
Oxidized High-Density Lipoprotein Induces Endothelial Fibrosis Promoting Hyperpermeability, Hypotension, and Increased Mortality
 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Human Artery Endothelial Cell Culture and mRNA and Protein Expression Determination
2.2. Oxidization of HDL
2.3. Small Interfering RNA and Transfection
2.4. Cell Death, Viability, Proliferation, and Migration
2.5. In Vitro Permeability Assay
2.6. Animals, Lipoprotein Administration, Blood Samples, and Parameter Recordings
2.7. Systolic Blood Pressure and Heart Rate
2.8. Primary Rat Mesenteric Artery Endothelial Cell (RMAEC) Isolation
2.9. In Vivo Permeability Assay
2.10. Antioxidant Diet
2.11. Patients and Volunteers
2.12. CECs Separation and Protein Expression Determination by Flow Cytometry
2.13. Reagents and Inhibitors
2.14. Statistical Analysis
3. Results
3.1. OxHDL Induces Endothelial Fibrosis through the LOX-1/NOX-2/ROS Pathway
3.2. OxHDL Induces Endothelial Fibrosis through the TGF-β 1/2 Secretion/ALK-5/Smad Protein Pathway
3.3. OxHDL Induces Endothelial Hyperpermeability through the LOX-1/NOX-2/ROS Pathway
3.4. In Vivo oxHDL Administration Induces Endothelial Fibrosis in Rats
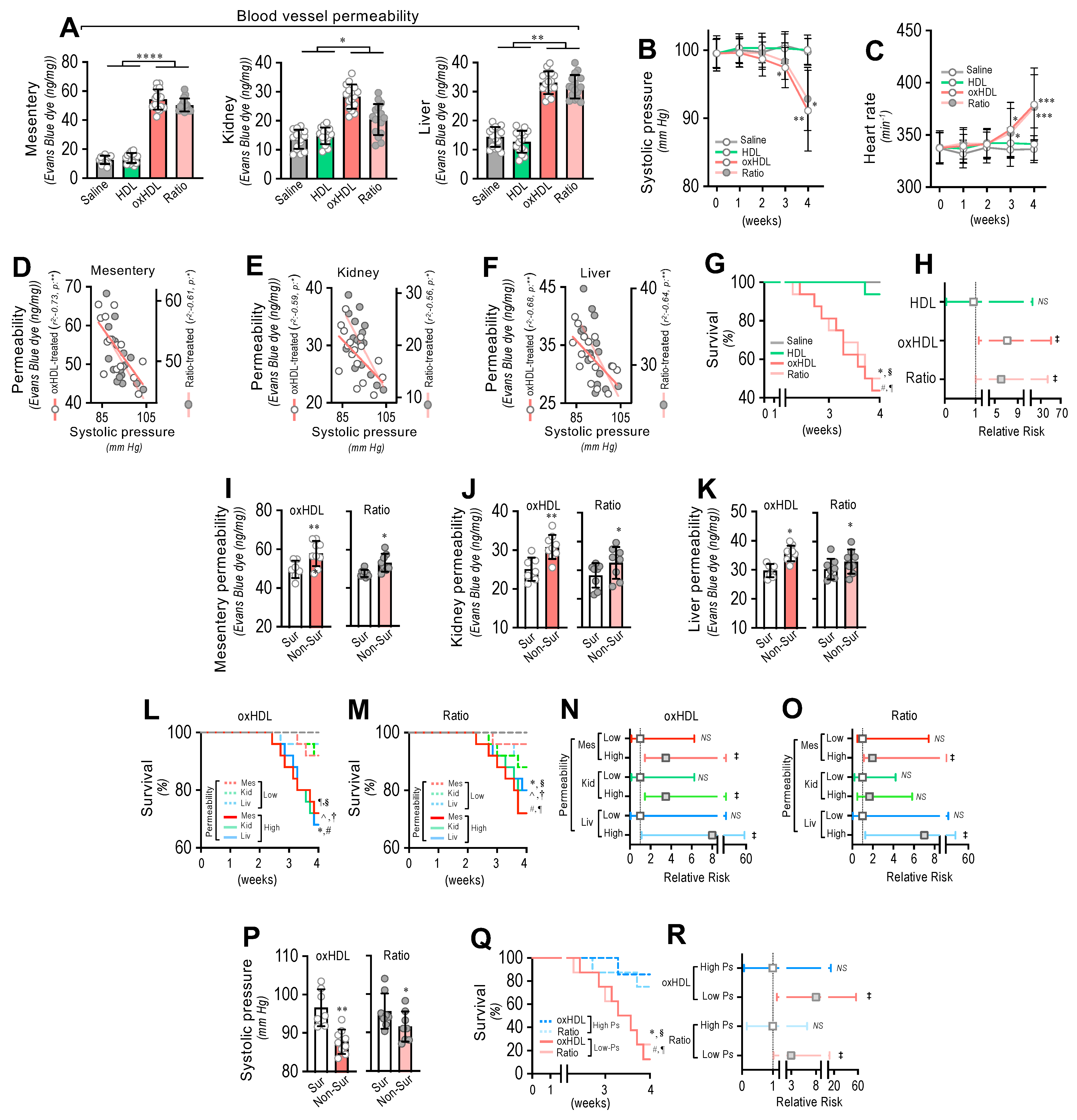
3.5. In Vivo oxHDL Administration Induces Blood Vessel Hyperpermeability, Hypotension, Increased Risk of Death and Decreased Survival in Rats
3.6. In Vivo Inhibition of oxHDL-Induced Endothelial Fibrosis Prevented Blood Vessel Hyperpermeability, Hypotension, Death, and Risk of Death in Rats
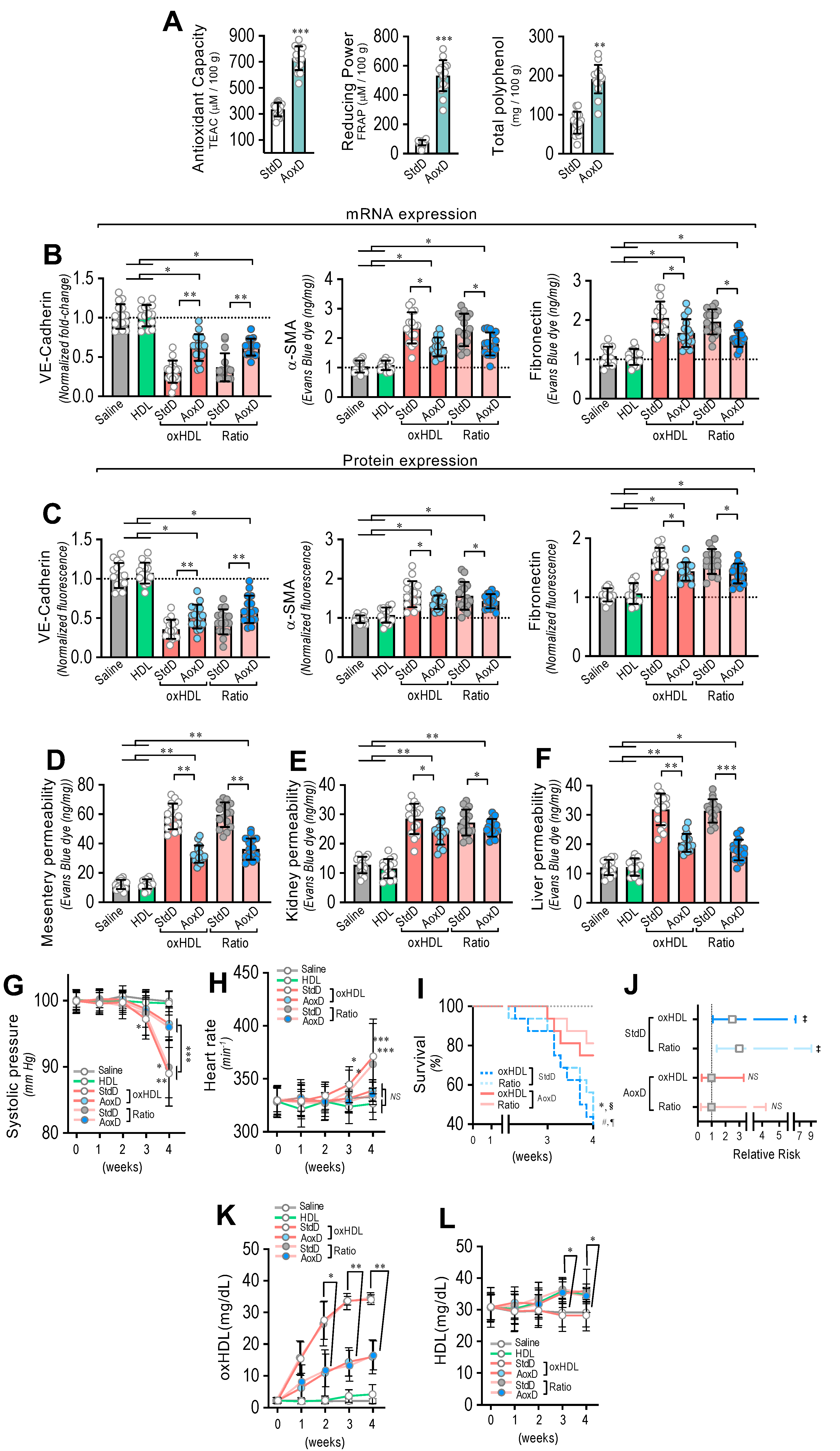
3.7. Antioxidant Diet Consumption Inhibits oxHDL-Induced Endothelial Fibrosis and Prevents Blood Vessel Hyperpermeability, Hypotension, Death, and Risk of Death in Rats
3.8. Circulating Endothelial Cells from ICU Patients Show a Fibrotic Expression Pattern, Which Correlates with Plasma oxHDL Levels Associated with Decreased Survival and Increased Risk of Death
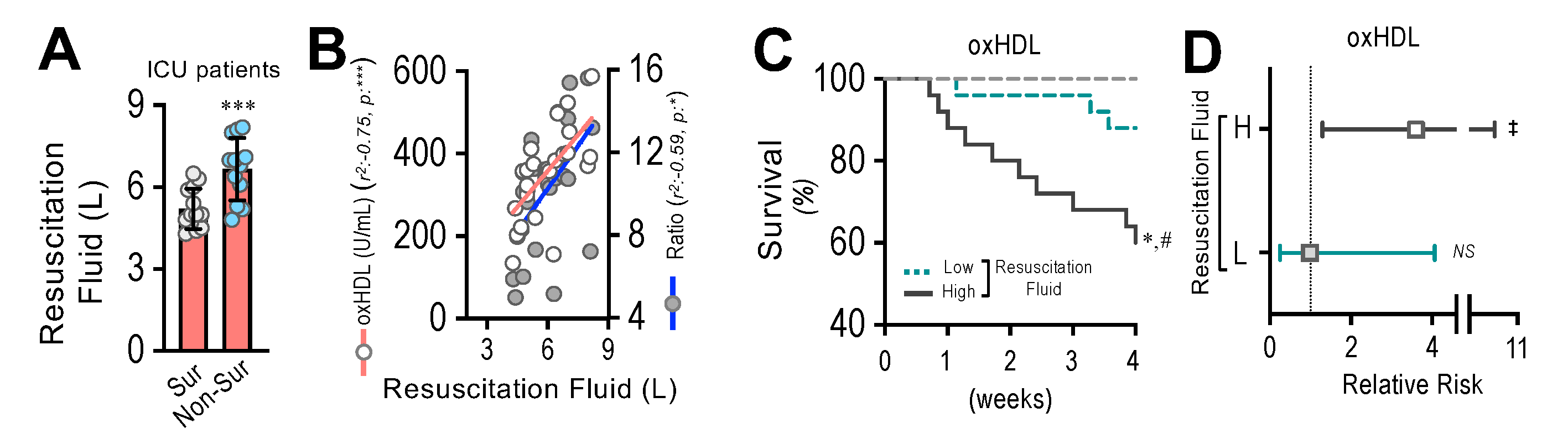
3.9. Resuscitation Fluid Dose Administered to ICU Patients Correlates with Plasma oxHDL and oxHDL/HDL Ratio Levels, Which Is Associated with Decreased Survival and Increased Risk of Death
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nestel, P.J. Dietary Cholesterol and Plasma Lipoproteins. Atherosclerosis 1994, 109, 87. [Google Scholar] [CrossRef]
- Miller, G.J. High-Density Lipoprotein, Low-Density Lipoprotein, and Coronary Heart Disease. Thorax 1978, 33, 137–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Connell, B.J.; Genest, J. High-Density Lipoproteins and Endothelial Function. Circulation 2001, 104, 1978–1983. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Xu, Y.; Chen, J.; Zhao, M.; Rye, K.-A. HDL and Endothelial Function. Adv. Exp. Med. Biol. 2022, 1377, 27–47. [Google Scholar] [PubMed]
- Norata, G.D.; Raselli, S.; Catapano, A.L. HDL and Endothelial Function: From Molecular Mechanisms to Clinical Observations. Future Lipidol. 2006, 1, 343–355. [Google Scholar] [CrossRef]
- Tran-Dinh, A.; Diallo, D.; Delbosc, S.; Varela-Perez, L.M.; Dang, Q.B.; Lapergue, B.; Burillo, E.; Michel, J.B.; Levoye, A.; Martin-Ventura, J.L.; et al. HDL and Endothelial Protection. Br. J. Pharmacol. 2013, 169, 493–511. [Google Scholar] [CrossRef] [Green Version]
- Pérez, L.; Muñoz-Durango, N.; Riedel, C.A.; Echeverría, C.; Kalergis, A.M.; Cabello-Verrugio, C.; Simon, F. Endothelial-to-Mesenchymal Transition: Cytokine-Mediated Pathways That Determine Endothelial Fibrosis under Inflammatory Conditions. Cytokine Growth Factor Rev. 2017, 33, 41–54. [Google Scholar] [CrossRef]
