
SLC7A11 as a Gateway of Metabolic Perturbation and Ferroptosis Vulnerability in Cancer

Abstract
: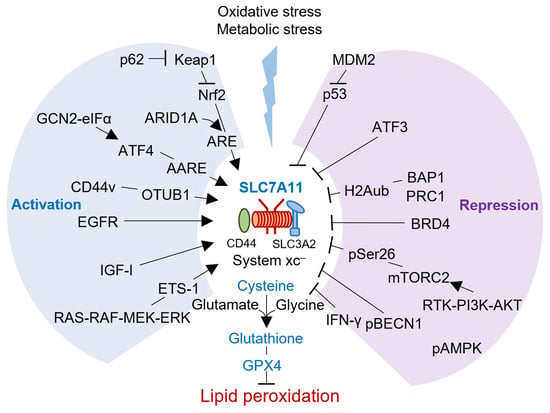
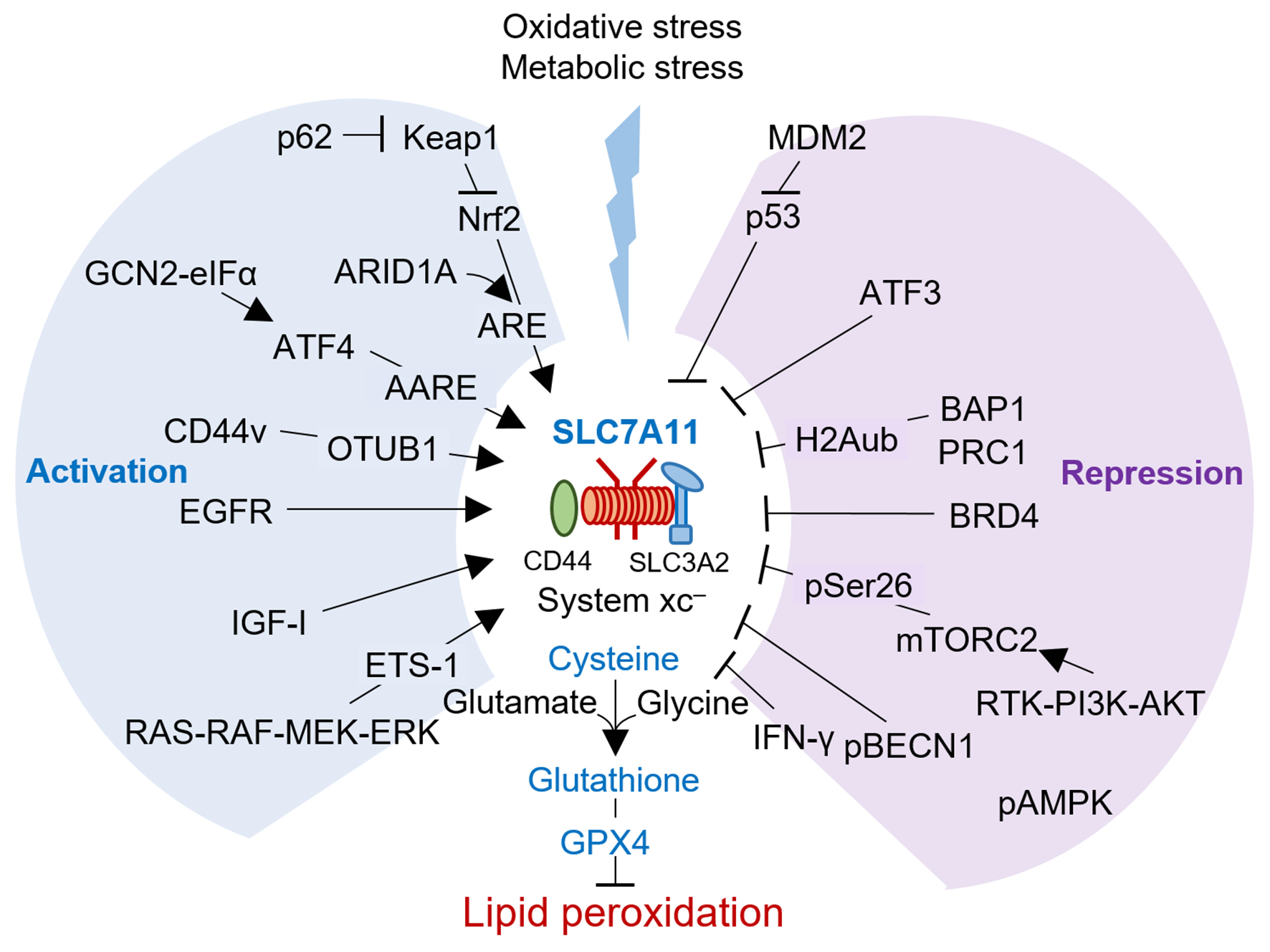{kind=link}
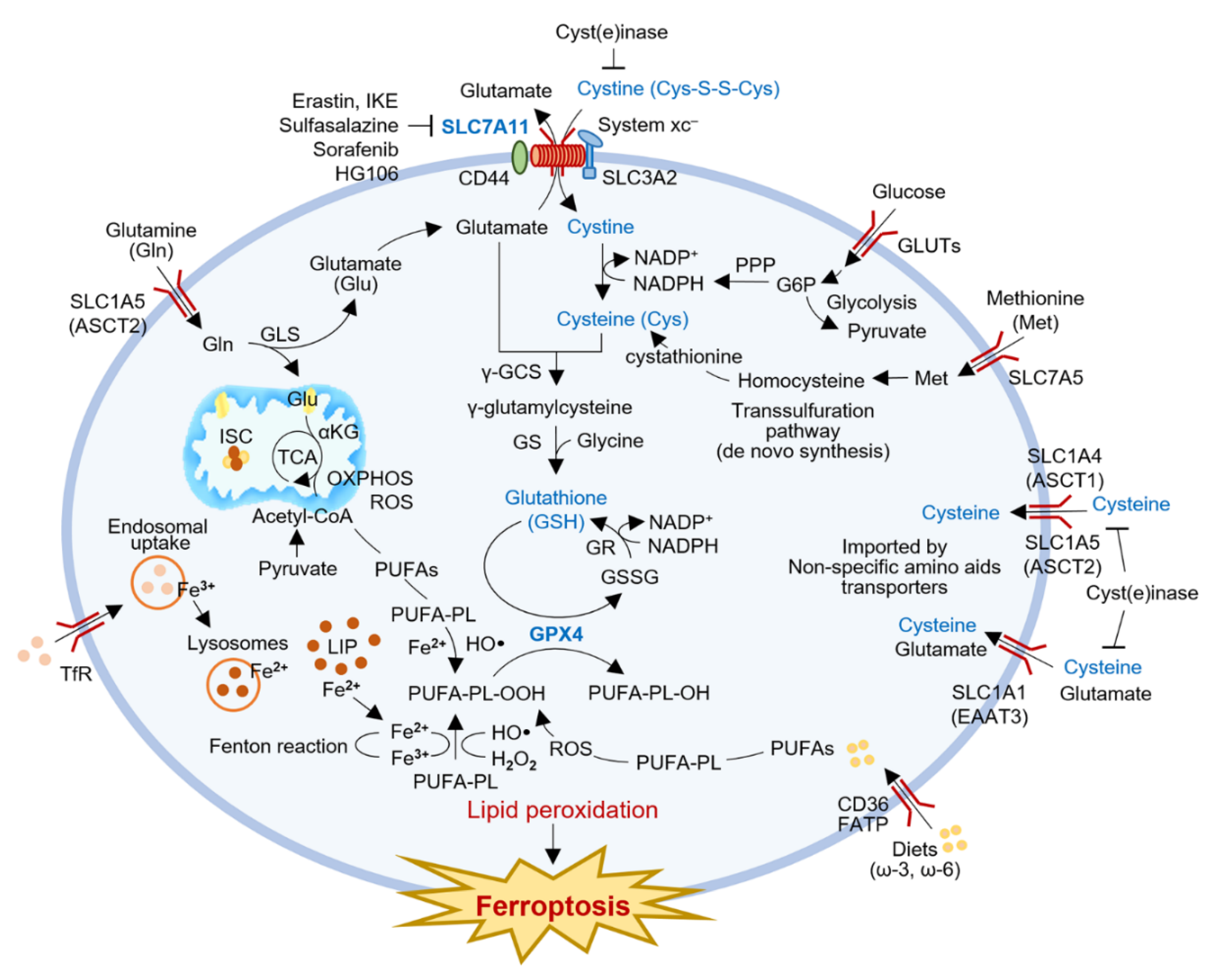{kind=link}
{kind=link}
1. Introduction
2. Role of Cysteine in Antioxidant and Redox Signaling
3. Regulators of Cysteine Bioavailability in Cancer Cells
4. Role of SLC7A11 Linking Cysteine Redox Signaling to Cancer Metabolism
4.1. SLC7A11 Promotes Tumorigenesis via Inhibiting Ferroptosis
4.2. SLC7A11 Promotes Immune Evasion, Invasion, and Metastasis in Human Cancers
4.3. SLC7A11 Induces Nutrient Dependency and Metabolic Vulnerability in Cancer
4.4. SLC7A11 Has a Role in Cancer Therapeutic Resistance
5. Targeting SLC7A11 for Novel Cancer Therapeutics
6. Conclusions and Perspectives
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Pompella, A.; Visvikis, A.; Paolicchi, A.; De Tata, V.; Casini, A.F. The changing faces of glutathione, a cellular protagonist. Biochem. Pharmacol. 2003, 66, 1499–1503. [Google Scholar] [CrossRef] [PubMed]
- Aquilano, K.; Baldelli, S.; Ciriolo, M.R. Glutathione: New roles in redox signaling for an old antioxidant. Front. Pharmacol. 2014, 5, 196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo, M.; Wang, Y.Z.; Gout, P.W. The x(c)- cystine/glutamate antiporter: A potential target for therapy of cancer and other diseases. J. Cell. Physiol. 2008, 215, 593–602. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Trachootham, D.; Liu, J.; Chen, G.; Pelicano, H.; Garcia-Prieto, C.; Lu, W.; Burger, J.A.; Croce, C.M.; Plunkett, W.; et al. Stromal control of cystine metabolism promotes cancer cell survival in chronic lymphocytic leukaemia. Nat. Cell Biol. 2012, 14, 276–286. [Google Scholar] [CrossRef] [Green Version]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascón, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef] [Green Version]
- Feng, H.; Stockwell, B.R. Unsolved mysteries: How does lipid peroxidation cause ferroptosis? PLoS Biol. 2018, 16, e2006203. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [Green Version]
- Li, F.J.; Long, H.Z.; Zhou, Z.W.; Luo, H.Y.; Xu, S.G.; Gao, L.C. System X(c) (-)/GSH/GPX4 axis: An important antioxidant system for the ferroptosis in drug-resistant solid tumor therapy. Front. Pharmacol. 2022, 13, 910292. [Google Scholar] [CrossRef]
- Chen, H.; Wang, C.; Liu, Z.; He, X.; Tang, W.; He, L.; Feng, Y.; Liu, D.; Yin, Y.; Li, T. Ferroptosis and Its Multifaceted Role in Cancer: Mechanisms and Therapeutic Approach. Antioxidants 2022, 11, 1504. [Google Scholar] [CrossRef]
- Sato, H.; Tamba, M.; Kuriyama-Matsumura, K.; Okuno, S.; Bannai, S. Molecular cloning and expression of human xCT, the light chain of amino acid transport system xc. Antioxid. Redox Signal. 2000, 2, 665–671. [Google Scholar] [CrossRef]
- Zhang, H.F.; Klein Geltink, R.I.; Parker, S.J.; Sorensen, P.H. Transsulfuration, minor player or crucial for cysteine homeostasis in cancer. Trends Cell Biol. 2022, 32, 800–814. [Google Scholar] [CrossRef] [PubMed]
- Koppula, P.; Zhuang, L.; Gan, B. Cystine transporter SLC7A11/xCT in cancer: Ferroptosis, nutrient dependency, and cancer therapy. Protein Cell 2021, 12, 599–620. [Google Scholar] [CrossRef] [PubMed]
