
Stevia and Stevioside Attenuate Liver Steatosis through PPARα-Mediated Lipophagy in db/db Mice Hepatocytes
 , ,
, ,
Abstract
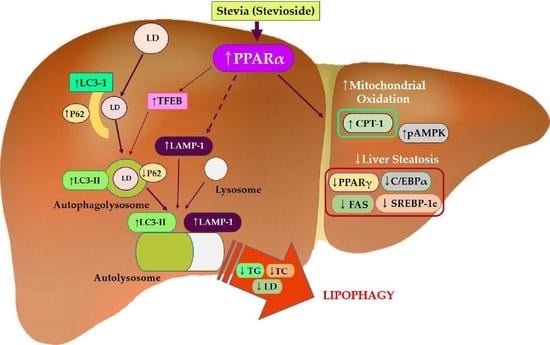
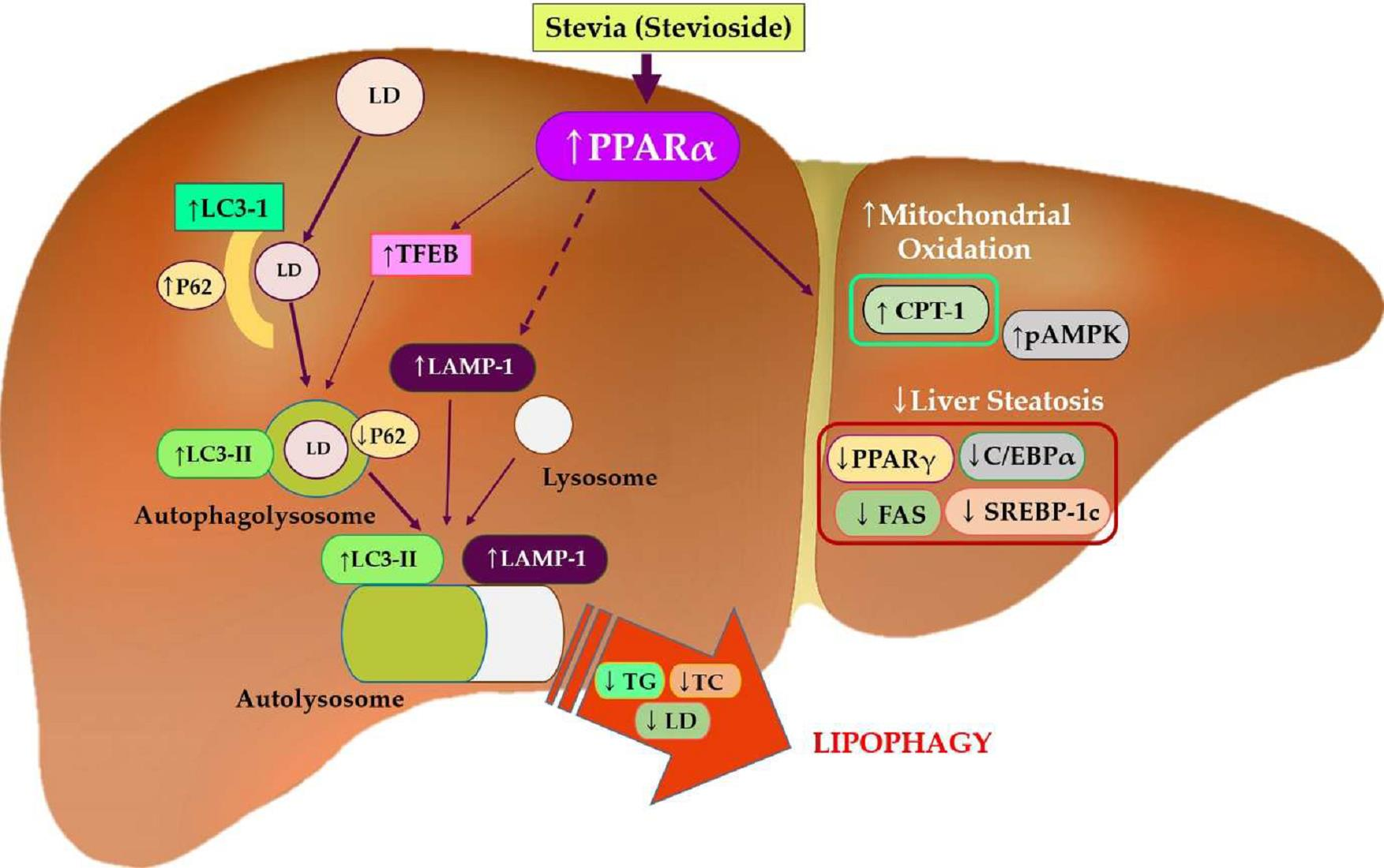
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. HepG2 Cells Culture, Steatosis Induction, and Oil Red O Staining
2.3. Mouse Models and Diets
2.4. Liver Tissue Histological Evaluation and Oil Red O Staining
2.5. Real-Time PCR Quantification of Gene Expression
2.6. Western Blotting
2.7. Detection of Autophagy with CYTO-ID® Green
2.8. Small Interfering RNAs (siRNAs) and Transfection
2.9. Statistical Analysis
3. Results
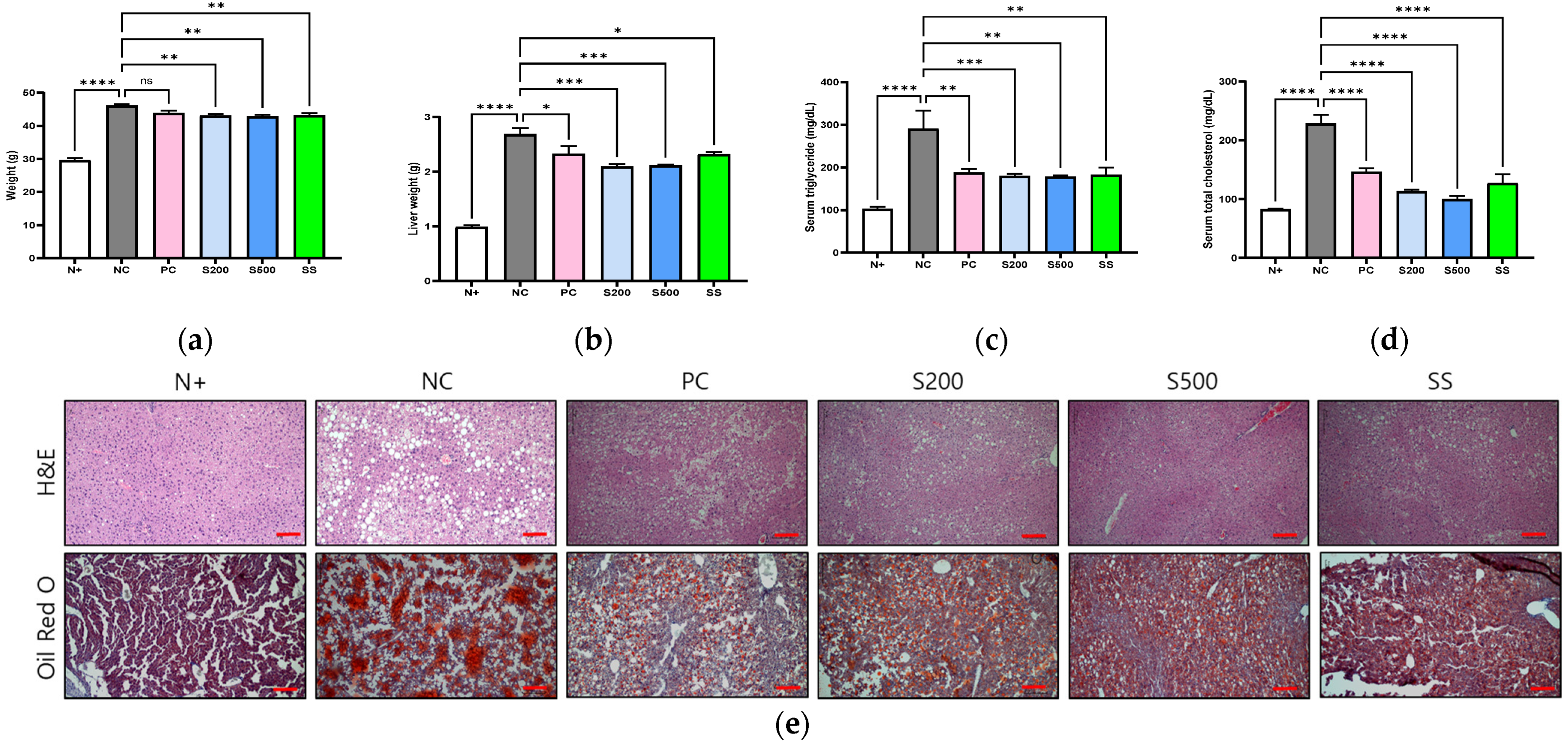
3.1. S and SS Attenuated Liver Steatosis in db/db Mice
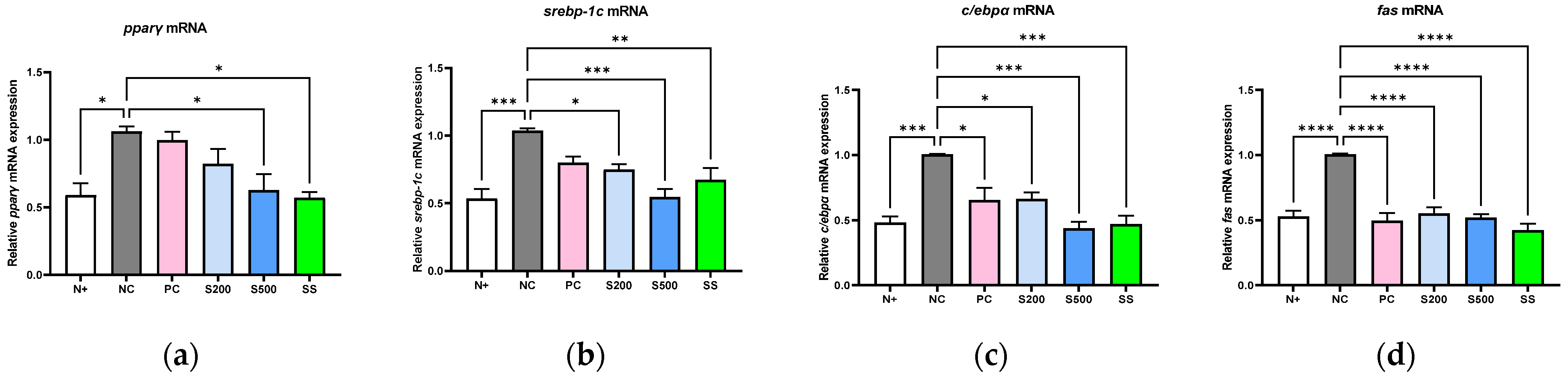
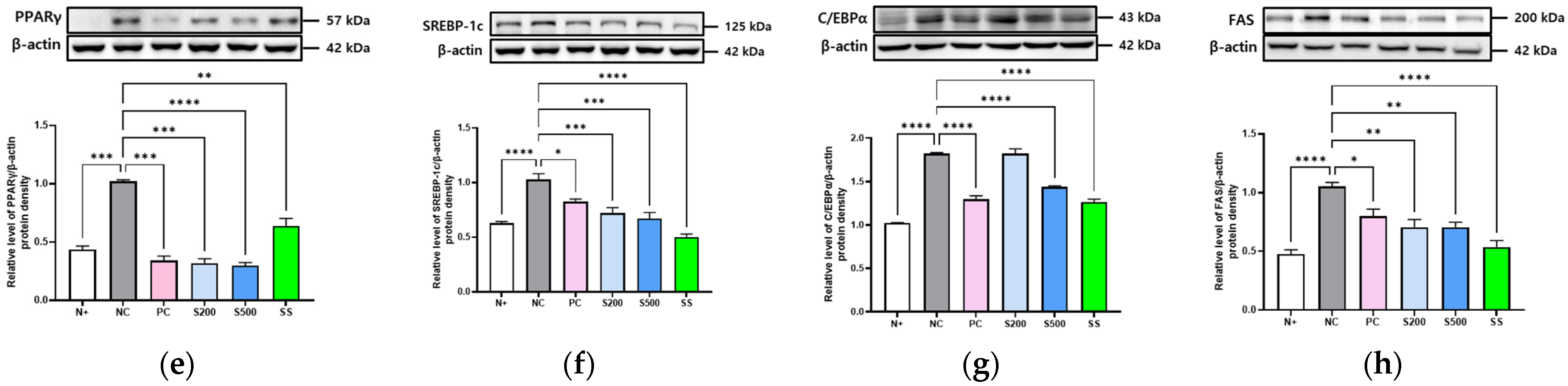
3.2. S and SS Attenuated Expressions of Hepatic Lipid Genes in db/db Mice
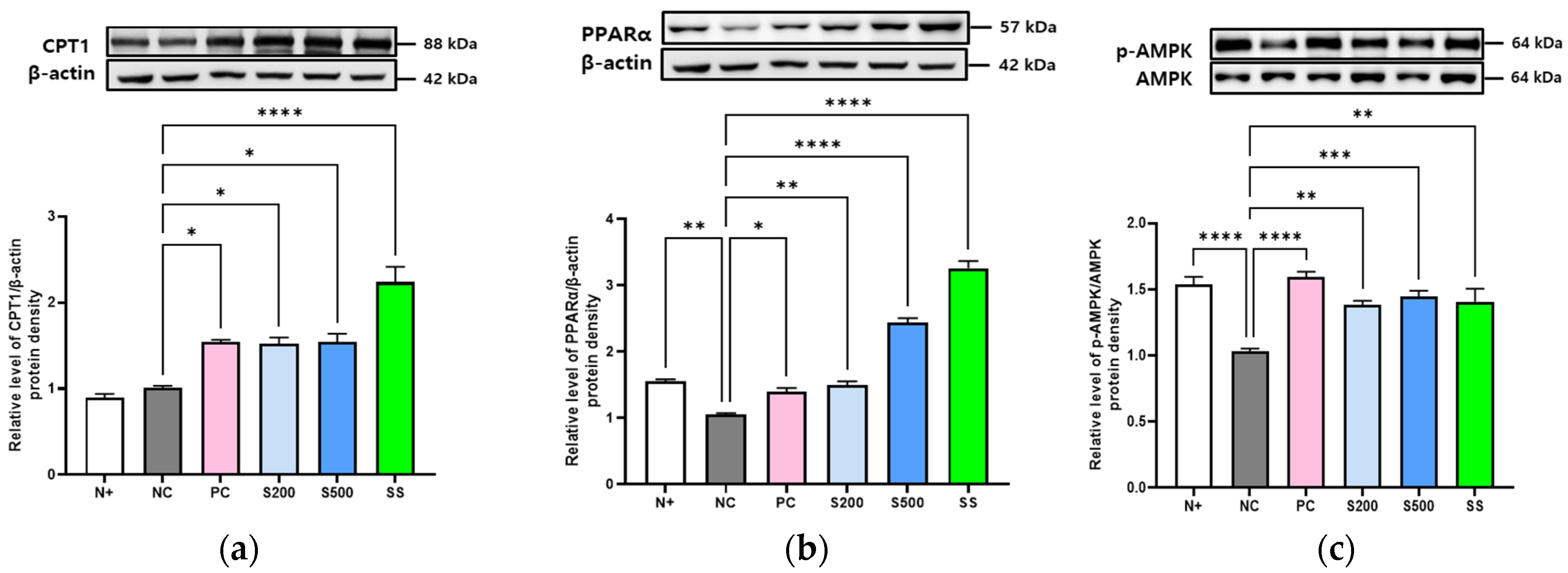
3.3. S and SS Activated Lipid Metabolism and AMPK Phosphorylation in db/db Mice
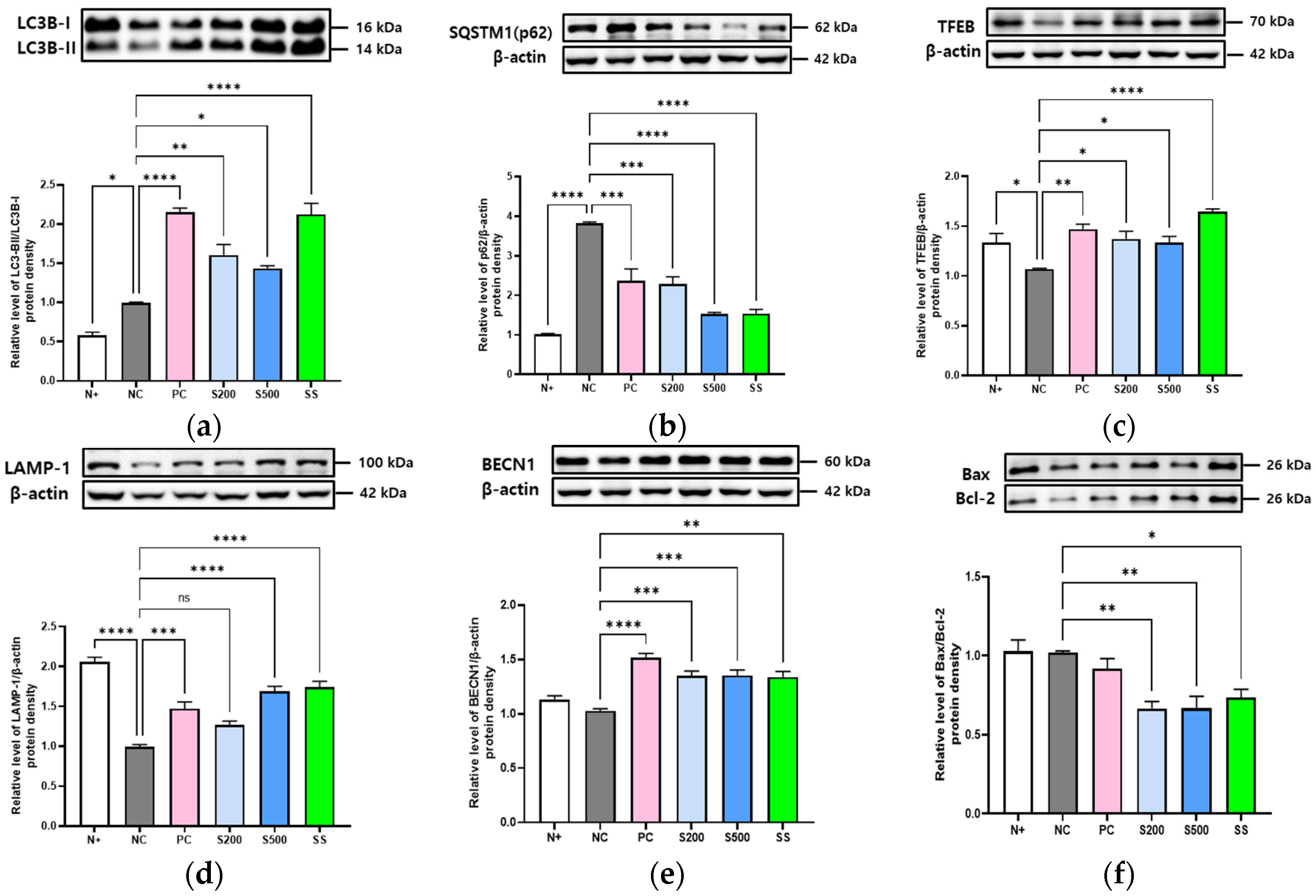
3.4. S- and SS-Induced Autophagy in Liver of db/db Mice
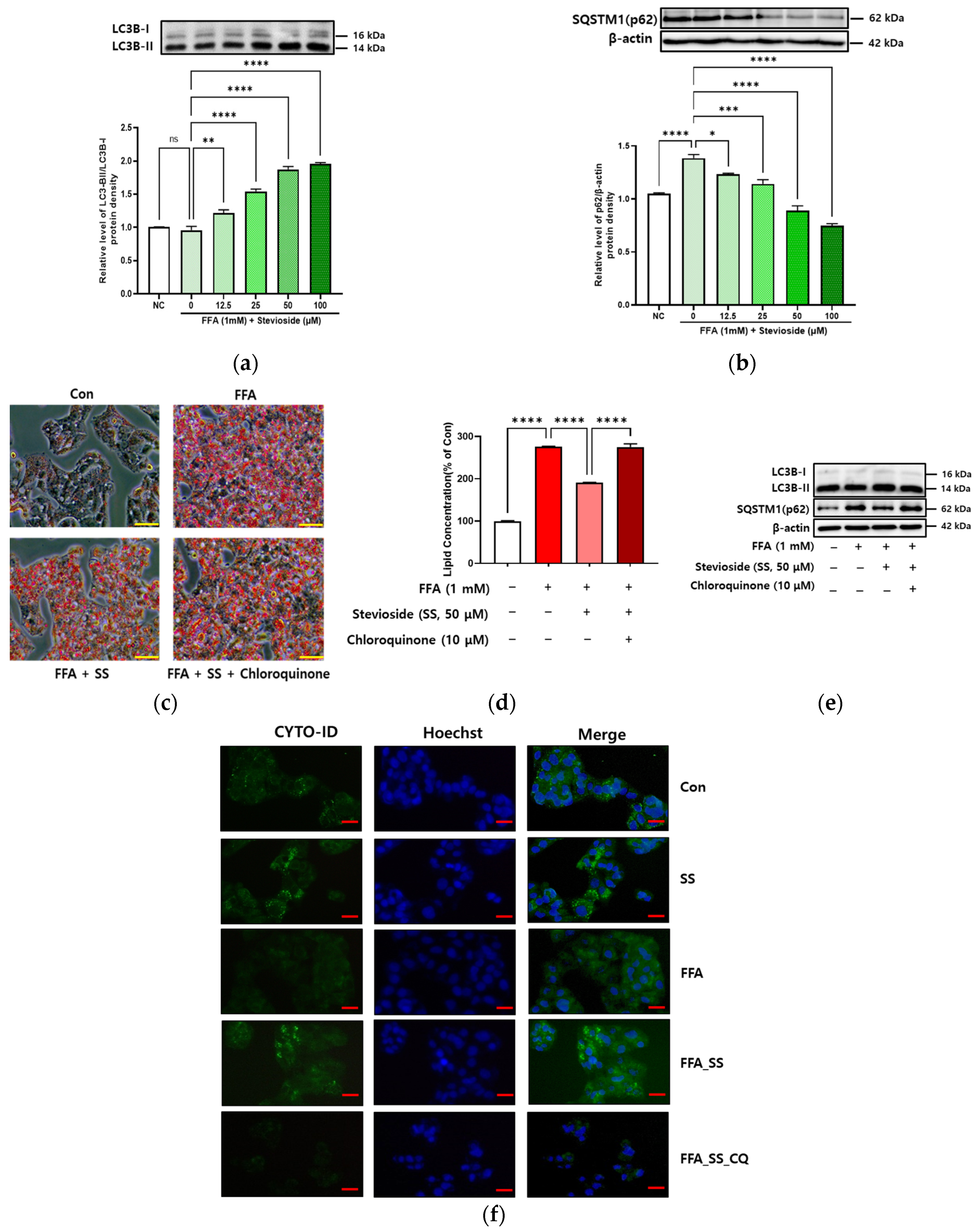
3.5. SS-Induced Autophagy in Steatosis-Induced Hepatocytes
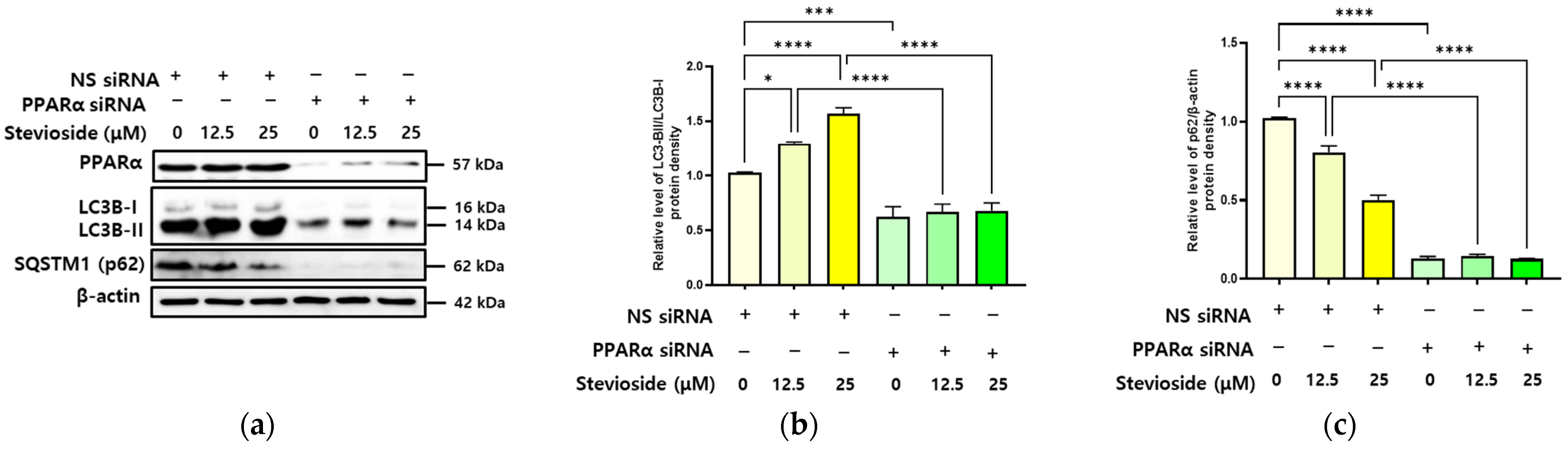
3.6. SS-Induced Autophagy Is Dependent on PPARα in Hepatocytes
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Abd El-Kader, S.M.; El-Den Ashmawy, E.M.S. Non-alcoholic fatty liver disease: The diagnosis and management. World J. Hepatol. 2015, 7, 846–858. [Google Scholar] [CrossRef]
- Nassir, F.; Rector, R.S.; Hammoud, G.M.; Ibdah, J.A. Pathogenesis and Prevention of Hepatic Steatosis. Gastroenterol. Hepatol. 2015, 11, 167–175. [Google Scholar]