- Domínguez-Álvarez, M.; Gea, J.; Barreiro, E. Inflammatory Events and Oxidant Production in the Diaphragm, Gastrocnemius, and Blood of Rats Exposed to Chronic Intermittent Hypoxia: Therapeutic Strategies: Muscle and Systemic Hypoxia-induced Inflammation and Ros. J. Cell. Physiol. 2016, 232, 1165–1175. [Google Scholar] [CrossRef] [Green Version]
- Higashi, Y.; Noma, K.; Yoshizumi, M.; Kihara, Y. Endothelial Function and Oxidative Stress in Cardiovascular Diseases. Circ. J. 2009, 73, 411–418. [Google Scholar] [CrossRef] [Green Version]
- Closa, D.; Folch-Puy, E. Oxygen Free Radicals and the Systemic Inflammatory Response. IUBMB Life (Int. Union Biochem. Mol. Biol. Life) 2004, 56, 185–191. [Google Scholar] [CrossRef]
- Tsimikas, S.; Miller, Y.I. Oxidative Modification of Lipoproteins: Mechanisms, Role in Inflammation and Potential Clinical Applications in Cardiovascular Disease. Curr. Pharm. Des. 2011, 17, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Pirillo, A.; Norata, G.D.; Catapano, A.L. LOX-1, OxLDL, and Atherosclerosis. Mediat. Inflamm. 2013, 2013, 152786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakano, T.; Nagata, A. Oxidative Susceptibility of Apolipoprotein AI in Serum. Clin. Chim. Acta. 2005, 362, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Speer, T.; Rohrer, L.; Blyszczuk, P.; Shroff, R.; Kuschnerus, K.; Kränkel, N.; Kania, G.; Zewinger, S.; Akhmedov, A.; Shi, Y.; et al. Abnormal High-Density Lipoprotein Induces Endothelial Dysfunction via Activation of Toll-like Receptor-2. Immunity 2013, 38, 754–768. [Google Scholar] [CrossRef] [Green Version]
- Shroff, R.; Speer, T.; Colin, S.; Charakida, M.; Zewinger, S.; Staels, B.; Chinetti-Gbaguidi, G.; Hettrich, I.; Rohrer, L.; O’Neill, F.; et al. HDL in Children with CKD Promotes Endothelial Dysfunction and an Abnormal Vascular Phenotype. J. Am. Soc. Nephrol. 2014, 25, 2658–2668. [Google Scholar] [CrossRef] [Green Version]
- Pérez, L.; Vallejos, A.; Echeverría, C.; Varela, D.; Cabello-Verrugio, C.; Simon, F. OxHDL Controls LOX-1 Expression and Plasma Membrane Localization through a Mechanism Dependent on NOX/ROS/NF-ΚB Pathway on Endothelial Cells. Lab. Invest. 2019, 99, 421–437. [Google Scholar] [CrossRef]
- Matsunaga, T.; Hokari, S.; Koyama, I.; Harada, T.; Komoda, T. NF-KB Activation in Endothelial Cells Treated with Oxidized High-Density Lipoprotein. Biochem. Biophys. Res. Commun. 2003, 303, 313–319. [Google Scholar] [CrossRef]
- Pirillo, A.; Reduzzi, A.; Ferri, N.; Kuhn, H.; Corsini, A.; Catapano, A.L. Upregulation of Lectin-like Oxidized Low-Density Lipoprotein Receptor-1 (LOX-1) by 15-Lipoxygenase-Modified LDL in Endothelial Cells. Atherosclerosis 2011, 214, 331–337. [Google Scholar] [CrossRef]
- Pirillo, A.; Uboldi, P.; Ferri, N.; Corsini, A.; Kuhn, H.; Catapano, A.L. Upregulation of Lectin-like Oxidized Low Density Lipoprotein Receptor 1 (LOX-1) Expression in Human Endothelial Cells by Modified High Density Lipoproteins. Biochem. Biophys. Res. Commun. 2012, 428, 230–233. [Google Scholar] [CrossRef]
- Zeisberg, E.M.; Tarnavski, O.; Zeisberg, M.; Dorfman, A.L.; McMullen, J.R.; Gustafsson, E.; Chandraker, A.; Yuan, X.; Pu, W.T.; Roberts, A.B.; et al. Endothelial-to-Mesenchymal Transition Contributes to Cardiac Fibrosis. Nat. Med. 2007, 13, 952–961. [Google Scholar] [CrossRef]
