neonatal. Med. 2006, 11, 309–316. [Google Scholar] [CrossRef]
- Bauduer, F. C282Y/H63D hemochromatosis mutations and microevolution: Speculations concerning the Basque population. Homo 2017, 68, 38–41. [Google Scholar] [CrossRef]
- Nekhai, S.; Xu, M.; Foster, A.; Kasvosve, I.; Diaz, S.; Machado, R.F.; Castro, O.L.; Kato, G.J.; Taylor, J.G.; Gordeuk, V.R. Reduced sensitivity of the ferroportin Q248H mutant to physiological concentrations of hepcidin. Haematologica 2013, 98, 455–463. [Google Scholar] [CrossRef]
- Wu, Y.H.; Sakamoto, H.; Kanenishi, K.; Li, J.; Khatun, R.; Hata, T. Transferrin microheterogeneity in pregnancies with preeclampsia. Clin. Chim. Acta 2003, 332, 103–110. [Google Scholar] [CrossRef]
- Koenig, M.D.; Tussing-Humphreys, L.; Day, J.; Cadwell, B.; Nemeth, E. Hepcidin and Iron Homeostasis during Pregnancy. Nutrients 2014, 6, 3062–3083. [Google Scholar] [CrossRef]
- Kehrer, J.P. The Haber-Weiss reaction and mechanisms of toxicity. Toxicology 2000, 149, 43–50. [Google Scholar] [CrossRef]
- Zhang, H.; He, Y.; Wang, J.X.; Chen, M.H.; Xu, J.J.; Jiang, M.H.; Feng, Y.L.; Gu, Y.F. miR-30-5p-mediated ferroptosis of trophoblasts is implicated in the pathogenesis of preeclampsia. Redox Biol. 2020, 29, 101402. [Google Scholar] [CrossRef]
- Lee, D.C.; Romero, R.; Kim, J.S.; Tarca, A.L.; Montenegro, D.; Pineles, B.L.; Kim, E.; Lee, J.; Kim, S.Y.; Draghici, S.; et al. miR-210 targets iron-sulfur cluster scaffold homologue in human trophoblast cell lines: Siderosis of interstitial trophoblasts as a novel pathology of preterm preeclampsia and small-for-gestational-age pregnancies. Am. J. Pathol. 2011, 179, 590–602. [Google Scholar] [CrossRef]
- Yang, N.; Wang, Q.; Ding, B.; Gong, Y.; Wu, Y.; Sun, J.; Wang, X.; Liu, L.; Zhang, F.; Du, D.; et al. Expression profiles and functions of ferroptosis-related genes in the placental tissue samples of early- and late-onset preeclampsia patients. BMC Pregnancy Childbirth 2022, 22, 87. [Google Scholar]
- Burton, G.J.; Woods, A.W.; Jauniaux, E.; Kingdom, J.C.P. Rheological and Physiological Consequences of Conversion of the Maternal Spiral Arteries for Uteroplacental Blood Flow during Human Pregnancy. Placenta 2009, 30, 473–482. [Google Scholar] [CrossRef] [Green Version]
- Rana, S.; Lemoine, E.; Granger, J.; Karumanchi, S.A. Preeclampsia Pathophysiology, Challenges, and Perspectives. Circ. Res. 2019, 124, 1094–1112. [Google Scholar] [CrossRef]
- Zur, R.L.; Kingdom, J.C.; Parks, W.T.; Hobson, S.R. The Placental Basis of Fetal Growth Restriction. Obstet. Gyn. Clin. N. Am. 2020, 47, 81. [Google Scholar] [CrossRef]
- Leanos-Miranda, A.; Campos-Galicia, I.; Berumen-Lechuga, M.G.; Molina-Perez, C.J.; Garcia-Paleta, Y.; Isordia-Salas, I.; Ramirez-Valenzuela, K.L. Circulating Angiogenic Factors and the Risk of Preeclampsia in Systemic Lupus Erythematosus Pregnancies. J. Rheumatol. 2015, 42, 1141–1149. [Google Scholar] [CrossRef] [PubMed]
- Levine, R.J.; Lam, C.; Qian, C.; Yu, K.F.; Maynard, S.E.; Sachs, B.P.; Sibai, B.M.; Epstein, F.H.; Romero, R.; Thadhani, R.; et al. Soluble endoglin and other circulating antiangiogenic factors in preeclampsia. N. Engl. J. Med. 2006, 355, 992–1005. [Google Scholar] [CrossRef]
- Chappell, L.C.; Cluver, C.A.; Kingdom, J.; Tong, S. Pre-eclampsia. Lancet 2021, 398, 341–354. [Google Scholar] [CrossRef]
- Roberts, J.; Redman, C. Pre-eclampsia: More than pregnancy-induced hypertension. Lancet 1993, 341, 1447–1451. [Google Scholar] [CrossRef]