- Benedict, M.; Zhang, X. Non-alcoholic fatty liver disease: An expanded review. World J. Hepatol. 2017, 9, 715–732. [Google Scholar] [CrossRef]
- Portillo-Sanchez, P.; Bril, F.; Maximos, M.; Lomonaco, R.; Biernacki, D.; Orsak, B.; Subbarayan, S.; Webb, A.; Hecht, J.; Cusi, K. High Prevalence of Nonalcoholic Fatty Liver Disease in Patients With Type 2 Diabetes Mellitus and Normal Plasma Aminotransferase Levels. J. Clin. Endocrinol. Metab. 2015, 100, 2231–2238. [Google Scholar] [CrossRef]
- Mao, Y.; Yu, F.; Wang, J.; Guo, C.; Fan, X. Autophagy: A new target for nonalcoholic fatty liver disease therapy. Hepatic Med. Evid. Res. 2016, 8, 27–37. [Google Scholar] [CrossRef] [Green Version]
- Sinha, R.A.; Farah, B.L.; Singh, B.K.; Siddique, M.M.; Li, Y.; Wu, Y.; Ilkayeva, O.R.; Gooding, J.; Ching, J.; Zhou, J.; et al. Caffeine stimulates hepatic lipid metabolism by the autophagy-lysosomal pathway in mice. Hepatology 2014, 59, 1366–1380. [Google Scholar] [CrossRef]
- Rogov, V.; Dötsch, V.; Johansen, T.; Kirkin, V. Interactions between autophagy receptors and ubiquitin-like proteins form the molecular basis for selective autophagy. Mol. Cell 2014, 53, 167–178. [Google Scholar] [CrossRef] [Green Version]
- Cole, N.B.; Murphy, D.D.; Grider, T.; Rueter, S.; Brasaemle, D.; Nussbaum, R.L. Lipid droplet binding and oligomerization properties of the Parkinson's disease protein alpha-synuclein. J. Biol. Chem. 2002, 277, 6344–6352. [Google Scholar] [CrossRef] [Green Version]
- Filipe, A.; McLauchlan, J. Hepatitis C virus and lipid droplets: Finding a niche. Trends Mol. Med. 2015, 21, 34–42. [Google Scholar] [CrossRef]
- Martinez-Lopez, N.; Singh, R. Autophagy and Lipid Droplets in the Liver. Annu. Rev. Nutr. 2015, 35, 215–237. [Google Scholar] [CrossRef]