- Maleszewska, M.; Moonen, J.-R.A.J.; Huijkman, N.; van de Sluis, B.; Krenning, G.; Harmsen, M.C. IL-1β and TGFβ2 Synergistically Induce Endothelial to Mesenchymal Transition in an NFκB-Dependent Manner. Immunobiology 2013, 218, 443–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Echeverría, C.; Montorfano, I.; Tapia, P.; Riedel, C.; Cabello-Verrugio, C.; Simon, F. Endotoxin-Induced Endothelial Fibrosis Is Dependent on Expression of Transforming Growth Factors Β1 and Β2. Infect. Immun. 2014, 82, 3678–3686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Echeverría, C.; Montorfano, I.; Sarmiento, D.; Becerra, A.; Nuñez-Villena, F.; Figueroa, X.F.; Cabello-Verrugio, C.; Elorza, A.A.; Riedel, C.; Simon, F. Lipopolysaccharide Induces a Fibrotic-like Phenotype in Endothelial Cells. J. Cell. Mol. Med. 2013, 17, 800–814. [Google Scholar] [CrossRef] [PubMed]
- Zeisberg, E.M.; Potenta, S.; Xie, L.; Zeisberg, M.; Kalluri, R. Discovery of Endothelial to Mesenchymal Transition as a Source for Carcinoma-Associated Fibroblasts. Cancer Res. 2007, 67, 10123–10128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trzeciak, S.; Dellinger, R.P.; Parrillo, J.E.; Guglielmi, M.; Bajaj, J.; Abate, N.L.; Arnold, R.C.; Colilla, S.; Zanotti, S.; Hollenberg, S.M. Early Microcirculatory Perfusion Derangements in Patients with Severe Sepsis and Septic Shock: Relationship to Hemodynamics, Oxygen Transport, and Survival. Ann. Emerg. Med. 2007, 49, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Levy, M.M.; Evans, L.E.; Rhodes, A. The Surviving Sepsis Campaign Bundle: 2018 Update. Intensive Care Med. 2018, 44, 925–928. [Google Scholar] [CrossRef] [Green Version]
- Levy, M.M.; Evans, L.E.; Rhodes, A. The Surviving Sepsis Campaign Bundle: 2018 Update. Crit. Care Med. 2018, 46, 997–1000. [Google Scholar] [CrossRef]
- Hollenberg, S.M.; Ahrens, T.S.; Annane, D.; Astiz, M.E.; Chalfin, D.B.; Dasta, J.F.; Heard, S.O.; Martin, C.; Napolitano, L.M.; Susla, G.M.; et al. Practice Parameters for Hemodynamic Support of Sepsis in Adult Patients: 2004 Update. Crit. Care Med. 2004, 32, 1928–1948. [Google Scholar] [CrossRef]
- Gatica, S.; Villegas, V.; Vallejos, A.; Olivares, P.; Aballai, V.; Lagos-Meza, F.; Echeverría, C.; Cabello-Verrugio, C.; Varela, D.; Simon, F. TRPM7 Mediates Kidney Injury, Endothelial Hyperpermeability and Mortality during Endotoxemia. Lab. Invest. 2020, 100, 234–249. [Google Scholar] [CrossRef]
- Tapia, P.; Gatica, S.; Cortés-Rivera, C.; Otero, C.; Becerra, A.; Riedel, C.A.; Cabello-Verrugio, C.; Kalergis, A.M.; Simon, F. Circulating Endothelial Cells From Septic Shock Patients Convert to Fibroblasts Are Associated With the Resuscitation Fluid Dose and Are Biomarkers for Survival Prediction. Crit. Care Med. 2019, 47, 942–950. [Google Scholar] [CrossRef]
- Scoccia, A.E.; Molinuevo, M.S.; McCarthy, A.D.; Cortizo, A.M. A Simple Method to Assess the Oxidative Susceptibility of Low Density Lipoproteins. BMC Clin. Pathol. 2001, 1, 20963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.-L.; Lai, T.W. Optimization of Evans Blue Quantitation in Limited Rat Tissue Samples. Sci. Rep. 2014, 4, 6588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, P.; Singh, R.P. Evaluation of Antioxidant Activity in Foods with Special Reference to TEAC Method. Am. J. Food Technol. 2013, 8, 83–101. [Google Scholar] [CrossRef] [Green Version]
- Obón, J.M.; Castellar, M.R.; Cascales, J.A.; Fernández-López, J.A. Assessment of the TEAC Method for Determining the Antioxidant Capacity of Synthetic Red Food Colorants. Food Res. Int. 2005, 38, 843–845. [Google Scholar] [CrossRef]