- Koppula, P.; Zhang, Y.; Shi, J.; Li, W.; Gan, B. The glutamate/cystine antiporter SLC7A11/xCT enhances cancer cell dependency on glucose by exporting glutamate. J. Biol. Chem. 2017, 292, 14240–14249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Lu, Z.; Sun, R.; Guo, S.; Gao, F.; Cao, B.; Aa, J. The Role of SLC7A11 in Cancer: Friend or Foe? Cancers 2022, 14, 3059. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Xia, X.; Huang, P. xCT: A Critical Molecule That Links Cancer Metabolism to Redox Signaling. Mol. Ther. J. Am. Soc. Gene Ther. 2020, 28, 2358–2366. [Google Scholar] [CrossRef] [PubMed]
- Borges, C.R.; Sherma, N.D. Techniques for the analysis of cysteine sulfhydryls and oxidative protein folding. Antioxid. Redox Signal. 2014, 21, 511–531. [Google Scholar] [CrossRef] [Green Version]
- Poole, L.B. The basics of thiols and cysteines in redox biology and chemistry. Free Radic. Biol. Med. 2015, 80, 148–157. [Google Scholar] [CrossRef] [Green Version]
- Stipanuk, M.H. Metabolism of Sulfur-Containing Amino Acids: How the Body Copes with Excess Methionine, Cysteine, and Sulfide. J. Nutr. 2020, 150, 2494s–2505s. [Google Scholar] [CrossRef]
- Fra, A.; Yoboue, E.D.; Sitia, R. Cysteines as Redox Molecular Switches and Targets of Disease. Front. Mol. Neurosci. 2017, 10, 167. [Google Scholar] [CrossRef] [Green Version]
- Schmitt, B.; Vicenzi, M.; Garrel, C.; Denis, F.M. Effects of N-acetylcysteine, oral glutathione (GSH) and a novel sublingual form of GSH on oxidative stress markers: A comparative crossover study. Redox Biol. 2015, 6, 198–205. [Google Scholar] [CrossRef]
- Paulsen, C.E.; Carroll, K.S. Cysteine-mediated redox signaling: Chemistry, biology, and tools for discovery. Chem. Rev. 2013, 113, 4633–4679. [Google Scholar] [CrossRef] [PubMed]
- Xiao, W.; Loscalzo, J. Metabolic Responses to Reductive Stress. Antioxid. Redox Signal. 2020, 32, 1330–1347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scalise, M.; Pochini, L.; Pingitore, P.; Hedfalk, K.; Indiveri, C. Cysteine is not a substrate but a specific modulator of human ASCT2 (SLC1A5) transporter. FEBS Lett. 2015, 589, 3617–3623. [Google Scholar] [CrossRef] [PubMed]
- Bayır, H.; Anthonymuthu, T.S.; Tyurina, Y.Y.; Patel, S.J.; Amoscato, A.A.; Lamade, A.M.; Yang, Q.; Vladimirov, G.K.; Philpott, C.C.; Kagan, V.E. Achieving Life through Death: Redox Biology of Lipid Peroxidation in Ferroptosis. Cell Chem. Biol. 2020, 27, 387–408. [Google Scholar] [CrossRef] [PubMed]
- Schafer, F.Q.; Buettner, G.R. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic. Biol. Med. 2001, 30, 1191–1212. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, Y.; Zhuang, L.; Olszewski, K.; Gan, B. NADPH debt drives redox bankruptcy: SLC7A11/xCT-mediated cystine uptake as a double-edged sword in cellular redox regulation. Genes Dis. 2021, 8, 731–745. [Google Scholar] [CrossRef]
- Lewerenz, J.; Hewett, S.J.; Huang, Y.; Lambros, M.; Gout, P.W.; Kalivas, P.W.; Massie, A.; Smolders, I.; Methner, A.; Pergande, M.; et al. The cystine/glutamate antiporter system x(c)(-) in health and disease: From molecular mechanisms to novel therapeutic opportunities. Antioxid. Redox Signal. 2013, 18, 522–555. [Google Scholar] [CrossRef] [Green Version]
- Al-Nasser, M.N.; Mellor, I.R.; Carter, W.G. Is L-Glutamate Toxic to Neurons and Thereby Contributes to Neuronal Loss and Neurodegeneration? A Systematic Review. Brain Sci. 2022, 12, 577. [Google Scholar] [CrossRef]
