
Role of Erythrocytes in Nitric Oxide Metabolism and Paracrine Regulation of Endothelial Function


{kind=link}
Abstract
:1. Human Erythrocytes as the Storage Pool of Amino Acids
2. RBC—The Importance of Transmembrane Translocases
3. Transport of the Selected Nitric Oxide Metabolic Pathway Intermediates between Erythrocytes and Plasma
4. Intraerythrocytic Metabolism of L-Arg and Its Regulation
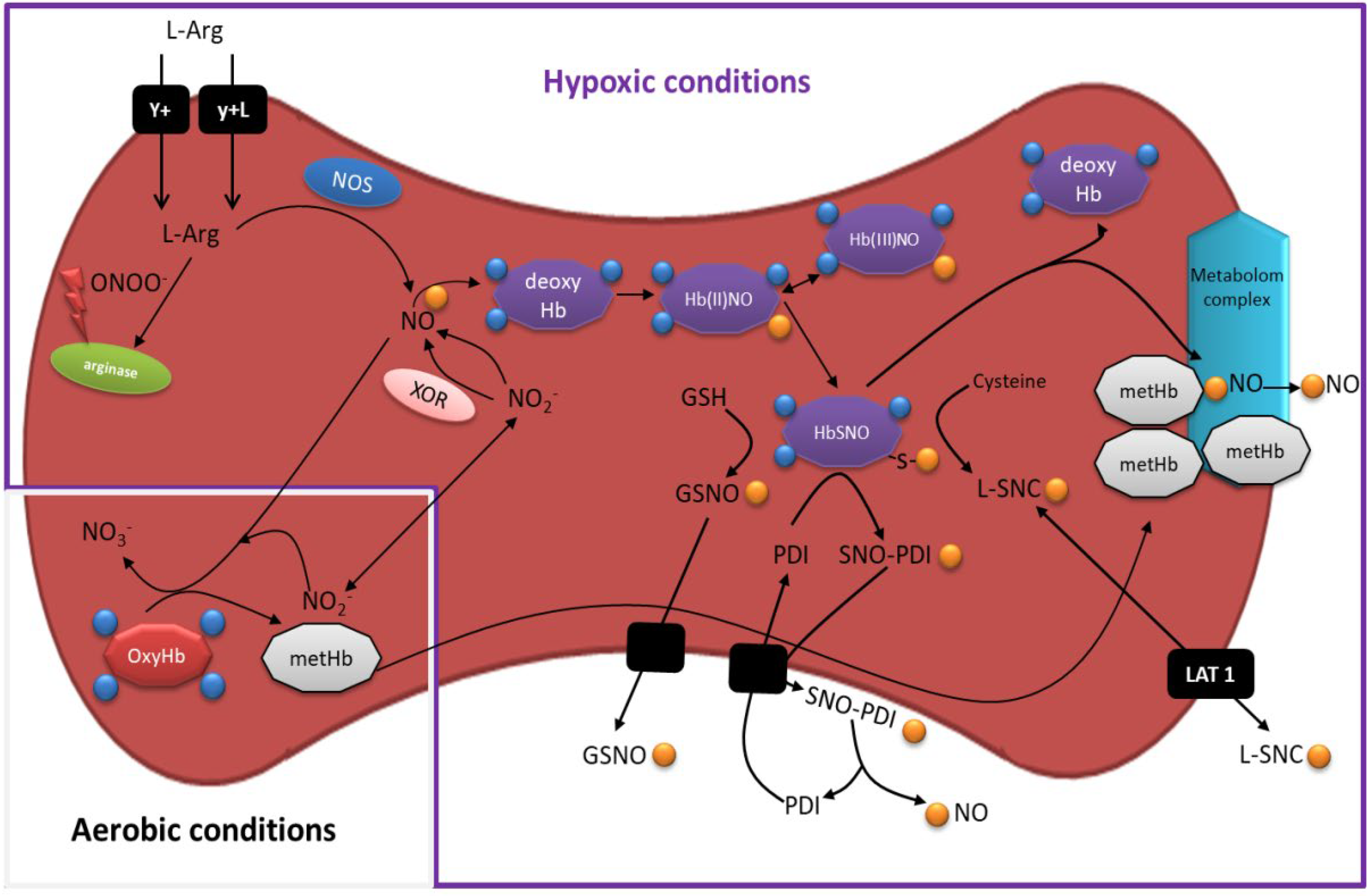
5. RBCs as a Source of NO. Mechanisms Underlying Homeostasis of Its Bioavailability
6. Erythrocyte-Dependent Paracrine Regulation of the Vascular NO Bioavailability
7. Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bryk, A.H.; Wiśniewski, J.R.W. Quantitative Analysis of Human Red Blood Cell Proteome. J. Proteome Res. 2017, 16, 2752–2761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agli, A.N.; Schaefer, A.; Geny, B.; Piquard, F.; Haberey, P. Erythrocytes Participate Significantly in Blood Transport of Amino Acids during the Post Absorptive State in Normal Humans. Eur. J. Appl. Physiol. Occup. Physiol. 1998, 78, 502–508. [Google Scholar] [CrossRef] [PubMed]
- Thorn, B.; Dunstan, R.H.; Macdonald, M.M.; Borges, N.; Roberts, T.K. Evidence That Human and Equine Erythrocytes Could Have Significant Roles in the Transport and Delivery of Amino Acids to Organs and Tissues. Amino Acids 2020, 52, 711–724. [Google Scholar] [CrossRef] [Green Version]
- Strobel, J.; Mieth, M.; Endreß, B.; Auge, D.; König, J.; Fromm, M.F.; Maas, R. Interaction of the Cardiovascular Risk Marker Asymmetric Dimethylarginine (ADMA) with the Human Cationic Amino Acid Transporter 1 (CAT1). J. Mol. Cell. Cardiol. 2012, 53, 392–400. [Google Scholar] [CrossRef] [PubMed]
- Franchi Gazzola, R.; Sala, R.; Bussolati, O.; Visigalli, R.; Dall’Asta, V.; Ganapathy, V.; Gazzola, G.C. The Adaptive Regulation of Amino Acid Transport System A Is Associated to Changes in ATA2 Expression. FEBS Lett. 2001, 490, 11–14. [Google Scholar] [CrossRef] [Green Version]
- Gräf, P.; Förstermann, U.; Closs, E.I. The Transport Activity of the Human Cationic Amino Acid Transporter HCAT-1 Is Downregulated by Activation of Protein Kinase C. Br. J. Pharmacol. 2001, 132, 1193–1200. [Google Scholar] [CrossRef] [Green Version]
- Fernandez, J.; Bode, B.; Koromilas, A.; Alan Diehl, J.; Krukovets, I.; Snider, M.D.; Hatzoglou, M. Translation Mediated by the Internal Ribosome Entry Site of the Cat-1 MRNA Is Regulated by Glucose Availability in a PERK Kinase-Dependent Manner. J. Biol. Chem. 2002, 277, 11780–11787. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharyya, S.N.; Habermacher, R.; Martine, U.; Closs, E.I.; Filipowicz, W. Relief of MicroRNA-Mediated Translational Repression in Human Cells Subjected to Stress. Cell 2006, 125, 1111–1124. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.-G.; Zhang, Q.-J.; Li, B.-W.; Li, L.-H.; Song, X.-H.; Xiong, C.-M.; Zou, Y.-B.; Liu, B.-Y.; Han, J.-Q.; Xiu, R.-J. The Circulating Level of MiR-122 Is a Potential Risk Factor for Endothelial Dysfunction in Young Patients with Essential Hypertension. Hypertens. Res. 2020, 43, 511–517. [Google Scholar] [CrossRef]
- Wang, L.; Chen, H. Correlation between Serum MiR-122 and Myocardial Damage and Ventricular Function in Patients with Essential Hypertension. J. Thorac. Dis. 2021, 13, 4999–5006. [Google Scholar] [CrossRef]
- Gaddam, R.R.; Dhuri, K.; Kim, Y.-R.; Jacobs, J.S.; Kumar, V.; Li, Q.; Irani, K.; Bahal, R.; Vikram, A. γ Peptide Nucleic Acid-Based MiR-122 Inhibition Rescues Vascular Endothelial Dysfunction in Mice Fed a High-Fat Diet. J. Med. Chem. 2022, 65, 3332–3342. [Google Scholar] [CrossRef] [PubMed]