- Benzie, I.F.F.; Strain, J.J. The Ferric Reducing Ability of Plasma (FRAP) as a Measure of “Antioxidant Power”: The FRAP Assay. Anal. Biochem. 1996, 239, 70–76. [Google Scholar] [CrossRef] [Green Version]
- Ring, J.; Chaung, M. The FRAP Assay: Allowing Students to Assess the Anti-Oxidizing Ability of Green Tea and More. J. Stud. Sci. Technol. 2015, 8, 23–26. [Google Scholar] [CrossRef] [Green Version]
- Cheung, L.M.; Cheung, P.C.K.; Ooi, V.E.C. Antioxidant Activity and Total Phenolics of Edible Mushroom Extracts. Food Chem. 2003, 81, 249–255. [Google Scholar] [CrossRef]
- Estes, M.L.; Mund, J.A.; Ingram, D.A.; Case, J. Identification of Endothelial Cells and Progenitor Cell Subsets in Human Peripheral Blood. Curr. Protoc. Cytom. 2001, 9, 9.33.1–9.33.11. [Google Scholar] [CrossRef]
- Simon, F.; Fernández, R. Early Lipopolysaccharide-Induced Reactive Oxygen Species Production Evokes Necrotic Cell Death in Human Umbilical Vein Endothelial Cells. J. Hypertens. 2009, 27, 1202–1216. [Google Scholar] [CrossRef]
- Simon, F.; Stutzin, A. Protein Kinase C-Mediated Phosphorylation of P47phox Modulates Platelet-Derived Growth Factor-Induced H2O2 Generation and Cell Proliferation in Human Umbilical Vein Endothelial Cells. Endothelium 2008, 15, 175–188. [Google Scholar] [CrossRef]
- Becerra, A.; Rojas, M.; Vallejos, A.; Villegas, V.; Pérez, L.; Cabello-Verrugio, C.; Simon, F. Endothelial Fibrosis Induced by Suppressed STAT3 Expression Mediated by Signaling Involving the TGF-Β1/ALK5/Smad Pathway. Lab. Invest. 2017, 97, 1033–1046. [Google Scholar] [CrossRef]
- Santibañez, J.F.; Quintanilla, M.; Bernabeu, C. TGF-β/TGF-β Receptor System and Its Role in Physiological and Pathological Conditions. Clin. Sci. 2011, 121, 233–251. [Google Scholar] [CrossRef] [Green Version]
- Vallejos, A.; Olivares, P.; Gatica, S.; Villegas, V.; Echeverría, C.; Cabello-Verrugio, C.; Simon, F. Endotoxemia-Induced Endothelial Fibrosis Inhibition Improves Hypotension, Tachycardia, Multiple Organ Dysfunction Syndrome, Cytokine Response, Oxidative Stress, and Survival. Lab. Invest. 2019, 99, 1173–1192. [Google Scholar] [CrossRef]
- Gellibert, F.; de Gouville, A.-C.; Woolven, J.; Mathews, N.; Nguyen, V.-L.; Bertho-Ruault, C.; Patikis, A.; Grygielko, E.T.; Laping, N.J.; Huet, S. Discovery of 4-{4-[3-(Pyridin-2-Yl)-1 H-Pyrazol-4-Yl]Pyridin-2-Yl}- N-(Tetrahydro-2 H- Pyran-4-Yl)Benzamide (GW788388): A Potent, Selective, and Orally Active Transforming Growth Factor-β Type I Receptor Inhibitor. J. Med. Chem. 2006, 49, 2210–2221. [Google Scholar] [CrossRef] [PubMed]
- Petersen, M.; Thorikay, M.; Deckers, M.; van Dinther, M.; Grygielko, E.T.; Gellibert, F.; de Gouville, A.C.; Huet, S.; ten Dijke, P.; Laping, N.J. Oral Administration of GW788388, an Inhibitor of TGF-Beta Type I and II Receptor Kinases, Decreases Renal Fibrosis. Kidney Int. 2008, 73, 705–715. [Google Scholar] [CrossRef] [Green Version]
- Holy, E.W.; Besler, C.; Reiner, M.F.; Camici, G.G.; Manz, J.; Beer, J.H.; Luescher, T.F.; Landmesser, U.; Tanner, F.C. High-Density Lipoprotein from Patients with Coronary Heart Disease Loses Anti-Thrombotic Effects on Endothelial Cells: Impact on Arterial Thrombus Formation. Thromb. Haemost. 2014, 112, 1024–1035. [Google Scholar] [CrossRef]
- Spillmann, F.; Miteva, K.; Pieske, B.; Tschöpe, C.; Linthout, S.V. High-Density Lipoproteins Reduce Endothelial-to-Mesenchymal Transition. Arterioscler. Thromb. Vasc. Biol. 2018, 35, 1774–1777. [Google Scholar] [CrossRef] [Green Version]