- Bonifácio, V.D.B.; Pereira, S.A.; Serpa, J.; Vicente, J.B. Cysteine metabolic circuitries: Druggable targets in cancer. Br. J. Cancer 2021, 124, 862–879. [Google Scholar] [CrossRef]
- Kilberg, M.S.; Shan, J.; Su, N. ATF4-dependent transcription mediates signaling of amino acid limitation. Trends Endocrinol. Metab. TEM 2009, 20, 436–443. [Google Scholar] [CrossRef]
- Chen, D.; Fan, Z.; Rauh, M.; Buchfelder, M.; Eyupoglu, I.Y.; Savaskan, N. ATF4 promotes angiogenesis and neuronal cell death and confers ferroptosis in a xCT-dependent manner. Oncogene 2017, 36, 5593–5608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rojo de la Vega, M.; Chapman, E.; Zhang, D.D. NRF2 and the Hallmarks of Cancer. Cancer Cell 2018, 34, 21–43. [Google Scholar] [CrossRef]
- Cao, J.Y.; Poddar, A.; Magtanong, L.; Lumb, J.H.; Mileur, T.R.; Reid, M.A.; Dovey, C.M.; Wang, J.; Locasale, J.W.; Stone, E.; et al. A Genome-wide Haploid Genetic Screen Identifies Regulators of Glutathione Abundance and Ferroptosis Sensitivity. Cell Rep. 2019, 26, 1544–1556.e1548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anandhan, A.; Dodson, M.; Schmidlin, C.J.; Liu, P.; Zhang, D.D. Breakdown of an Ironclad Defense System: The Critical Role of NRF2 in Mediating Ferroptosis. Cell Chem. Biol. 2020, 27, 436–447. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Kon, N.; Li, T.; Wang, S.J.; Su, T.; Hibshoosh, H.; Baer, R.; Gu, W. Ferroptosis as a p53-mediated activity during tumour suppression. Nature 2015, 520, 57–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Liu, Y.; Du, T.; Yang, H.; Lei, L.; Guo, M.; Ding, H.F.; Zhang, J.; Wang, H.; Chen, X.; et al. ATF3 promotes erastin-induced ferroptosis by suppressing system Xc(). Cell Death Differ. 2020, 27, 662–675. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Zhang, S.; Gong, X.; Tam, S.; Xiao, D.; Liu, S.; Tao, Y. The epigenetic regulators and metabolic changes in ferroptosis-associated cancer progression. Mol. Cancer 2020, 19, 39. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Shi, J.; Liu, X.; Feng, L.; Gong, Z.; Koppula, P.; Sirohi, K.; Li, X.; Wei, Y.; Lee, H.; et al. BAP1 links metabolic regulation of ferroptosis to tumour suppression. Nat. Cell Biol. 2018, 20, 1181–1192. [Google Scholar] [CrossRef]
- Sui, S.; Zhang, J.; Xu, S.; Wang, Q.; Wang, P.; Pang, D. Ferritinophagy is required for the induction of ferroptosis by the bromodomain protein BRD4 inhibitor (+)-JQ1 in cancer cells. Cell Death Dis. 2019, 10, 331. [Google Scholar] [CrossRef] [Green Version]
- Ogiwara, H.; Takahashi, K.; Sasaki, M.; Kuroda, T.; Yoshida, H.; Watanabe, R.; Maruyama, A.; Makinoshima, H.; Chiwaki, F.; Sasaki, H.; et al. Targeting the Vulnerability of Glutathione Metabolism in ARID1A-Deficient Cancers. Cancer Cell 2019, 35, 177–190.e178. [Google Scholar] [CrossRef]
- Liu, T.; Jiang, L.; Tavana, O.; Gu, W. The Deubiquitylase OTUB1 Mediates Ferroptosis via Stabilization of SLC7A11. Cancer Res. 2019, 79, 1913–1924. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Albuquerque, C.P.; Braas, D.; Zhang, W.; Villa, G.R.; Bi, J.; Ikegami, S.; Masui, K.; Gini, B.; Yang, H.; et al. mTORC2 Regulates Amino Acid Metabolism in Cancer by Phosphorylation of the Cystine-Glutamate Antiporter xCT. Mol. Cell 2017, 67, 128–138.e127. [Google Scholar] [CrossRef] [Green Version]