- Hatzoglou, M.; Fernandez, J.; Yaman, I.; Closs, E. Regulation of Cationic Amino Acid Transport: The Story of the CAT-1 Transporter. Annu. Rev. Nutr. 2004, 24, 377–399. [Google Scholar] [CrossRef]
- Doss, J.F.; Corcoran, D.L.; Jima, D.D.; Telen, M.J.; Dave, S.S.; Chi, J.T. A Comprehensive Joint Analysis of the Long and Short RNA Transcriptomes of Human Erythrocytes. BMC Genom. 2015, 16, 952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kabanova, S.; Kleinbongard, P.; Volkmer, J.; Andrée, B.; Kelm, M.; Jax, T.W. Gene Expression Analysis of Human Red Blood Cells. Int. J. Med. Sci. 2009, 6, 156–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cortese-Krott, M.M.; Kelm, M. Endothelial Nitric Oxide Synthase in Red Blood Cells: Key to a New Erythrocrine Function? Redox Biol. 2014, 2, 251–258. [Google Scholar] [CrossRef] [Green Version]
- Davids, M.; van Hell, A.J.; Visser, M.; Nijveldt, R.J.; van Leeuwen, P.A.M.; Teerlink, T. Role of the Human Erythrocyte in Generation and Storage of Asymmetric Dimethylarginine. Am. J. Physiol.-Heart Circ. Physiol. 2012, 302, 1762–1770. [Google Scholar] [CrossRef] [Green Version]
- Neelam, S.; Kakhniashvili, D.G.; Wilkens, S.; Levene, S.D.; Goodman, S.R. Functional 20S Proteasomes in Mature Human Red Blood Cells. Exp. Biol. Med. 2011, 236, 580–591. [Google Scholar] [CrossRef]
- Sitar, M.E.; Kayacelebi, A.A.; Beckmann, B.; Kielstein, J.T.; Tsikas, D. Asymmetric Dimethylarginine (ADMA) in Human Blood: Effects of Extended Haemodialysis in the Critically Ill Patient with Acute Kidney Injury, Protein Binding to Human Serum Albumin and Proteolysis by Thermolysin. Amino Acids 2015, 47, 1983–1993. [Google Scholar] [CrossRef]
- Yokoro, M.; Suzuki, M.; Murota, K.; Otsuka, C.; Yamashita, H.; Takahashi, Y.; Tsuji, H.; Kimoto, M. Asymmetric Dimethylarginine, an Endogenous NOS Inhibitor, Is Actively Metabolized in Rat Erythrocytes. Biosci. Biotechnol. Biochem. 2012, 76, 1334–1342. [Google Scholar] [CrossRef]
- Kang, E.S.; Cates, T.B.; Harper, D.N.; Chiang, T.M.; Myers, L.K.; Acchiardo, S.R.; Kimoto, M. An Enzyme Hydrolyzing Methylated Inhibitors of Nitric Oxide Synthase Is Present in Circulating Human Red Blood Cells. Free Radic. Res. 2001, 35, 693–707. [Google Scholar] [CrossRef]
- Bentur, O.S.; Schwartz, D.; Chernichovski, T.; Ingbir, M.; Weinstein, T.; Chernin, G.; Schwartz, I.F. Estradiol Augments While Progesterone Inhibits Arginine Transport in Human Endothelial Cells through Modulation of Cationic Amino Acid Transporter-1. Am. J. Physiol. Integr. Comp. Physiol. 2015, 309, R421–R427. [Google Scholar] [CrossRef] [PubMed]
- Toral, M.; Jimenez, R.; Montoro-Molina, S.; Romero, M.; Wangensteen, R.; Duarte, J.; Vargas, F. Thyroid Hormones Stimulate L-Arginine Transport in Human Endothelial Cells. J. Endocrinol. 2018, 239, 49–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kietadisorn, R.; Juni, R.P.; Moens, A.L. Tackling Endothelial Dysfunction by Modulating NOS Uncoupling: New Insights into Its Pathogenesis and Therapeutic Possibilities. Am. J. Physiol. Metab. 2012, 302, E481–E495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roe, N.D.; Ren, J. Nitric Oxide Synthase Uncoupling: A Therapeutic Target in Cardiovascular Diseases. Vascul. Pharmacol. 2012, 57, 168–172. [Google Scholar] [CrossRef]
- Zou, M.-H.; Shi, C.; Cohen, R.A. Oxidation of the Zinc-Thiolate Complex and Uncoupling of Endothelial Nitric Oxide Synthase by Peroxynitrite. J. Clin. Investig. 2002, 109, 817–826. [Google Scholar] [CrossRef]