- Kazakov, A.; Müller, P.; Jagoda, P.; Semenov, A.; Böhm, M.; Laufs, U. Endothelial Nitric Oxide Synthase of the Bone Marrow Regulates Myocardial Hypertrophy, Fibrosis, and Angiogenesis. Cardiovasc. Res. 2012, 93, 397–405. [Google Scholar] [CrossRef] [Green Version]
- Yao, Y.; Wang, Y.; Zhang, Y.; Liu, C. Klotho Ameliorates Oxidized Low Density Lipoprotein (Ox-LDL)-Induced Oxidative Stress via Regulating LOX-1 and PI3K/Akt/ENOS Pathways. Lipids Health Dis. 2017, 16, 77. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, A.; Joseph, B.; Anil, S. Nitric Oxide in the Management of Respiratory Consequences in COVID-19: A Scoping Review of a Different Treatment Approach. Cureus 2022, 14, e23852. [Google Scholar] [CrossRef]
- Nofer, J.-R. High Density Lipoproteins, From Biological Understanding to Clinical Exploitation; Handbook of Experimental Pharmacology; Springer: Berlin/Heidelberg, Germany, 2014; Volume 224, pp. 229–256. [Google Scholar]
- Barreto, J.; Karathanasis, S.K.; Remaley, A.; Sposito, A.C. Role of LOX-1 (Lectin-Like Oxidized Low-Density Lipoprotein Receptor 1) as a Cardiovascular Risk Predictor: Mechanistic Insight and Potential Clinical Use. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 153–166. [Google Scholar] [CrossRef]
- Abe, R.J.; Abe, J.; Nguyen, M.T.H.; Olmsted-Davis, E.A.; Mamun, A.; Banerjee, P.; Cooke, J.P.; Fang, L.; Pownall, H.; Le, N.-T. Free Cholesterol Bioavailability and Atherosclerosis. Curr. Atheroscler. Rep. 2022, 24, 323–336. [Google Scholar] [CrossRef] [PubMed]
- Valiyaveettil, M.; Kar, N.; Ashraf, M.Z.; Byzova, T.V.; Febbraio, M.; Podrez, E.A. Oxidized High-Density Lipoprotein Inhibits Platelet Activation and Aggregation via Scavenger Receptor BI. Blood 2008, 111, 1962–1971. [Google Scholar] [CrossRef] [PubMed]
- Thorne, R.F.; Mhaidat, N.M.; Ralston, K.J.; Burns, G.F. CD36 Is a Receptor for Oxidized High Density Lipoprotein: Implications for the Development of Atherosclerosis. FEBS Lett. 2007, 581, 1227–1232. [Google Scholar] [CrossRef] [Green Version]
- Pirillo, A.; Catapano, A.L.; Norata, G.D. Biological Consequences of Dysfunctional HDL. Curr. Med. Chem. 2018, 25, 1–22. [Google Scholar] [CrossRef]
- Takaeko, Y.; Matsui, S.; Kajikawa, M.; Maruhashi, T.; Kishimoto, S.; Hashimoto, H.; Kihara, Y.; Hida, E.; Chayama, K.; Goto, C.; et al. Association of Extremely High Levels of High-Density Lipoprotein Cholesterol with Endothelial Dysfunction in Men. J. Clin. Lipidol. 2019, 13, 664–672. [Google Scholar] [CrossRef] [Green Version]
- Chiesa, S.T.; Charakida, M.; McLoughlin, E.; Nguyen, H.C.; Georgiopoulos, G.; Motran, L.; Elia, Y.; Marcovecchio, M.L.; Dunger, D.B.; Dalton, R.N.; et al. Elevated High-Density Lipoprotein in Adolescents with Type 1 Diabetes Is Associated with Endothelial Dysfunction in the Presence of Systemic Inflammation. Eur. Heart J. 2019, 40, 3559–3566. [Google Scholar] [CrossRef] [Green Version]
- Peterson, S.J.; Shapiro, J.I.; Thompson, E.; Singh, S.; Liu, L.; Weingarten, J.A.; O’Hanlon, K.; Bialczak, A.; Bhesania, S.R.; Abraham, N.G. Oxidized HDL, Adipokines, and Endothelial Dysfunction: A Potential Biomarker Profile for Cardiovascular Risk in Women with Obesity. Obesity 2019, 27, 87–93. [Google Scholar] [CrossRef] [Green Version]
- Chien, J.-Y.; Jerng, J.-S.; Yu, C.-J.; Yang, P.-C. Low Serum Level of High-Density Lipoprotein Cholesterol Is a Poor Prognostic Factor for Severe Sepsis. Crit. Care Med. 2005, 33, 1688–1693. [Google Scholar] [CrossRef]