- Kounakis, K.; Chaniotakis, M.; Markaki, M.; Tavernarakis, N. Emerging Roles of Lipophagy in Health and Disease. Front. Cell Dev. Biol. 2019, 7, 185. [Google Scholar] [CrossRef]
- Zhang, S.; Peng, X.; Yang, S.; Li, X.; Huang, M.; Wei, S.; Liu, J.; He, G.; Zheng, H.; Yang, L.; et al. The regulation, function, and role of lipophagy, a form of selective autophagy, in metabolic disorders. Cell Death Dis. 2022, 13, 132. [Google Scholar] [CrossRef]
- Salehi, B.; López, M.D.; Martínez-López, S.; Victoriano, M.; Sharifi-Rad, J.; Martorell, M.; Rodrigues, C.F.; Martins, N. Stevia rebaudiana Bertoni bioactive effects: From in vivo to clinical trials towards future therapeutic approaches. Phytother. Res. PTR 2019, 33, 2904–2917. [Google Scholar] [CrossRef]
- Iatridis, N.; Kougioumtzi, A.; Vlataki, K.; Papadaki, S.; Magklara, A. Anti-Cancer Properties of Stevia rebaudiana; More than a Sweetener. Molecules 2022, 27, 1362. [Google Scholar] [CrossRef]
- Purkayastha, S.; Markosyan, A.; Prakash, I.; Bhusari, S.; Pugh, G., Jr.; Lynch, B.; Roberts, A. Steviol glycosides in purified stevia leaf extract sharing the same metabolic fate. Regul. Toxicol. Pharmacol. RTP 2016, 77, 125–133. [Google Scholar] [CrossRef]
- González, C.; Tapia, M.; Pérez, E.; Pallet, D.; Dornier, M. Main properties of steviol glycosides and their potential in the food industry: A review. Fruits 2014, 69, 127–141. [Google Scholar] [CrossRef] [Green Version]
- Alavala, S.; Nalban, N.; Sangaraju, R.; Kuncha, M.; Jerald, M.K.; Kilari, E.K.; Sistla, R. Anti-inflammatory effect of stevioside abates Freund’s complete adjuvant (FCA)-induced adjuvant arthritis in rats. Inflammopharmacology 2020, 28, 1579–1597. [Google Scholar] [CrossRef]