- Mahdi, A.; Tengbom, J.; Alvarsson, M.; Wernly, B.; Zhou, Z.; Pernow, J. Red Blood Cell Peroxynitrite Causes Endothelial Dysfunction in Type 2 Diabetes Mellitus via Arginase. Cells 2020, 9, 1712. [Google Scholar] [CrossRef]
- Zhou, Z.; Mahdi, A.; Tratsiakovich, Y.; Zahorán, S.; Kövamees, O.; Nordin, F.; Uribe Gonzalez, A.E.; Alvarsson, M.; Östenson, C.G.; Andersson, D.C.; et al. Erythrocytes from Patients with Type 2 Diabetes Induce Endothelial Dysfunction Via Arginase I. J. Am. Coll. Cardiol. 2018, 72, 769–780. [Google Scholar] [CrossRef]
- Pernow, J.; Mahdi, A.; Yang, J.; Zhou, Z. Red Blood Cell Dysfunction: A New Player in Cardiovascular Disease. Cardiovasc. Res. 2019, 115, 1596–1605. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Gonon, A.T.; Sjoquist, P.-O.; Lundberg, J.O.; Pernow, J. Arginase Regulates Red Blood Cell Nitric Oxide Synthase and Export of Cardioprotective Nitric Oxide Bioactivity. Proc. Natl. Acad. Sci. USA 2013, 110, 15049–15054. [Google Scholar] [CrossRef] [Green Version]
- Mahdi, A.; Kövamees, O.; Pernow, J. Improvement in Endothelial Function in Cardiovascular Disease-Is Arginase the Target? Int. J. Cardiol. 2020, 301, 207–214. [Google Scholar] [CrossRef]
- Romero, M.J.; Platt, D.H.; Tawfik, H.E.; Labazi, M.; El-Remessy, A.B.; Bartoli, M.; Caldwell, R.B.; Caldwell, R.W. Diabetes-Induced Coronary Vascular Dysfunction Involves Increased Arginase Activity. Circ. Res. 2008, 102, 95–102. [Google Scholar] [CrossRef] [PubMed]
- El-Remessy, A.B.; Tawfik, H.E.; Matragoon, S.; Pillai, B.; Caldwell, R.B.; Caldwell, R.W. Peroxynitrite Mediates Diabetes-Induced Endothelial Dysfunction: Possible Role of Rho Kinase Activation. Exp. Diabetes Res. 2010, 2010, 247861. [Google Scholar] [CrossRef] [PubMed]
- Santhanam, L.; Lim, H.K.; Lim, H.K.; Miriel, V.; Brown, T.; Patel, M.; Balanson, S.; Ryoo, S.; Anderson, M.; Irani, K.; et al. Inducible NO Synthase–Dependent S -Nitrosylation and Activation of Arginase1 Contribute to Age-Related Endothelial Dysfunction. Circ. Res. 2007, 101, 692–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, P.S.; Iyer, R.K.; Lu, K.V.; Yu, H.; Karimi, A.; Kern, R.M.; Tai, D.K.; Cederbaum, S.D.; Grody, W.W. Expression of the Liver Form of Arginase in Erythrocytes. Mol. Genet. Metab. 2002, 76, 100–110. [Google Scholar] [CrossRef]
- Duckles, S.P.; Miller, V.M. Hormonal Modulation of Endothelial NO Production. Pflügers Arch.-Eur. J. Physiol. 2010, 459, 841–851. [Google Scholar] [CrossRef] [Green Version]
- Vona, R.; Gambardella, L.; Ortona, E.; Santulli, M.; Malorni, W.; Carè, A.; Pietraforte, D.; Straface, E. Functional Estrogen Receptors of Red Blood Cells. Do They Influence Intracellular Signaling? Cell. Physiol. Biochem. 2019, 53, 186–199. [Google Scholar] [CrossRef]
- Liu, X.; Miller, M.J.S.; Joshi, M.S.; Sadowska-Krowicka, H.; Clark, D.A.; Lancaster, J.R. Diffusion-Limited Reaction of Free Nitric Oxide with Erythrocytes. J. Biol. Chem. 1998, 273, 18709–18713. [Google Scholar] [CrossRef] [Green Version]
- Helms, C.C.; Gladwin, M.T.; Kim-Shapiro, D.B. Erythrocytes and Vascular Function: Oxygen and Nitric Oxide. Front. Physiol. 2018, 9, 125. [Google Scholar] [CrossRef] [Green Version]
- Vaughn, M.W.; Huang, K.-T.; Kuo, L.; Liao, J.C. Erythrocytes Possess an Intrinsic Barrier to Nitric Oxide Consumption. J. Biol. Chem. 2000, 275, 2342–2348. [Google Scholar] [CrossRef] [Green Version]
- Chebbi, R. A Two-Zone Shear-Induced Red Blood Cell Migration Model for Blood Flow in Microvessels. Front. Phys. 2019, 7, 206. [Google Scholar] [CrossRef] [Green Version]