- Feng, H.; Stockwell, B.R. Unsolved mysteries: How does lipid peroxidation cause ferroptosis? PLoS Biol. 2018, 16, e2006203. [Google Scholar] [CrossRef]
- Patil, S.B.; Kodliwadmath, M.V.; Kodliwadmath, M. Lipid peroxidation and antioxidant activity in complicated pregnancies. Clin. Exp. Obstet. Gynecol. 2009, 36, 110–112. [Google Scholar]
- Sarandol, E.; Safak, O.; Dirican, M.; Uncu, G. Oxidizability of apolipoprotein B-containing lipoproteins and serum paraoxonase/arylesterase activities in preeclampsia. Clin. Biochem. 2004, 37, 990–996. [Google Scholar] [CrossRef]
- Ahmadi, R.; Rahimi, Z.; Vaisi-Raygani, A.; Kiani, A.; Jalilian, N.; Rahimi, Z. Apolipoprotein E genotypes, lipid peroxidation, and antioxidant status among mild and severe preeclamptic women from western Iran: Protective role of apolipoprotein epsilon2 allele in severe preeclampsia. Hypertens Pregnancy 2012, 31, 405–418. [Google Scholar] [CrossRef]
- Zhou, X.B.; Han, T.L.; Chen, H.; Baker, P.N.; Qi, H.B.; Zhang, H. Impaired mitochondrial fusion, autophagy, biogenesis and dysregulated lipid metabolism is associated with preeclampsia. Exp. Cell Res. 2017, 359, 195–204. [Google Scholar] [CrossRef] [Green Version]
- Staff, A.C.; Johnsen, G.M.; Dechend, R.; Redman, C.W.G. Preeclampsia and uteroplacental acute atherosis: Immune and inflammatory factors. J. Reprod. Immunol. 2014, 101, 120–126. [Google Scholar] [CrossRef]
- Alahari, S.; Post, M.; Rolfo, A.; Weksberg, R.; Caniggia, I. Compromised JMJD6 Histone Demethylase Activity Affects VHL Gene Repression in Preeclampsia. J. Clin. Endocrinol. Metab. 2018, 103, 1545–1557. [Google Scholar] [CrossRef] [PubMed]
- Vanderlelie, J.; Venardos, K.; Clifton, V.L.; Gude, N.M.; Clarke, F.M.; Perkins, A.V. Increased biological oxidation and reduced anti-oxidant enzyme activity in pre-eclamptic placentae. Placenta 2005, 26, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Chamy, V.M.; Lepe, J.; Catalan, A.; Retamal, D.; Escobar, J.A.; Madrid, E.M. Oxidative stress is closely related to clinical severity of pre-eclampsia. Biol. Res. 2006, 39, 229–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, C.; Huang, P.Z.; Lyu, M.L.; Dong, J.F. Oxidative Stress and Preeclampsia-Associated Prothrombotic State. Antioxidants 2020, 9, 1139. [Google Scholar] [CrossRef]
- Vaughan, J.E.; Walsh, S.W. Oxidative stress reproduces placental abnormalities of preeclampsia. Hypertens Pregnancy 2002, 21, 205–223. [Google Scholar] [CrossRef]
- Bensaad, K.; Favaro, E.; Lewis, C.A.; Peck, B.; Lord, S.; Collins, J.M.; Pinnick, K.E.; Wigfield, S.; Buffa, F.M.; Li, J.L.; et al. Fatty acid uptake and lipid storage induced by HIF-1alpha contribute to cell growth and survival after hypoxia-reoxygenation. Cell Rep. 2014, 9, 349–365. [Google Scholar] [CrossRef] [Green Version]
- Huang, D.; Li, T.T.; Li, X.H.; Zhang, L.; Sun, L.C.; He, X.P.; Zhong, X.Y.; Jia, D.Y.; Song, L.B.; Semenza, G.L.; et al. HIF-1-Mediated Suppression of Acyl-CoA Dehydrogenases and Fatty Acid Oxidation Is Critical for Cancer Progression. Cell Rep. 2014, 8, 1930–1942. [Google Scholar] [CrossRef] [Green Version]
- Caniggia, I.; Mostachfi, H.; Winter, J.; Gassmann, M.; Lye, S.J.; Kuliszewski, M.; Post, M. Hypoxia-inducible factor-1 mediates the biological effects of oxygen on human trophoblast differentiation through TGF beta(3). J. Clin. Investig. 2000, 105, 577–587. [Google Scholar] [CrossRef] [Green Version]