- Pariwat, P.; Homvisasevongsa, S.; Muanprasat, C.; Chatsudthipong, V. A natural plant-derived dihydroisosteviol prevents cholera toxin-induced intestinal fluid secretion. J. Pharmacol. Exp. Ther. 2008, 324, 798–805. [Google Scholar] [CrossRef] [Green Version]
- Ferri, L.A.; Alves-Do-Prado, W.; Yamada, S.S.; Gazola, S.; Batista, M.R.; Bazotte, R.B. Investigation of the antihypertensive effect of oral crude stevioside in patients with mild essential hypertension. Phytother. Res. PTR 2006, 20, 732–736. [Google Scholar] [CrossRef]
- Ahmad, U.; Ahmad, R.S.; Arshad, M.S.; Mushtaq, Z.; Hussain, S.M.; Hameed, A. Antihyperlipidemic efficacy of aqueous extract of Stevia rebaudiana Bertoni in albino rats. Lipids Health Dis. 2018, 17, 175. [Google Scholar] [CrossRef] [Green Version]
- Nishina, P.M.; Lowe, S.; Wang, J.; Paigen, B. Characterization of plasma lipids in genetically obese mice: The mutants obese, diabetes, fat, tubby, and lethal yellow. Metabolism 1994, 43, 549–553. [Google Scholar] [CrossRef]
- Parzych, K.R.; Klionsky, D.J. An overview of autophagy: Morphology, mechanism, and regulation. Antioxid. Redox Signal. 2014, 20, 460–473. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, P.; Leray, V.; Diez, M.; Serisier, S.; Bloc’h, J.L.; Siliart, B.; Dumon, H. Liver lipid metabolism. J. Anim. Physiol. Anim. Nutr. 2008, 92, 272–283. [Google Scholar] [CrossRef]
- Mashek, D.G. Hepatic lipid droplets: A balancing act between energy storage and metabolic dysfunction in NAFLD. Mol. Metab. 2021, 50, 101115. [Google Scholar] [CrossRef]
- Xie, Y.; Li, J.; Kang, R.; Tang, D. Interplay Between Lipid Metabolism and Autophagy. Front. Cell Dev. Biol. 2020, 8, 431. [Google Scholar] [CrossRef]