- Deonikar, P.; Kavdia, M. A Computational Model for Nitric Oxide, Nitrite and Nitrate Biotransport in the Microcirculation: Effect of Reduced Nitric Oxide Consumption by Red Blood Cells and Blood Velocity. Microvasc. Res. 2010, 80, 464–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, J.C.; Hein, T.W.; Vaughn, M.W.; Huang, K.-T.; Kuo, L. Intravascular Flow Decreases Erythrocyte Consumption of Nitric Oxide. Proc. Natl. Acad. Sci. USA 1999, 96, 8757–8761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagababu, E.; Ramasamy, S.; Abernethy, D.R.; Rifkind, J.M. Active Nitric Oxide Produced in the Red Cell under Hypoxic Conditions by Deoxyhemoglobin-Mediated Nitrite Reduction. J. Biol. Chem. 2003, 278, 46349–46356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stamler, J.S.; Jia, L.; Eu, J.P.; McMahon, T.J.; Demchenko, I.T.; Bonaventura, J.; Gernert, K.; Piantadosi, C.A. Blood Flow Regulation by S -Nitrosohemoglobin in the Physiological Oxygen Gradient. Science 1997, 276, 2034–2037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fago, A.; Crumbliss, A.L.; Hendrich, M.P.; Pearce, L.L.; Peterson, J.; Henkens, R.; Bonaventura, C. Oxygen Binding to Partially Nitrosylated Hemoglobin. Biochim. Biophys. Acta.-Proteins Proteom. 2013, 1834, 1894–1900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rifkind, J.M.; Nagababu, E.; Ramasamy, S. The Quaternary Hemoglobin Conformation Regulates the Formation of the Nitrite-Induced Bioactive Intermediate and the Dissociation of Nitric Oxide from This Intermediate. Nitric Oxide 2011, 24, 102–109. [Google Scholar] [CrossRef] [Green Version]
- Jia, L.; Bonaventura, C.; Bonaventura, J.; Stamler, J.S. S-Nitrosohaemoglobin: A Dynamic Activity of Blood Involved in Vascular Control. Transfus. Med. Rev. 1997, 11, 82. [Google Scholar] [CrossRef]
- Baylis, C.; Vallance, P. Measurement of Nitrite and Nitrate Levels in Plasma and Urine—What Does This Measure Tell Us about the Activity of the Endogenous Nitric Oxite System? Curr. Opin. Nephrol. Hypertens. 1998, 7, 59–62. [Google Scholar] [CrossRef]
- Cosby, K.; Partovi, K.S.; Crawford, J.H.; Patel, R.P.; Reiter, C.D.; Martyr, S.; Yang, B.K.; Waclawiw, M.A.; Zalos, G.; Xu, X.; et al. Nitrite Reduction to Nitric Oxide by Deoxyhemoglobin Vasodilates the Human Circulation. Nat. Med. 2003, 9, 1498–1505. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, H.H.H.W.; Feelisch, M. Red Blood Cell–Derived Nitric Oxide Bioactivity and Hypoxic Vasodilation. Circulation 2019, 139, 2664–2667. [Google Scholar] [CrossRef]
- Dejam, A. Erythrocytes Are the Major Intravascular Storage Sites of Nitrite in Human Blood. Blood 2005, 106, 734–739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rathod, K.S.; Webb, A.J.; Lovell, M.J.; Lecomte, F.; Ahluwalia, A. Nitrite Is Reduced to Nitric Oxide by Xanthine Oxidoreductase and Nitric Oxide Synthase in the Erythrocyte Membrane in Hypoxemia. Blood 2006, 108, 1560. [Google Scholar] [CrossRef]
- Webb, A.J.; Milsom, A.B.; Rathod, K.S.; Chu, W.L.; Qureshi, S.; Lovell, M.J.; Lecomte, F.M.J.; Perrett, D.; Raimondo, C.; Khoshbin, E.; et al. Mechanisms Underlying Erythrocyte and Endothelial Nitrite Reduction to Nitric Oxide in Hypoxia. Circ. Res. 2008, 103, 957–964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dejam, A.; Hunter, C.J.; Tremonti, C.; Pluta, R.M.; Yuen, Y.H.; Hon, Y.; Grimes, G.; Partovi, K.; Pelletier, M.M.; Oldfield, E.H.; et al. Nitrite Infusion in Humans and Nonhuman Primates Endocrine Effects, Pharmacokinetics and Tolerance Formation. Circulation 2007, 116, 1821–1831. [Google Scholar] [CrossRef]