- Rajakumar, A.; Doty, K.; Daftary, A.; Harger, G.; Conrad, K.P. Impaired oxygen-dependent reduction of HIF-1alpha and -2alpha proteins in pre-eclamptic placentae. Placenta 2003, 24, 199–208. [Google Scholar] [CrossRef]
- Cargnello, M.; Roux, P.P. Activation and Function of the MAPKs and Their Substrates, the MAPK-Activated Protein Kinases. Microbiol. Mol. Biol. R 2011, 75, 50–83. [Google Scholar] [CrossRef] [Green Version]
- Conte, M.; Franceschi, C.; Sandri, M.; Salvioli, S. Perilipin 2 and Age-Related Metabolic Diseases: A New Perspective. Trends Endocrin. Met. 2016, 27, 893–903. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.T.; Zhong, M.; Tian, J.W.; Hou, F.F. Higher plasma AOPP is associated with increased proteinuria excretion and decreased glomerular filtration rate in pre-eclamptic women. Pregnancy Hypertens 2013, 3, 16–20. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Ma, Y.Y.; Shao, Q.Q.; Liang, J.; Qi, T.T.; Huang, Y.; Wang, Q.J. ROS/p53/miR3355p/Sp1 axis modulates the migration and epithelial to mesenchymal transition of JEG3 cells. Mol. Med. Rep. 2020, 21, 1208–1216. [Google Scholar] [PubMed]
- Boutet, M.; Roland, L.; Thomas, N.; Bilodeau, J.F. Specific systemic antioxidant response to preeclampsia in late pregnancy: The study of intracellular glutathione peroxidases in maternal and fetal blood. Am. J. Obstet. Gynecol. 2009, 200, 530.e1–530.e7. [Google Scholar] [CrossRef]
- Liu, N.; Lin, X.L.; Huang, C.Y. Activation of the reverse transsulfuration pathway through NRF2/CBS confers erastin-induced ferroptosis resistance. Brit. J. Cancer 2020, 122, 279–292. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, L.; Zhou, X.X. Activation of Nrf2 signaling protects hypoxia-induced HTR-8/SVneo cells against ferroptosis. J. Obstet. Gynaecol. Res. 2021, 47, 3797–3806. [Google Scholar] [CrossRef]
- Kwon, H.S.; Hwang, H.S.; Sohn, I.S.; Park, S.H. Expression of DJ-1 proteins in placentas from women with severe preeclampsia. Eur. J. Obstet. Gynecol. Reprod. Biol. 2013, 168, 40–44. [Google Scholar] [CrossRef]
- Bellezza, I.; Giambanco, I.; Minelli, A.; Donato, R. Nrf2-Keap1 signaling in oxidative and reductive stress. BBA-Mol. Cell Res. 2018, 1865, 721–733. [Google Scholar] [CrossRef]
- Liao, T.; Xu, X.; Ye, X.; Yan, J. DJ-1 upregulates the Nrf2/GPX4 signal pathway to inhibit trophoblast ferroptosis in the pathogenesis of preeclampsia. Sci. Rep. 2022, 12, 2934. [Google Scholar] [CrossRef]
- Rizzardi, N.; Liparulo, I.; Antonelli, G.; Orsini, F.; Riva, A.; Bergamini, C.; Fato, R. Coenzyme Q10 Phytosome Formulation Improves CoQ10 Bioavailability and Mitochondrial Functionality in Cultured Cells. Antioxidants 2021, 10, 927. [Google Scholar] [CrossRef]
- Garrido-Maraver, J.; Cordero, M.D.; Oropesa-Avila, M.; Vega, A.F.; de la Mata, M.; Pavon, A.D.; Alcocer-Gomez, E.; Calero, C.P.; Paz, M.V.; Alanis, M.; et al. Clinical applications of coenzyme Q10. Front. Biosci.-Landmrk 2014, 19, 619–633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noia, G.; Littarru, G.P.; De Santis, M.; Oradei, A.; Mactromarino, C.; Trivellini, C.; Caruso, A. Coenzyme Q10 in pregnancy. Fetal Diagn. Ther. 1996, 11, 264–270. [Google Scholar] [CrossRef]