- Song, X.; Zhu, S.; Chen, P.; Hou, W.; Wen, Q.; Liu, J.; Xie, Y.; Liu, J.; Klionsky, D.J.; Kroemer, G.; et al. AMPK-Mediated BECN1 Phosphorylation Promotes Ferroptosis by Directly Blocking System X(c)(-) Activity. Curr. Biol. CB 2018, 28, 2388–2399.e2385. [Google Scholar] [CrossRef] [Green Version]
- Tsuchihashi, K.; Okazaki, S.; Ohmura, M.; Ishikawa, M.; Sampetrean, O.; Onishi, N.; Wakimoto, H.; Yoshikawa, M.; Seishima, R.; Iwasaki, Y.; et al. The EGF Receptor Promotes the Malignant Potential of Glioma by Regulating Amino Acid Transport System xc(-). Cancer Res. 2016, 76, 2954–2963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Yee, D. IGF-I regulates redox status in breast cancer cells by activating the amino acid transport molecule xC. Cancer Res. 2014, 74, 2295–2305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gout, P.W.; Kang, Y.J.; Buckley, D.J.; Bruchovsky, N.; Buckley, A.R. Increased cystine uptake capability associated with malignant progression of Nb2 lymphoma cells. Leukemia 1997, 11, 1329–1337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- DeNicola, G.M.; Karreth, F.A.; Humpton, T.J.; Gopinathan, A.; Wei, C.; Frese, K.; Mangal, D.; Yu, K.H.; Yeo, C.J.; Calhoun, E.S.; et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 2011, 475, 106–109. [Google Scholar] [CrossRef] [Green Version]
- Lim, J.K.M.; Leprivier, G. The impact of oncogenic RAS on redox balance and implications for cancer development. Cell Death Dis. 2019, 10, 955. [Google Scholar] [CrossRef] [Green Version]
- Lim, J.K.M.; Delaidelli, A.; Minaker, S.W.; Zhang, H.F.; Colovic, M.; Yang, H.; Negri, G.L.; von Karstedt, S.; Lockwood, W.W.; Schaffer, P.; et al. Cystine/glutamate antiporter xCT (SLC7A11) facilitates oncogenic RAS transformation by preserving intracellular redox balance. Proc. Natl. Acad. Sci. USA 2019, 116, 9433–9442. [Google Scholar] [CrossRef]
- Hu, K.; Li, K.; Lv, J.; Feng, J.; Chen, J.; Wu, H.; Cheng, F.; Jiang, W.; Wang, J.; Pei, H.; et al. Suppression of the SLC7A11/glutathione axis causes synthetic lethality in KRAS-mutant lung adenocarcinoma. J. Clin. Investig. 2020, 130, 1752–1766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, D.S.; Duong, C.P.; Haupt, S.; Montgomery, K.G.; House, C.M.; Azar, W.J.; Pearson, H.B.; Fisher, O.M.; Read, M.; Guerra, G.R.; et al. Inhibiting the system x(C)(-)/glutathione axis selectively targets cancers with mutant-p53 accumulation. Nat. Commun. 2017, 8, 14844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, T.; Kon, N.; Jiang, L.; Tan, M.; Ludwig, T.; Zhao, Y.; Baer, R.; Gu, W. Tumor suppression in the absence of p53-mediated cell-cycle arrest, apoptosis, and senescence. Cell 2012, 149, 1269–1283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.J.; Li, D.; Ou, Y.; Jiang, L.; Chen, Y.; Zhao, Y.; Gu, W. Acetylation Is Crucial for p53-Mediated Ferroptosis and Tumor Suppression. Cell Rep. 2016, 17, 366–373. [Google Scholar] [CrossRef] [Green Version]
- Chu, B.; Kon, N.; Chen, D.; Li, T.; Liu, T.; Jiang, L.; Song, S.; Tavana, O.; Gu, W. ALOX12 is required for p53-mediated tumour suppression through a distinct ferroptosis pathway. Nat. Cell Biol. 2019, 21, 579–591. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhuang, L.; Gan, B. BAP1 suppresses tumor development by inducing ferroptosis upon SLC7A11 repression. Mol. Cell. Oncol. 2019, 6, 1536845. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Green, M.; Choi, J.E.; Gijón, M.; Kennedy, P.D.; Johnson, J.K.; Liao, P.; Lang, X.; Kryczek, I.; Sell, A.; et al. CD8(+) T cells regulate tumour ferroptosis during cancer immunotherapy. Nature 2019, 569, 270–274. [Google Scholar] [CrossRef]