- Ulker, P.; Yaras, N.; Yalcin, O.; Celik-Ozenci, C.; Johnson, P.C.; Meiselman, H.J.; Baskurt, O.K. Shear Stress Activation of Nitric Oxide Synthase and Increased Nitric Oxide Levels in Human Red Blood Cells. Nitric Oxide 2011, 24, 184–191. [Google Scholar] [CrossRef] [Green Version]
- Ulker, P.; Sati, L.; Celik-Ozenci, C.; Meiselman, H.J.; Baskurt, O.K. Mechanical Stimulation of Nitric Oxide Synthesizing Mechanisms in Erythrocytes. Biorheology 2009, 46, 121–132. [Google Scholar] [CrossRef]
- Feron, O.; Saldana, F.; Michel, J.B.; Michel, T. The Endothelial Nitric-Oxide Synthase-Caveolin Regulatory Cycle. J. Biol. Chem. 1998, 273, 3125–3128. [Google Scholar] [CrossRef] [Green Version]
- Ulker, P.; Meiselman, H.J.; Baskurt, O.K. Nitric Oxide Generation in Red Blood Cells Induced by Mechanical Stress. Clin. Hemorheol. Microcirc. 2010, 45, 169–175. [Google Scholar] [CrossRef]
- Fleming, I.; Busse, R. Molecular Mechanisms Involved in the Regulation of the Endothelial Nitric Oxide Synthase. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2003, 284, R1–R12. [Google Scholar] [CrossRef] [Green Version]
- Bor-Kucukatay, M.; Wenby, R.B.; Meiselman, H.J.; Baskurt, O.K. Effects of Nitric Oxide on Red Blood Cell Deformability. Am. J. Physiol.-Heart Circ. Physiol. 2003, 284, H1577–H1584. [Google Scholar] [CrossRef] [Green Version]
- Cortese-Krott, M.M.; Mergia, E.; Kramer, C.M.; Lückstädt, W.; Yang, J.; Wolff, G.; Panknin, C.; Bracht, T.; Sitek, B.; Pernow, J.; et al. Identification of a Soluble Guanylate Cyclase in RBCs: Preserved Activity in Patients with Coronary Artery Disease. Redox Biol. 2018, 14, 328–337. [Google Scholar] [CrossRef] [PubMed]
- Adragna, N.C.; Fulvio, M.D.; Lauf, P.K. Regulation of K-Cl Cotransport: From Function to Genes. J. Membr. Biol. 2004, 201, 109–137. [Google Scholar] [CrossRef] [PubMed]
- Dreischer, P.; Duszenko, M.; Stein, J.; Wieder, T. Eryptosis: Programmed Death of Nucleus-Free, Iron-Filled Blood Cells. Cells 2022, 11, 503. [Google Scholar] [CrossRef] [PubMed]
- Brun, J.-F.; Varlet-Marie, E.; Myzia, J.; de Mauverger, E.R.; Pretorius, E. Metabolic Influences Modulating Erythrocyte Deformability and Eryptosis. Metabolites 2021, 12, 4. [Google Scholar] [CrossRef] [PubMed]
- Föller, M.; Feil, S.; Ghoreschi, K.; Koka, S.; Gerling, A.; Thunemann, M.; Hofmann, F.; Schuler, B.; Vogel, J.; Pichler, B.; et al. Anemia and Splenomegaly in CGKI-Deficient Mice. Proc. Natl. Acad. Sci. USA 2008, 105, 6771–6776. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.; Piknova, B.; Pittman, R.N.; Schechter, A.N.; Popel, A.S. Nitric Oxide from Nitrite Reduction by Hemoglobin in the Plasma and Erythrocytes. Nitric Oxide 2008, 18, 47–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandmann, J.; Schwedhelm, K.S.; Tsikas, D. Specific Transport of S -Nitrosocysteine in Human Red Blood Cells: Implications for Formation of S -Nitrosothiols and Transport of NO Bioactivity within the Vasculature. FEBS Lett. 2005, 579, 4119–4124. [Google Scholar] [CrossRef] [Green Version]
- Pawloski, J.R.; Hess, D.T.; Stamler, J.S. Export by Red Blood Cells of Nitric Oxide Bioactivity. Nature 2001, 409, 622–626. [Google Scholar] [CrossRef]
- Chu, H.; McKenna, M.M.; Krump, N.A.; Zheng, S.; Mendelsohn, L.; Thein, S.L.; Garrett, L.J.; Bodine, D.M.; Low, P.S. Reversible Binding of Hemoglobin to Band 3 Constitutes the Molecular Switch That Mediates O2 Regulation of Erythrocyte Properties. Blood 2016, 128, 2708–2716. [Google Scholar] [CrossRef]