- Teran, E.; Racines-Orbe, M.; Vivero, S.; Escudero, C.; Molina, G.; Calle, A. Preeclampsia is associated with a decrease in plasma coenzyme Q10 levels. Free Radical. Biol. Med. 2003, 35, 1453–1456. [Google Scholar] [CrossRef] [PubMed]
- Teran, E.; Hernandez, I.; Tana, L.; Teran, S.; Galaviz-Hernandez, C.; Sosa-Macias, M.; Molina, G.; Calle, A. Mitochondria and Coenzyme Q10 in the Pathogenesis of Preeclampsia. Front. Physiol. 2018, 9, 1561. [Google Scholar] [CrossRef] [PubMed]
- Teran, E.; Vivero, S.; Racines-Orbe, M.; Castellanos, A.; Chuncha, G.; Enriquez, G.; Moya, W. Coenzyme Q(10) is increased in placenta and cord blood during preeclampsia. Biofactors 2005, 25, 153–158. [Google Scholar] [CrossRef]
- Teran, E.; Chedraui, P.; Racines-Orbe, M.; Vivero, S.; Villena, F.; Duchicela, F.; Nacevilla, L.; Schwager, G.; Calle, A. Coenzyme Q(10) levels in women with preeclampsia living at different altitudes. Biofactors 2008, 32, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Teran, E.; Hernandez, I.; Nieto, B.; Tavara, R.; Ocampo, J.E.; Calle, A. Coenzyme Q10 supplementation during pregnancy reduces the risk of pre-eclampsia. Int. J. Gynecol. Obstet. 2009, 105, 43–45. [Google Scholar] [CrossRef]
- Hernandez, I.; Vivero, S.; Racines-Orbe, M.; Calle, A.; Molina, G.; Teran, E. Placental and mitochondrial Q10 content after CoQ10 supplementation during pregnancy. Free Radical. Biol. Med. 2017, 108, S77. [Google Scholar] [CrossRef]
- Rana, S.; Rajakumar, A.; Geahchan, C.; Salahuddin, S.; Cerdeira, A.S.; Burke, S.D.; George, E.M.; Granger, J.P.; Karumanchi, S.A. Ouabain inhibits placental sFlt1 production by repressing HSP27-dependent HIF-1 alpha pathway. FASEB J. 2014, 28, 4324–4334. [Google Scholar] [CrossRef] [Green Version]
- Roberge, S.; Bujold, E.; Nicolaides, K.H. Aspirin for the prevention of preterm and term preeclampsia: Systematic review and metaanalysis. Am. J. Obstet. Gynecol. 2018, 218, 287–293.e1. [Google Scholar] [CrossRef] [Green Version]
- Rolnik, D.L.; Wright, D.; Poon, L.C.; O’Gorman, N.; Syngelaki, A.; Matallana, C.D.; Akolekar, R.; Cicero, S.; Janga, D.; Singh, M.; et al. Aspirin Versus Placebo in Pregnancies at High Risk for Preterm Preeclampsia. Obstet. Gynecol. Surv. 2018, 73, 11–12. [Google Scholar] [CrossRef]
- LeFevre, M.L.; U.S. Preventive Services Task Force. Low-Dose Aspirin Use for the Prevention of Morbidity and Mortality from Preeclampsia: US Preventive Services Task Force Recommendation Statement. Ann. Intern. Med. 2014, 326, 1186–1191. [Google Scholar] [CrossRef]
- Cruz-Lemini, M.; Vazquez, J.C.; Ullmo, J.; Llurba, E. Low-molecular-weight heparin for prevention of preeclampsia and other placenta-mediated complications: A systematic review and meta-analysis. Am. J. Obstet. Gynecol. 2022, 226, S1126–S1144.e17. [Google Scholar] [CrossRef] [PubMed]
- Katsi, V.; Kanellopoulou, T.; Makris, T.; Nihoyannopoulos, P.; Nomikou, E.; Tousoulis, D. Aspirin vs Heparin for the Prevention of Preeclampsia. Curr. Hypertens Rep. 2016, 18, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Roberts, J.M.; Myatt, L.; Spong, C.Y.; Thom, E.A.; Hauth, J.C.; Leveno, K.J.; Pearson, G.D.; Wapner, R.J.; Varner, M.W.; Thorp, J.M.; et al. Vitamins C and E to Prevent Complications of Pregnancy-Associated Hypertension. N. Engl. J. Med. 2010, 362, 1282–1291. [Google Scholar] [CrossRef] [Green Version]
- Poston, L.; Briley, A.L.; Seed, P.T.; Kelly, F.J.; Shennan, A.H.; Consortium, V.T. Vitamin C and vitamin E in pregnant women at risk for pre-eclampsia (VIP trial): Randomised placebo-controlled trial. Lancet 2006, 367, 1145–1154. [Google Scholar] [CrossRef]