- Levring, T.B.; Kongsbak, M.; Rode, A.K.; Woetmann, A.; Ødum, N.; Bonefeld, C.M.; Geisler, C. Human CD4+ T cells require exogenous cystine for glutathione and DNA synthesis. Oncotarget 2015, 6, 21853–21864. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, M.K.; Sinha, P.; Clements, V.K.; Rodriguez, P.; Ostrand-Rosenberg, S. Myeloid-derived suppressor cells inhibit T-cell activation by depleting cystine and cysteine. Cancer Res. 2010, 70, 68–77. [Google Scholar] [CrossRef] [Green Version]
- Pacheco, R.; Oliva, H.; Martinez-Navío, J.M.; Climent, N.; Ciruela, F.; Gatell, J.M.; Gallart, T.; Mallol, J.; Lluis, C.; Franco, R. Glutamate released by dendritic cells as a novel modulator of T cell activation. J. Immunol. 2006, 177, 6695–6704. [Google Scholar] [CrossRef]
- Long, Y.; Tao, H.; Karachi, A.; Grippin, A.J.; Jin, L.; Chang, Y.E.; Zhang, W.; Dyson, K.A.; Hou, A.Y.; Na, M.; et al. Dysregulation of Glutamate Transport Enhances Treg Function That Promotes VEGF Blockade Resistance in Glioblastoma. Cancer Res. 2020, 80, 499–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, X.; Xiao, L.; Liu, L.; Ye, L.; Su, P.; Bi, E.; Wang, Q.; Yang, M.; Qian, J.; Yi, Q. CD36-mediated ferroptosis dampens intratumoral CD8(+) T cell effector function and impairs their antitumor ability. Cell Metab. 2021, 33, 1001–1012.e1005. [Google Scholar] [CrossRef] [PubMed]
- Sharbeen, G.; McCarroll, J.A.; Akerman, A.; Kopecky, C.; Youkhana, J.; Kokkinos, J.; Holst, J.; Boyer, C.; Erkan, M.; Goldstein, D.; et al. Cancer-Associated Fibroblasts in Pancreatic Ductal Adenocarcinoma Determine Response to SLC7A11 Inhibition. Cancer Res. 2021, 81, 3461–3479. [Google Scholar] [CrossRef] [PubMed]
- Sugano, K.; Maeda, K.; Ohtani, H.; Nagahara, H.; Shibutani, M.; Hirakawa, K. Expression of xCT as a predictor of disease recurrence in patients with colorectal cancer. Anticancer Res. 2015, 35, 677–682. [Google Scholar]
- Lee, J.R.; Roh, J.L.; Lee, S.M.; Park, Y.; Cho, K.J.; Choi, S.H.; Nam, S.Y.; Kim, S.Y. Overexpression of cysteine-glutamate transporter and CD44 for prediction of recurrence and survival in patients with oral cavity squamous cell carcinoma. Head Neck 2018, 40, 2340–2346. [Google Scholar] [CrossRef]
- Lyons, S.A.; Chung, W.J.; Weaver, A.K.; Ogunrinu, T.; Sontheimer, H. Autocrine glutamate signaling promotes glioma cell invasion. Cancer Res. 2007, 67, 9463–9471. [Google Scholar] [CrossRef] [Green Version]
- Sato, M.; Onuma, K.; Domon, M.; Hasegawa, S.; Suzuki, A.; Kusumi, R.; Hino, R.; Kakihara, N.; Kanda, Y.; Osaki, M.; et al. Loss of the cystine/glutamate antiporter in melanoma abrogates tumor metastasis and markedly increases survival rates of mice. Int. J. Cancer 2020, 147, 3224–3235. [Google Scholar] [CrossRef]
- Wang, X.; Li, Y.; Li, Z.; Lin, S.; Wang, H.; Sun, J.; Lan, C.; Wu, L.; Sun, D.; Huang, C.; et al. Mitochondrial Calcium Uniporter Drives Metastasis and Confers a Targetable Cystine Dependency in Pancreatic Cancer. Cancer Res. 2022, 82, 2254–2268. [Google Scholar] [CrossRef]
- Dornier, E.; Rabas, N.; Mitchell, L.; Novo, D.; Dhayade, S.; Marco, S.; Mackay, G.; Sumpton, D.; Pallares, M.; Nixon, C.; et al. Glutaminolysis drives membrane trafficking to promote invasiveness of breast cancer cells. Nat. Commun. 2017, 8, 2255. [Google Scholar] [CrossRef] [Green Version]