- Gladwin, M.T.; Schechter, A.N.; Kim-Shapiro, D.B.; Patel, R.P.; Hogg, N.; Shiva, S.; Cannon, R.O.; Kelm, M.; Wink, D.A.; Espey, M.G.; et al. The Emerging Biology of the Nitrite Anion. Nat. Chem. Biol. 2005, 1, 308–314. [Google Scholar] [CrossRef]
- Zhao, Y.; Wang, X.; Wang, R.; Chen, D.; Noviana, M.; Zhu, H. Nitric Oxide Inhibits Hypoxia-induced Impairment of Human RBC Deformability through Reducing the Cross-linking of Membrane Protein Band 3. J. Cell. Biochem. 2019, 120, 305–320. [Google Scholar] [CrossRef] [Green Version]
- Jennings, M.L. Cell Physiology and Molecular Mechanism of Anion Transport by Erythrocyte Band 3/AE1. Am. J. Physiol. Physiol. 2021, 321, C1028–C1059. [Google Scholar] [CrossRef] [PubMed]
- Dosier, L.B.M.; Premkumar, V.J.; Zhu, H.; Akosman, I.; Wempe, M.F.; McMahon, T.J. Antagonists of the System L Neutral Amino Acid Transporter (LAT) Promote Endothelial Adhesivity of Human Red Blood Cells. Thromb. Haemost. 2017, 117, 1402–1411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verrey, F.; Closs, E.I.; Wagner, C.A.; Palacin, M.; Endou, H.; Kanai, Y. CATs and HATs: The SLC7 Family of Amino Acid Transporters. Pflügers Arch.-Eur. J. Physiol. 2004, 447, 532–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kallakunta, V.M.; Slama-Schwok, A.; Mutus, B. Protein Disulfide Isomerase May Facilitate the Efflux of Nitrite Derived S-Nitrosothiols from Red Blood Cells. Redox Biol. 2013, 1, 373–380. [Google Scholar] [CrossRef]
- Ramachandran, N.; Root, P.; Jiang, X.-M.; Hogg, P.J.; Mutus, B. Mechanism of Transfer of NO from Extracellular S-Nitrosothiols into the Cytosol by Cell-Surface Protein Disulfide Isomerase. Proc. Natl. Acad. Sci. USA 2001, 98, 9539–9544. [Google Scholar] [CrossRef] [Green Version]
- Bergfeld, G.R.; Forrester, T. Release of ATP from Human Erythrocytes in Response to a Brief Period of Hypoxia and Hypercapnia. Cardiovasc. Res. 1992, 26, 40–47. [Google Scholar] [CrossRef]
- Ellsworth, M.L.; Forrester, T.; Ellis, C.G.; Dietrich, H.H. The Erythrocyte as a Regulator of Vascular Tone. Am. J. Physiol. Circ. Physiol. 1995, 269, H2155–H2161. [Google Scholar] [CrossRef]
- Locovei, S.; Bao, L.; Dahl, G. Pannexin 1 in Erythrocytes: Function without a Gap. Proc. Natl. Acad. Sci. USA 2006, 103, 7655–7659. [Google Scholar] [CrossRef] [Green Version]
- Ransford, G.A.; Fregien, N.; Qiu, F.; Dahl, G.; Conner, G.E.; Salathe, M. Pannexin 1 Contributes to ATP Release in Airway Epithelia. Am. J. Respir. Cell Mol. Biol. 2009, 41, 525–534. [Google Scholar] [CrossRef] [Green Version]
- Di Virgilio, F.; Chiozzi, P.; Ferrari, D.; Falzoni, S.; Sanz, J.M.; Morelli, A.; Torboli, M.; Bolognesi, G.; Baricordi, O.R. Nucleotide Receptors: An Emerging Family of Regulatory Molecules in Blood Cells. Blood 2001, 97, 587–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Öhman, J.; Kudira, R.; Albinsson, S.; Olde, B.; Erlinge, D. Ticagrelor Induces Adenosine Triphosphate Release from Human Red Blood Cells. Biochem. Biophys. Res. Commun. 2012, 418, 754–758. [Google Scholar] [CrossRef] [PubMed]
- Nylander, S.; Femia, E.A.; Scavone, M.; Berntsson, P.; Asztély, A.-K.; Nelander, K.; Löfgren, L.; Nilsson, R.G.; Cattaneo, M. Ticagrelor Inhibits Human Platelet Aggregation via Adenosine in Addition to P2Y 12 Antagonism. J. Thromb. Haemost. 2013, 11, 1867–1876. [Google Scholar] [CrossRef] [PubMed]
- Kirby, B.S.; Sparks, M.A.; Lazarowski, E.R.; Domowicz, D.A.L.; Zhu, H.; McMahon, T.J. Integrative Cardiovascular Physiology and Pathophysiology: Pannexin 1 Channels Control the Hemodynamic Response to Hypoxia by Regulating O2-Sensitive Extracellular ATP in Blood. Am. J. Physiol.-Heart Circ. Physiol. 2021, 320, H1055. [Google Scholar] [CrossRef]