- Vaka, V.R.; McMaster, K.M.; Cunningham, M.W.; Ibrahim, T.; Hazlewood, R.; Usry, N.; Cornelius, D.C.; Amaral, L.M.; LaMarca, B. Role of Mitochondrial Dysfunction and Reactive Oxygen Species in Mediating Hypertension in the Reduced Uterine Perfusion Pressure Rat Model of Preeclampsia. Hypertension 2018, 72, 703–711. [Google Scholar] [CrossRef]
- Covarrubias, A.E.; Lecarpentier, E.; Lo, A.; Salahuddin, S.; Gray, K.J.; Karumanchi, S.A.; Zsengeller, Z.K. AP39, a Modulator of Mitochondrial Bioenergetics, Reduces Antiangiogenic Response and Oxidative Stress in Hypoxia-Exposed Trophoblasts: Relevance for Preeclampsia Pathogenesis. Am. J. Pathol. 2019, 189, 104–114. [Google Scholar] [CrossRef] [Green Version]
- Wei, J.; Liao, J.K.; Bairey Merz, C.N. Challenging Statin Pleiotropy: Preeclampsia. Circulation 2021, 144, 680–683. [Google Scholar] [CrossRef]
- Tricarico, P.M.; Crovella, S.; Celsi, F. Mevalonate Pathway Blockade, Mitochondrial Dysfunction and Autophagy: A Possible Link. Int. J. Mol. Sci. 2015, 16, 16067–16084. [Google Scholar] [CrossRef] [Green Version]
- Esteve-Valverde, E.; Ferrer-Oliveras, R.; Gil-Aliberas, N.; Baraldes-Farre, A.; Llurba, E.; Alijotas-Reig, J. Pravastatin for Preventing and Treating Preeclampsia: A Systematic Review. Obstet. Gynecol. Surv. 2018, 73, 40–55. [Google Scholar] [CrossRef] [PubMed]
- Otten, L.A.; van der Ven, K.; Kuhr, M.; Gembruch, U.; Merz, W.M. Pravastatin for prevention of HELLP syndrome: A case report. Medicine 2017, 96, 42–45. [Google Scholar] [CrossRef] [PubMed]
- Nourollahpour, S.M.; Cassinerio, E.; Modarres, M.; Zareiyan, A.; Hamzehgardeshi, Z.; Behboodi Moghadam, Z. Reproductive health issues in female patients with beta-thalassaemia major: A narrative literature review. J. Obstet. Gynaecol. 2020, 40, 902–911. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Author (Ref) | Year | Related Pathway | Target | Findings |
|---|---|---|---|---|
| Boutet M [157] | 2008 | GSH synthesis pathway | GPX4 | Over expression of GPX4 in PE group |
| Peng XG [33] | 2016 | GSH synthesis pathway | GPX4 | rs713041 in GPX4 gene and the C allele has the higher risk for pathogenesis of PE |
| Zhang H [128] | 2020 | GSH synthesis and iron metabolism pathway | FPN1 | Downregulation of Cys2/glutamate antiporter, PAX3 and GSH levels, upregulation of labile Fe2+ due to miR-30b-5p |
| Wang Y [159] | 2021 | GSH synthesis and iron metabolism pathway | System Xc− GPX4 FPN1 | Activation of Nrf2/HO-1 signaling pathway and overexpression of SLC7A11, GPX4 and FPN1 due to Nrf2 overexpression |
| Liao TT [162] | 2022 | GSH synthesis pathway | GPX4 | Upregulation of Nrf2/GPX4 signaling pathway and resist ferroptosis in the pathogenesis of PE |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Z.; Gan, J.; Zhang, M.; Du, Y.; Zhao, H. Ferroptosis and Its Emerging Role in Pre-Eclampsia. Antioxidants 2022, 11, 1282. https://doi.org/10.3390/antiox11071282
Chen Z, Gan J, Zhang M, Du Y, Zhao H. Ferroptosis and Its Emerging Role in Pre-Eclampsia. Antioxidants. 2022; 11(7):1282. https://doi.org/10.3390/antiox11071282
Chicago/Turabian StyleChen, Zhixian, Jianfeng Gan, Mo Zhang, Yan Du, and Hongbo Zhao. 2022. "Ferroptosis and Its Emerging Role in Pre-Eclampsia" Antioxidants 11, no. 7: 1282. https://doi.org/10.3390/antiox11071282