- He, Q.; Liu, M.; Huang, W.; Chen, X.; Zhang, B.; Zhang, T.; Wang, Y.; Liu, D.; Xie, M.; Ji, X.; et al. IL-1β-Induced Elevation of Solute Carrier Family 7 Member 11 Promotes Hepatocellular Carcinoma Metastasis Through Up-regulating Programmed Death Ligand 1 and Colony-Stimulating Factor 1. Hepatology 2021, 74, 3174–3193. [Google Scholar] [CrossRef]
- Sharma, M.K.; Seidlitz, E.P.; Singh, G. Cancer cells release glutamate via the cystine/glutamate antiporter. Biochem. Biophys. Res. Commun. 2010, 391, 91–95. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Olszewski, K.; Zhang, Y.; Lim, E.W.; Shi, J.; Zhang, X.; Zhang, J.; Lee, H.; Koppula, P.; Lei, G.; et al. Cystine transporter regulation of pentose phosphate pathway dependency and disulfide stress exposes a targetable metabolic vulnerability in cancer. Nat. Cell Biol. 2020, 22, 476–486. [Google Scholar] [CrossRef] [PubMed]
- Goji, T.; Takahara, K.; Negishi, M.; Katoh, H. Cystine uptake through the cystine/glutamate antiporter xCT triggers glioblastoma cell death under glucose deprivation. J. Biol. Chem. 2017, 292, 19721–19732. [Google Scholar] [CrossRef] [Green Version]
- Joly, J.H.; Delfarah, A.; Phung, P.S.; Parrish, S.; Graham, N.A. A synthetic lethal drug combination mimics glucose deprivation-induced cancer cell death in the presence of glucose. J. Biol. Chem. 2020, 295, 1350–1365. [Google Scholar] [CrossRef]
- Chen, M.C.; Hsu, L.L.; Wang, S.F.; Hsu, C.Y.; Lee, H.C.; Tseng, L.M. ROS Mediate xCT-Dependent Cell Death in Human Breast Cancer Cells under Glucose Deprivation. Cells 2020, 9, 1598. [Google Scholar] [CrossRef]
- Yamaguchi, I.; Yoshimura, S.H.; Katoh, H. High cell density increases glioblastoma cell viability under glucose deprivation via degradation of the cystine/glutamate transporter xCT (SLC7A11). J. Biol. Chem. 2020, 295, 6936–6945. [Google Scholar] [CrossRef] [Green Version]
- Bhutia, Y.D.; Babu, E.; Ramachandran, S.; Ganapathy, V. Amino Acid transporters in cancer and their relevance to “glutamine addiction”: Novel targets for the design of a new class of anticancer drugs. Cancer Res. 2015, 75, 1782–1788. [Google Scholar] [CrossRef] [Green Version]
- Muir, A.; Danai, L.V.; Gui, D.Y.; Waingarten, C.Y.; Lewis, C.A.; Vander Heiden, M.G. Environmental cystine drives glutamine anaplerosis and sensitizes cancer cells to glutaminase inhibition. eLife 2017, 6, e27713. [Google Scholar] [CrossRef]
- Shin, C.S.; Mishra, P.; Watrous, J.D.; Carelli, V.; D’Aurelio, M.; Jain, M.; Chan, D.C. The glutamate/cystine xCT antiporter antagonizes glutamine metabolism and reduces nutrient flexibility. Nat. Commun. 2017, 8, 15074. [Google Scholar] [CrossRef] [Green Version]
- Hu, N.; Hu, W.H.; Zhou, S.L.; Yang, Z.; Liang, W.L.; Yang, R.Y.; Li, M.H.; Jing, Z.; Li, Z.A.; Fu, X.D.; et al. SLC7A11 negatively associates with mismatch repair gene expression and endows glioblastoma cells sensitive to radiation under low glucose conditions. Neoplasma 2021, 68, 1147–1156. [Google Scholar] [CrossRef]
- Okazaki, S.; Umene, K.; Yamasaki, J.; Suina, K.; Otsuki, Y.; Yoshikawa, M.; Minami, Y.; Masuko, T.; Kawaguchi, S.; Nakayama, H.; et al. Glutaminolysis-related genes determine sensitivity to xCT-targeted therapy in head and neck squamous cell carcinoma. Cancer Sci. 2019, 110, 3453–3463. [Google Scholar] [CrossRef] [PubMed]