- von Kügelgen, I.; Wetter, A. Molecular Pharmacology of P2Y-Receptors. Naunyn. Schmiedebergs. Arch. Pharmacol. 2000, 362, 310–323. [Google Scholar] [CrossRef]
- Forsberg, E.J.; Feuerstein, G.; Shohami, E.; Pollard, H.B. Adenosine Triphosphate Stimulates Inositol Phospholipid Metabolism and Prostacyclin Formation in Adrenal Medullary Endothelial Cells by Means of P2-Purinergic Receptors. Proc. Natl. Acad. Sci. USA 1987, 84, 5630–5634. [Google Scholar] [CrossRef] [Green Version]
- Burnstock, G. Purinergic Signaling in the Cardiovascular System. Circ. Res. 2017, 120, 207–228. [Google Scholar] [CrossRef] [Green Version]
- Sridharan, M.; Adderley, S.P.; Bowles, E.A.; Egan, T.M.; Stephenson, A.H.; Ellsworth, M.L.; Sprague, R.S. Pannexin 1 Is the Conduit for Low Oxygen Tension-Induced ATP Release from Human Erythrocytes. Am. J. Physiol. Circ. Physiol. 2010, 299, H1146–H1152. [Google Scholar] [CrossRef] [Green Version]
- Sridharan, M.; Bowles, E.A.; Richards, J.P.; Krantic, M.; Davis, K.L.; Dietrich, K.A.; Stephenson, A.H.; Ellsworth, M.L.; Sprague, R.S. Prostacyclin Receptor-Mediated ATP Release from Erythrocytes Requires the Voltage-Dependent Anion Channel. Am. J. Physiol.-Heart Circ. Physiol. 2012, 302, H553–H559. [Google Scholar] [CrossRef]
- Bouyer, G.; Cueff, A.; Egée, S.; Kmiecik, J.; Maksimova, Y.; Glogowska, E.; Gallagher, P.G.; Thomas, S.L.Y. Erythrocyte Peripheral Type Benzodiazepine Receptor/Voltage-Dependent Anion Channels Are Upregulated by Plasmodium Falciparum. Blood 2011, 118, 2305–2312. [Google Scholar] [CrossRef] [Green Version]
- Marginedas-Freixa, I.; Alvarez, C.L.; Moras, M.; Leal Denis, M.F.; Hattab, C.; Halle, F.; Bihel, F.; Mouro-Chanteloup, I.; Lefevre, S.D.; Le Van Kim, C.; et al. Human Erythrocytes Release ATP by a Novel Pathway Involving VDAC Oligomerization Independent of Pannexin-1. Sci. Rep. 2018, 8, 11384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srihirun, S.; Sriwantana, T.; Unchern, S.; Kittikool, D.; Noulsri, E.; Pattanapanyasat, K.; Fucharoen, S.; Piknova, B.; Schechter, A.N.; Sibmooh, N. Platelet Inhibition by Nitrite Is Dependent on Erythrocytes and Deoxygenation. PLoS ONE 2012, 7, e30380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leo, F.; Suvorava, T.; Heuser, S.K.; Li, J.; Lobue, A.; Barbarino, F.; Piragine, E.; Schneckmann, R.; Hutzler, B.; Good, M.E.; et al. Red Blood Cell and Endothelial ENOS Independently Regulate Circulating Nitric Oxide Metabolites and Blood Pressure. Circulation 2021, 144, 870–889. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Venardos, K.; Jones, E.; Morris, B.J.; Chin-Dusting, J.; Kaye, D.M. Identification of a Novel Polymorphism in the 3′UTR of the L-Arginine Transporter Gene SLC7A1. Circulation 2007, 115, 1269–1274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grupper, A.; Shashar, M.; Bahry, D.; Pri-Paz, Y.; Tur, O.B.; Levi, S.; Chernichovski, T.; Chernin, G.; Schwartz, I.F. Cyclosporine Attenuates Arginine Transport, in Human Endothelial Cells, through Modulation of Cationic Amino Acid Transporter-1. Am. J. Nephrol. 2013, 37, 612–618. [Google Scholar] [CrossRef]
- Grau, M.; Pauly, S.; Ali, J.; Walpurgis, K.; Thevis, M.; Bloch, W.; Suhr, F. RBC-NOS-Dependent S-Nitrosylation of Cytoskeletal Proteins Improves RBC Deformability. PLoS ONE 2013, 8, e56759. [Google Scholar] [CrossRef] [Green Version]