- Ebara, S.; Marumo, M.; Yamabata, C.; Nishibe, I.; Soneda, J.-I.; Mukai, J.; Ohki, M.; Uchida, K.; Wakabayashi, I. Inverse Associations of HDL Cholesterol and Oxidized HDL with D-Dimer in Patients with Type 2 Diabetes Mellitus. Thromb. Res. 2017, 155, 12–15. [Google Scholar] [CrossRef]
- Ebara, S.; Marumo, M.; Mukai, J.; Ohki, M.; Uchida, K.; Wakabayashi, I. Relationships of Oxidized HDL with Blood Coagulation and Fibrinolysis in Patients with Type 2 Diabetes Mellitus. J. Thromb. Thrombolysis 2018, 45, 200–205. [Google Scholar] [CrossRef]
- Marsillach, J.; Adorni, M.P.; Zimetti, F.; Papotti, B.; Zuliani, G.; Cervellati, C. HDL Proteome and Alzheimer’s Disease: Evidence of a Link. Antioxidants 2020, 9, 1224. [Google Scholar] [CrossRef]
- Zimetti, F.; Adorni, M.P.; Marsillach, J.; Marchi, C.; Trentini, A.; Valacchi, G.; Cervellati, C. Connection between the Altered HDL Antioxidant and Anti-Inflammatory Properties and the Risk to Develop Alzheimer’s Disease: A Narrative Review. Oxid. Med. Cell Longev. 2021, 2021, 6695796. [Google Scholar] [CrossRef]
- Zuin, M.; Trentini, A.; Marsillach, J.; D’Amuri, A.; Bosi, C.; Roncon, L.; Passaro, A.; Zuliani, G.; Mackness, M.; Cervellati, C. Paraoxonase-1 (PON-1) Arylesterase Activity Levels in Patients with Coronary Artery Disease: A Meta-Analysis. Dis. Markers 2022, 2022, 4264314. [Google Scholar] [CrossRef]
- Montorfano, I.; Becerra, A.; Cerro, R.; Echeverría, C.; Sáez, E.; Morales, M.G.; Fernández, R.; Cabello-Verrugio, C.; Simon, F. Oxidative Stress Mediates the Conversion of Endothelial Cells into Myofibroblasts via a TGF-Β1 and TGF-Β2-Dependent Pathway. Lab. Invest. 2014, 94, 1068–1082. [Google Scholar] [CrossRef] [Green Version]
- Gouville, A.-C.; Boullay, V.; Krysa, G.; Pilot, J.; Brusq, J.-M.; Loriolle, F.; Gauthier, J.-M.; Papworth, S.A.; Laroze, A.; Gellibert, F.; et al. Inhibition of TGF- Βsignaling by an ALK5 Inhibitor Protects Rats from Dimethylnitrosamine-Induced Liver Fibrosis. Br. J. Pharmacol. 2005, 145, 166–177. [Google Scholar] [CrossRef] [Green Version]
- Kunsch, C.; Medford, R.M. Oxidative Stress as a Regulator of Gene Expression in the Vasculature. Circ. Res. 1999, 85, 753–766. [Google Scholar] [CrossRef]
- Huet, O.; Dupic, L.; Harrois, A.; Duranteau, J. Oxidative Stress and Endothelial Dysfunction during Sepsis. Front. Biosci. 2011, 16, 1986–1995. [Google Scholar] [CrossRef]
- Crimi, E.; Sica, V.; Slutsky, A.S.; Zhang, H.; Williams-Ignarro, S.; Ignarro, L.J.; Napoli, C. Role of Oxidative Stress in Experimental Sepsis and Multisystem Organ Dysfunction. Free Radic. Res. 2006, 40, 665–672. [Google Scholar] [CrossRef]
- Singer, P.; Singer, J.A.; Shapiro, H.; Lev, S. Oxidative Stress in the ICU. Curr. Nutr. Food Sci. 2007, 3, 209–215. [Google Scholar] [CrossRef]
- Ayala, J.C.; Grismaldo, A.; Sequeda-Castañeda, L.G.; Aristizábal-Pachón, A.F.; Morales, L. Oxidative Stress in ICU Patients: ROS as Mortality Long-Term Predictor. Antioxidants 2021, 10, 1912. [Google Scholar] [CrossRef]
- Karapetsa, M.; Pitsika, M.; Goutzourelas, N.; Stagos, D.; Becker, A.T.; Zakynthinos, E. Oxidative Status in ICU Patients with Septic Shock. Food Chem. Toxicol. 2013, 61, 106–111. [Google Scholar] [CrossRef] [PubMed]