- Onal, G.; Kutlu, O.; Gozuacik, D.; Dokmeci Emre, S. Lipid Droplets in Health and Disease. Lipids Health Dis. 2017, 16, 128. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Zhang, X.; Zheng, L.; Kou, J.; Zhong, Z.; Jiang, Y.; Wang, W.; Dong, Z.; Liu, Z.; Han, X.; et al. Hypericin-mediated sonodynamic therapy induces autophagy and decreases lipids in THP-1 macrophage by promoting ROS-dependent nuclear translocation of TFEB. Cell Death Dis. 2016, 7, e2527. [Google Scholar] [CrossRef] [Green Version]
- Moruno-Manchon, J.F.; Uzor, N.E.; Kesler, S.R.; Wefel, J.S.; Townley, D.M.; Nagaraja, A.S.; Pradeep, S.; Mangala, L.S.; Sood, A.K.; Tsvetkov, A.S. TFEB ameliorates the impairment of the autophagy-lysosome pathway in neurons induced by doxorubicin. Aging 2016, 8, 3507–3519. [Google Scholar] [CrossRef] [Green Version]
- Settembre, C.; De Cegli, R.; Mansueto, G.; Saha, P.K.; Vetrini, F.; Visvikis, O.; Huynh, T.; Carissimo, A.; Palmer, D.; Klisch, T.J.; et al. TFEB controls cellular lipid metabolism through a starvation-induced autoregulatory loop. Nat. Cell Biol. 2013, 15, 647–658. [Google Scholar] [CrossRef] [Green Version]
- Montagner, A.; Polizzi, A.; Fouché, E.; Ducheix, S.; Lippi, Y.; Lasserre, F.; Barquissau, V.; Régnier, M.; Lukowicz, C.; Benhamed, F.; et al. Liver PPARα is crucial for whole-body fatty acid homeostasis and is protective against NAFLD. Gut 2016, 65, 1202–1214. [Google Scholar] [CrossRef] [Green Version]
- Sinha, R.A.; Rajak, S.; Singh, B.K.; Yen, P.M. Hepatic Lipid Catabolism via PPARα-Lysosomal Crosstalk. Int. J. Mol. Sci. 2020, 21, 2391. [Google Scholar] [CrossRef]