- Barbarino, F.; Wäschenbach, L.; Cavalho-Lemos, V.; Dillenberger, M.; Becker, K.; Gohlke, H.; Cortese-Krott, M.M. Targeting Spectrin Redox Switches to Regulate the Mechanoproperties of Red Blood Cells. Biol. Chem. 2021, 402, 317–331. [Google Scholar] [CrossRef]
- Porro, B.; Eligini, S.; Veglia, F.; Lualdi, A.; Squellerio, I.; Fiorelli, S.; Giovannardi, M.; Chiorino, E.; Dalla Cia, A.; Crisci, M.; et al. Nitric Oxide Synthetic Pathway in Patients with Microvascular Angina and Its Relations with Oxidative Stress. Oxid. Med. Cell. Longev. 2014, 2014, 726539. [Google Scholar] [CrossRef]
- Holowatz, L.A.; Kenney, W.L. Up-Regulation of Arginase Activity Contributes to Attenuated Reflex Cutaneous Vasodilatation in Hypertensive Humans. J. Physiol. 2007, 581, 863–872. [Google Scholar] [CrossRef]
- Pernow, J.; Jung, C. Arginase as a Potential Target in the Treatment of Cardiovascular Disease: Reversal of Arginine Steal? Cardiovasc. Res. 2013, 98, 334–343. [Google Scholar] [CrossRef] [Green Version]
- Quitter, F.; Figulla, H.R.; Ferrari, M.; Pernow, J.; Jung, C. Increased Arginase Levels in Heart Failure Represent a Therapeutic Target to Rescue Microvascular Perfusion. Clin. Hemorheol. Microcirc. 2013, 54, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Masuda, H. Significance of Nitric Oxide and Its Modulation Mechanisms by Endogenous Nitric Oxide Synthase Inhibitors and Arginase in the Micturition Disorders and Erectile Dysfunction. Int. J. Urol. 2008, 15, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Hein, T.W.; Zhang, C.; Wang, W.; Chang, C.-I.; Thengchaisri, N.; Kuo, L. Ischemia-reperfusion Selectively Impairs Nitric Oxide-Mediated Dilation in Coronary Arterioles: Counteracting Role of Arginase. FASEB J. 2003, 17, 2328–2330. [Google Scholar] [CrossRef]
- Jung, C.; Gonon, A.T.; Sjoquist, P.-O.; Lundberg, J.O.; Pernow, J. Arginase Inhibition Mediates Cardioprotection during Ischaemia-Reperfusion. Cardiovasc. Res. 2010, 85, 147–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonon, A.T.; Jung, C.; Katz, A.; Westerblad, H.; Shemyakin, A.; Sjöquist, P.-O.; Lundberg, J.O.; Pernow, J. Local Arginase Inhibition during Early Reperfusion Mediates Cardioprotection via Increased Nitric Oxide Production. PLoS ONE 2012, 7, e42038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kövamees, O.; Shemyakin, A.; Checa, A.; Wheelock, C.E.; Lundberg, J.O.; Östenson, C.-G.; Pernow, J. Arginase Inhibition Improves Microvascular Endothelial Function in Patients with Type 2 Diabetes Mellitus. J. Clin. Endocrinol. Metab. 2016, 101, 3952–3958. [Google Scholar] [CrossRef]
- Kövamees, O.; Shemyakin, A.; Pernow, J. Effect of Arginase Inhibition on Ischemia-Reperfusion Injury in Patients with Coronary Artery Disease with and without Diabetes Mellitus. PLoS ONE 2014, 9, e103260. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gajecki, D.; Gawryś, J.; Szahidewicz-Krupska, E.; Doroszko, A. Role of Erythrocytes in Nitric Oxide Metabolism and Paracrine Regulation of Endothelial Function. Antioxidants 2022, 11, 943. https://doi.org/10.3390/antiox11050943
Gajecki D, Gawryś J, Szahidewicz-Krupska E, Doroszko A. Role of Erythrocytes in Nitric Oxide Metabolism and Paracrine Regulation of Endothelial Function. Antioxidants. 2022; 11(5):943. https://doi.org/10.3390/antiox11050943
Chicago/Turabian StyleGajecki, Damian, Jakub Gawryś, Ewa Szahidewicz-Krupska, and Adrian Doroszko. 2022. "Role of Erythrocytes in Nitric Oxide Metabolism and Paracrine Regulation of Endothelial Function" Antioxidants 11, no. 5: 943. https://doi.org/10.3390/antiox11050943