- Ducastel, M.; Chenevier-Gobeaux, C.; Ballaa, Y.; Meritet, J.-F.; Brack, M.; Chapuis, N.; Pene, F.; Carlier, N.; Szwebel, T.-A.; Roche, N.; et al. Oxidative Stress and Inflammatory Biomarkers for the Prediction of Severity and ICU Admission in Unselected Patients Hospitalized with COVID-19. Int. J. Mol. Sci. 2021, 22, 7462. [Google Scholar] [CrossRef] [PubMed]
- Aird, W.C. Endothelium as an Organ System. Crit. Care Med. 2004, 32, S271–S279. [Google Scholar] [CrossRef]
- Lee, W.L.; Slutsky, A.S. Sepsis and Endothelial Permeability. N. Engl. J. Med. 2010, 363, 689–691. [Google Scholar] [CrossRef] [Green Version]
- Echeverría, C.; Eltit, F.; Santibañez, J.F.; Gatica, S.; Cabello-Verrugio, C.; Simon, F. Endothelial Dysfunction in Pregnancy Metabolic Disorders. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165414. [Google Scholar] [CrossRef]
- Siddall, E.; Khatri, M.; Radhakrishnan, J. Capillary Leak Syndrome: Etiologies, Pathophysiology, and Management. Kidney Int. 2017, 92, 37–46. [Google Scholar] [CrossRef] [Green Version]
- Tapia, P.; Chinchón, E.; Morales, D.; Stehberg, J.; Simon, F. Effectiveness of Short-Term 6-Hour High-Volume Hemofiltration during Refractory Severe Septic Shock. J. Trauma Acute Care Surg. 2012, 72, 1228–1238. [Google Scholar] [CrossRef]
- Spicer, A.; Calfee, C.S. Fixing the Leak: Targeting the Vascular Endothelium in Sepsis. Crit. Care. 2012, 16, 177. [Google Scholar] [CrossRef] [Green Version]
- Xiao, F.; Wang, D.; Kong, L.; Li, M.; Feng, Z.; Shuai, B.; Wang, L.; Wei, Y.; Li, H.; Wu, S.; et al. Intermedin Protects against Sepsis by Concurrently Re-Establishing the Endothelial Barrier and Alleviating Inflammatory Responses. Nat. Commun. 2018, 9, 2644. [Google Scholar] [CrossRef]
- Ziesmann, M.T.; Marshall, J.C. Multiple Organ Dysfunction: The Defining Syndrome of Sepsis. Surg. Infect. 2018, 19, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Englert, J.A.; Fink, M.P. The Multiple Organ Dysfunction Syndrome and Late-Phase Mortality in Sepsis. Curr. Infect. Dis. Rep. 2005, 7, 335–341. [Google Scholar] [CrossRef]
- Kalla, M.; Herring, N. Physiology of Shock and Volume Resuscitation. Surg. Oxf. 2013, 31, 545–551. [Google Scholar] [CrossRef]
- Mameledzija, M.; Nasrallah, E.; Hartrich, M. Hypotension Unresponsive to Fluid Resuscitation: A Case Report. Clin. Pract. Cases Emerg. Med. 2022, 6, 236–239. [Google Scholar] [CrossRef] [PubMed]
- Holt, D.B.; Lardaro, T.; Wang, A.Z.; Musey, P.I.; Trigonis, R.; Bucca, A.; Croft, A.; Glober, N.; Peterson, K.; Schaffer, J.T.; et al. Fluid Resuscitation and Progression to Renal Replacement Therapy in Patients With COVID-19. J. Emerg. Med. 2022, 62, 145–153. [Google Scholar] [CrossRef]
- De Oliveira, F.L.; Araújo-Jorge, T.C.; de Souza, E.M.; de Oliveira, G.M.; Degrave, W.M.; Feige, J.-J.; Bailly, S.; Waghabi, M.C. Oral Administration of GW788388, an Inhibitor of Transforming Growth Factor Beta Signaling, Prevents Heart Fibrosis in Chagas Disease. PLoS Negl. Trop. Dis. 2012, 6, e1696-14. [Google Scholar] [CrossRef]
- Ruemelin, A.; Jaehde, U.; Kerz, T.; Krämer, M.; Fauth, U. Early Postoperative Substitution Procedure of the Antioxidant Ascorbic Acid (AA) in Postoperative Intensive Care Unit (ICU) Patients. Crit. Care 2004, 8, 263. [Google Scholar] [CrossRef] [Green Version]
- Gajardo, A.I.; von Dessauer, B.; Molina, V.; Vera, S.; Libuy, M.; Rodrigo, R. Plasma Antioxidant Potential at Admission Is Associated with Length of ICU Stay in Child with Sepsis: A Pilot Study. Fetal Pediatr. Pathol. 2018, 37, 348–358. [Google Scholar] [CrossRef]