- Feige, J.N.; Gelman, L.; Michalik, L.; Desvergne, B.; Wahli, W. From molecular action to physiological outputs: Peroxisome proliferator-activated receptors are nuclear receptors at the crossroads of key cellular functions. Prog. Lipid Res. 2006, 45, 120–159. [Google Scholar] [CrossRef] [PubMed]
- Geuns, J.M. Stevioside. Phytochemistry 2003, 64, 913–921. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhu, W.; Cai, J.; Liu, W.; Akihisa, T.; Li, W.; Kikuchi, T.; Xu, J.; Feng, F.; Zhang, J. The role of metabolites of steviol glycosides and their glucosylated derivatives against diabetes-related metabolic disorders. Food Funct. 2021, 12, 8248–8259. [Google Scholar] [CrossRef]
- Xia, H.M.; Wang, J.; Xie, X.J.; Xu, L.J.; Tang, S.Q. Green tea polyphenols attenuate hepatic steatosis, and reduce insulin resistance and inflammation in high-fat diet-induced rats. Int. J. Mol. Med. 2019, 44, 1523–1530. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Zhu, M.-Z.; Zhang, Y.-B.; Wen, B.-B.; An, H.-M.; Ou, X.-C.; Xiong, Y.-F.; Lin, H.-Y.; Liu, Z.-H.; Huang, J.-A. Coadministration of epigallocatechin-3-gallate (EGCG) and caffeine in low dose ameliorates obesity and nonalcoholic fatty liver disease in obese rats. Phytother. Res. 2019, 33, 1019–1026. [Google Scholar] [CrossRef] [PubMed]
- Khoo, W.Y.; Chrisfield, B.J.; Sae-tan, S.; Lambert, J.D. Mitigation of nonalcoholic fatty liver disease in high-fat-fed mice by the combination of decaffeinated green tea extract and voluntary exercise. J. Nutr. Biochem. 2020, 76, 108262. [Google Scholar] [CrossRef]
- Park, M.; Yoo, J.H.; Lee, Y.S.; Lee, H.J. Lonicera caerulea Extract Attenuates Non-Alcoholic Fatty Liver Disease in Free Fatty Acid-Induced HepG2 Hepatocytes and in High Fat Diet-Fed Mice. Nutrients 2019, 11, 494. [Google Scholar] [CrossRef] [Green Version]