- Romero, R.; Sayin, V.I.; Davidson, S.M.; Bauer, M.R.; Singh, S.X.; LeBoeuf, S.E.; Karakousi, T.R.; Ellis, D.C.; Bhutkar, A.; Sánchez-Rivera, F.J.; et al. Keap1 loss promotes Kras-driven lung cancer and results in dependence on glutaminolysis. Nat. Med. 2017, 23, 1362–1368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Dai, Z.; Barbacioru, C.; Sadée, W. Cystine-glutamate transporter SLC7A11 in cancer chemosensitivity and chemoresistance. Cancer Res. 2005, 65, 7446–7454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyoshi, S.; Tsugawa, H.; Matsuzaki, J.; Hirata, K.; Mori, H.; Saya, H.; Kanai, T.; Suzuki, H. Inhibiting xCT Improves 5-Fluorouracil Resistance of Gastric Cancer Induced by CD44 Variant 9 Expression. Anticancer Res. 2018, 38, 6163–6170. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, M.; Tsuchihashi, K.; Ishimoto, T.; Yae, T.; Motohara, T.; Sugihara, E.; Onishi, N.; Masuko, T.; Yoshizawa, K.; Kawashiri, S.; et al. xCT inhibition depletes CD44v-expressing tumor cells that are resistant to EGFR-targeted therapy in head and neck squamous cell carcinoma. Cancer Res. 2013, 73, 1855–1866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wada, F.; Koga, H.; Akiba, J.; Niizeki, T.; Iwamoto, H.; Ikezono, Y.; Nakamura, T.; Abe, M.; Masuda, A.; Sakaue, T.; et al. High expression of CD44v9 and xCT in chemoresistant hepatocellular carcinoma: Potential targets by sulfasalazine. Cancer Sci. 2018, 109, 2801–2810. [Google Scholar] [CrossRef] [PubMed]
- Ogihara, K.; Kikuchi, E.; Okazaki, S.; Hagiwara, M.; Takeda, T.; Matsumoto, K.; Kosaka, T.; Mikami, S.; Saya, H.; Oya, M. Sulfasalazine could modulate the CD44v9-xCT system and enhance cisplatin-induced cytotoxic effects in metastatic bladder cancer. Cancer Sci. 2019, 110, 1431–1441. [Google Scholar] [CrossRef]
- Feng, L.; Zhao, K.; Sun, L.; Yin, X.; Zhang, J.; Liu, C.; Li, B. SLC7A11 regulated by NRF2 modulates esophageal squamous cell carcinoma radiosensitivity by inhibiting ferroptosis. J. Transl. Med. 2021, 19, 367. [Google Scholar] [CrossRef]
- Singer, E.; Judkins, J.; Salomonis, N.; Matlaf, L.; Soteropoulos, P.; McAllister, S.; Soroceanu, L. Reactive oxygen species-mediated therapeutic response and resistance in glioblastoma. Cell Death Dis. 2015, 6, e1601. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.; Jang, J.; Shin, T.H.; Bae, S.M.; Kim, J.S.; Kim, K.M.; Myung, S.J.; Choi, E.K.; Seo, H.R. Sulfasalazine attenuates evading anticancer response of CD133-positive hepatocellular carcinoma cells. J. Exp. Clin. Cancer Res. CR 2017, 36, 38. [Google Scholar] [CrossRef] [PubMed]
- Ke, Y.; Chen, X.; Su, Y.; Chen, C.; Lei, S.; Xia, L.; Wei, D.; Zhang, H.; Dong, C.; Liu, X.; et al. Low Expression of SLC7A11 Confers Drug Resistance and Worse Survival in Ovarian Cancer via Inhibition of Cell Autophagy as a Competing Endogenous RNA. Front. Oncol. 2021, 11, 744940. [Google Scholar] [CrossRef] [PubMed]
- Ge, C.; Cao, B.; Feng, D.; Zhou, F.; Zhang, J.; Yang, N.; Feng, S.; Wang, G.; Aa, J. The down-regulation of SLC7A11 enhances ROS induced P-gp over-expression and drug resistance in MCF-7 breast cancer cells. Sci. Rep. 2017, 7, 3791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.J.; Cao, B.; Lu, Z.Y.; Sun, R.B.; Guo, S.H.; Xie, Y.; Aa, J.Y.; Wang, G.J. Cystine supplementation rebalances the redox homeostasis of microenvironment in non-small cell lung cancer cells and reverses their resistance to docetaxel. Acta Pharmacol. Sin. 2021, 42, 2132–2143. [Google Scholar] [CrossRef]