- Herrera-Quintana, L.; Vázquez-Lorente, H.; Molina-López, J.; Gamarra-Morales, Y.; Planells, E. Selenium Levels and Antioxidant Activity in Critically Ill Patients with Systemic Inflammatory Response Syndrome. Metabolites 2022, 12, 274. [Google Scholar] [CrossRef]
- Luca, L.; Rogobete, A.F.; Bedreag, O.H. Oxidative Stress and Antioxidant Therapy in Critically Ill Polytrauma Patients with Severe Head Injury. J. Crit. Care Med. 2015, 1, 83–91. [Google Scholar] [CrossRef]
- Koekkoek, W.A.C.; van Zanten, A.R.H. Antioxidant Vitamins and Trace Elements in Critical Illness. Nutr. Clin. Pract. 2016, 31, 457–474. [Google Scholar] [CrossRef] [PubMed]
- Haenen, G.R.M.M.; Arts, M.J.T.J.; Bast, A.; Coleman, M.D. Structure and Activity in Assessing Antioxidant Activity in Vitro and in Vivo A Critical Appraisal Illustrated with the Flavonoids. Environ. Toxicol. Phar. 2006, 21, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Abdulla, K.A.; Um, C.Y.; Gross, M.D.; Bostick, R.M. Circulating γ-Tocopherol Concentrations Are Inversely Associated with Antioxidant Exposures and Directly Associated with Systemic Oxidative Stress and Inflammation in Adults. J. Nutr. 2018, 148, 1453–1461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheong, K.-L.; Yu, B.; Chen, J.; Zhong, S. A Comprehensive Review of the Cardioprotective Effect of Marine Algae Polysaccharide on the Gut Microbiota. Foods 2022, 11, 3550. [Google Scholar] [CrossRef] [PubMed]
- Vincent, J.-L.; Marshall, J.C.; Namendys-Silva, S.A.; François, B.; Martin-Loeches, I.; Lipman, J.; Reinhart, K.; Antonelli, M.; Pickkers, P.; Njimi, H.; et al. Assessment of the Worldwide Burden of Critical Illness: The Intensive Care over Nations (ICON) Audit. Lancet Respir. Med. 2014, 2, 380–386. [Google Scholar] [CrossRef] [PubMed]
- Tennilä, A.; Salmi, T.; Pettila, V.; Roine, R.O.; Varpula, T.; Takkunen, O. Early Signs of Critical Illness Polyneuropathy in ICU Patients with Systemic Inflammatory Response Syndrome or Sepsis. Intensive Care Med. 2000, 26, 1360–1363. [Google Scholar] [CrossRef]
- Granja, C.; Amaro, A.; Dias, C.; Costa-Pereira, A. Outcome of ICU Survivors. Acta Anaesth. Scand. 2012, 56, 1092–1103. [Google Scholar] [CrossRef]
- Kredentser, M.S.; Blouw, M.; Marten, N.; Sareen, J.; Bienvenu, O.J.; Ryu, J.; Beatie, B.E.; Logsetty, S.; Graff, L.A.; Eggertson, S.; et al. Preventing Posttraumatic Stress in ICU Survivors. Crit. Care Med. 2018, 46, 1914–1922. [Google Scholar] [CrossRef]
- Offor, O.L.; Ezeagu, R.; Olanipekun, T. ICU Survivors Have Increased Health Resource Utilization During the Post-ICU Period. Crit. Care Med. 2020, 48, e344. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rojas, M.; Prado, Y.; Tapia, P.; Carreño, L.J.; Cabello-Verrugio, C.; Simon, F. Oxidized High-Density Lipoprotein Induces Endothelial Fibrosis Promoting Hyperpermeability, Hypotension, and Increased Mortality. Antioxidants 2022, 11, 2469. https://doi.org/10.3390/antiox11122469
Rojas M, Prado Y, Tapia P, Carreño LJ, Cabello-Verrugio C, Simon F. Oxidized High-Density Lipoprotein Induces Endothelial Fibrosis Promoting Hyperpermeability, Hypotension, and Increased Mortality. Antioxidants. 2022; 11(12):2469. https://doi.org/10.3390/antiox11122469
Chicago/Turabian StyleRojas, Macarena, Yolanda Prado, Pablo Tapia, Leandro J. Carreño, Claudio Cabello-Verrugio, and Felipe Simon. 2022. "Oxidized High-Density Lipoprotein Induces Endothelial Fibrosis Promoting Hyperpermeability, Hypotension, and Increased Mortality" Antioxidants 11, no. 12: 2469. https://doi.org/10.3390/antiox11122469