- Park, M.; Yoo, J.H.; Lee, Y.S.; Park, E.J.; Lee, H.J. Ameliorative effects of black ginseng on nonalcoholic fatty liver disease in free fatty acid-induced HepG2 cells and high-fat/high-fructose diet-fed mice. J. Ginseng. Res. 2020, 44, 350–361. [Google Scholar] [CrossRef]
- Liu, Q.; Pan, R.; Ding, L.; Zhang, F.; Hu, L.; Ding, B.; Zhu, L.; Xia, Y.; Dou, X. Rutin exhibits hepatoprotective effects in a mouse model of non-alcoholic fatty liver disease by reducing hepatic lipid levels and mitigating lipid-induced oxidative injuries. Int. Immunopharmacol. 2017, 49, 132–141. [Google Scholar] [CrossRef]
- Gao, J.; Chen, S.; Qiu, Z.; Fang, L.; Zhang, L.; Guo, C.; Chen, T.; Qiu, L. Myricitrin ameliorates ethanol-induced steatosis in mouse AML12 liver cells by activating AMPK, and reducing oxidative stress and expression of inflammatory cytokines. Mol. Med. Rep. 2018, 17, 7381–7387. [Google Scholar] [CrossRef] [PubMed]
- Li, H.-Y.; Gan, R.-Y.; Shang, A.; Mao, Q.-Q.; Sun, Q.-C.; Wu, D.-T.; Geng, F.; He, X.-Q.; Li, H.-B. Plant-Based Foods and Their Bioactive Compounds on Fatty Liver Disease: Effects, Mechanisms, and Clinical Application. Oxid. Med. Cell Longev. 2021, 2021, 6621644. [Google Scholar] [CrossRef] [PubMed]
- Bagherniya, M.; Nobili, V.; Blesso, C.N.; Sahebkar, A. Medicinal plants and bioactive natural compounds in the treatment of non-alcoholic fatty liver disease: A clinical review. Pharmacol. Res. 2018, 130, 213–240. [Google Scholar] [CrossRef]
- Allaire, M.; Rautou, P.E.; Codogno, P.; Lotersztajn, S. Autophagy in liver diseases: Time for translation? J. Hepatol. 2019, 70, 985–998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwanten, W.J.; Martinet, W.; Michielsen, P.P.; Francque, S.M. Role of autophagy in the pathophysiology of nonalcoholic fatty liver disease: A controversial issue. World J. Gastroenterol. 