- Pratt, D.A.; Tallman, K.A.; Porter, N.A. Free radical oxidation of polyunsaturated lipids: New mechanistic insights and the development of peroxyl radical clocks. Acc. Chem. Res. 2011, 44, 458–467. [Google Scholar] [CrossRef] [Green Version]
- Liang, D.; Minikes, A.M.; Jiang, X. Ferroptosis at the intersection of lipid metabolism and cellular signaling. Mol. Cell 2022, 82, 2215–2227. [Google Scholar] [CrossRef]
- Kandasamy, P.; Gyimesi, G.; Kanai, Y.; Hediger, M.A. Amino acid transporters revisited: New views in health and disease. Trends Biochem. Sci. 2018, 43, 752–789. [Google Scholar] [CrossRef]
- Badgley, M.A.; Kremer, D.M.; Maurer, H.C.; DelGiorno, K.E.; Lee, H.J.; Purohit, V.; Sagalovskiy, I.R.; Ma, A.; Kapilian, J.; Firl, C.E.M.; et al. Cysteine depletion induces pancreatic tumor ferroptosis in mice. Science 2020, 368, 85–89. [Google Scholar] [CrossRef]
- Sato, H.; Shiiya, A.; Kimata, M.; Maebara, K.; Tamba, M.; Sakakura, Y.; Makino, N.; Sugiyama, F.; Yagami, K.; Moriguchi, T.; et al. Redox imbalance in cystine/glutamate transporter-deficient mice. J. Biol. Chem. 2005, 280, 37423–37429. [Google Scholar] [CrossRef] [Green Version]
- Yant, L.J.; Ran, Q.; Rao, L.; Van Remmen, H.; Shibatani, T.; Belter, J.G.; Motta, L.; Richardson, A.; Prolla, T.A. The selenoprotein GPX4 is essential for mouse development and protects from radiation and oxidative damage insults. Free Radic. Biol. Med. 2003, 34, 496–502. [Google Scholar] [CrossRef]
- Dolma, S.; Lessnick, S.L.; Hahn, W.C.; Stockwell, B.R. Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell 2003, 3, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Tan, H.; Daniels, J.D.; Zandkarimi, F.; Liu, H.; Brown, L.M.; Uchida, K.; O’Connor, O.A.; Stockwell, B.R. Imidazole Ketone Erastin Induces Ferroptosis and Slows Tumor Growth in a Mouse Lymphoma Model. Cell Chem. Biol. 2019, 26, 623–633.e629. [Google Scholar] [CrossRef] [PubMed]
- Roh, J.L.; Kim, E.H.; Jang, H.J.; Park, J.Y.; Shin, D. Induction of ferroptotic cell death for overcoming cisplatin resistance of head and neck cancer. Cancer Lett. 2016, 381, 96–103. [Google Scholar] [CrossRef]
- Roh, J.L.; Kim, E.H.; Jang, H.; Shin, D. Aspirin plus sorafenib potentiates cisplatin cytotoxicity in resistant head and neck cancer cells through xCT inhibition. Free Radic. Biol. Med. 2017, 104, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Cramer, S.L.; Saha, A.; Liu, J.; Tadi, S.; Tiziani, S.; Yan, W.; Triplett, K.; Lamb, C.; Alters, S.E.; Rowlinson, S.; et al. Systemic depletion of L-cyst(e)ine with cyst(e)inase increases reactive oxygen species and suppresses tumor growth. Nat. Med. 2017, 23, 120–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, J.; Roh, J.-L. SLC7A11 as a Gateway of Metabolic Perturbation and Ferroptosis Vulnerability in Cancer. Antioxidants 2022, 11, 2444. https://doi.org/10.3390/antiox11122444
Lee J, Roh J-L. SLC7A11 as a Gateway of Metabolic Perturbation and Ferroptosis Vulnerability in Cancer. Antioxidants. 2022; 11(12):2444. https://doi.org/10.3390/antiox11122444
Chicago/Turabian StyleLee, Jaewang, and Jong-Lyel Roh. 2022. "SLC7A11 as a Gateway of Metabolic Perturbation and Ferroptosis Vulnerability in Cancer" Antioxidants 11, no. 12: 2444. https://doi.org/10.3390/antiox11122444