2014, 20, 7325–7338. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.M.; Wagner, M.; Xiao, R.; Kim, K.H.; Feng, D.; Lazar, M.A.; Moore, D.D. Nutrient-sensing nuclear receptors coordinate autophagy. Nature 2014, 516, 112–115. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.H.; Kim, G.; Han, D.H.; Lee, M.; Kim, I.; Kim, B.; Kim, K.H.; Song, Y.M.; Yoo, J.E.; Wang, H.J.; et al. Ezetimibe ameliorates steatohepatitis via AMP activated protein kinase-TFEB-mediated activation of autophagy and NLRP3 inflammasome inhibition. Autophagy 2017, 13, 1767–1781. [Google Scholar] [CrossRef] [PubMed]
- Rusten, T.E.; Stenmark, H. p62, an autophagy hero or culprit? Nat. Cell Biol. 2010, 12, 207–209. [Google Scholar] [CrossRef]
- Kang, R.; Zeh, H.J.; Lotze, M.T.; Tang, D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011, 18, 571–580. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, M.; Sharma, A.; Baek, H.; Han, J.-Y.; Yu, J.; Lee, H.-J. Stevia and Stevioside Attenuate Liver Steatosis through PPARα-Mediated Lipophagy in db/db Mice Hepatocytes. Antioxidants 2022, 11, 2496. https://doi.org/10.3390/antiox11122496
Park M, Sharma A, Baek H, Han J-Y, Yu J, Lee H-J. Stevia and Stevioside Attenuate Liver Steatosis through PPARα-Mediated Lipophagy in db/db Mice Hepatocytes. Antioxidants. 2022; 11(12):2496. https://doi.org/10.3390/antiox11122496
Chicago/Turabian StylePark, Miey, Anshul Sharma, Hana Baek, Jin-Young Han, Junho Yu, and Hae-Jeung Lee. 2022. "Stevia and Stevioside Attenuate Liver Steatosis through PPARα-Mediated Lipophagy in db/db Mice Hepatocytes" Antioxidants 11, no. 12: 2496. https://doi.org/10.3390/antiox11122496