

Vitamin D3 and Ischemic Stroke: A Narrative Review

Abstract
:1. Introduction
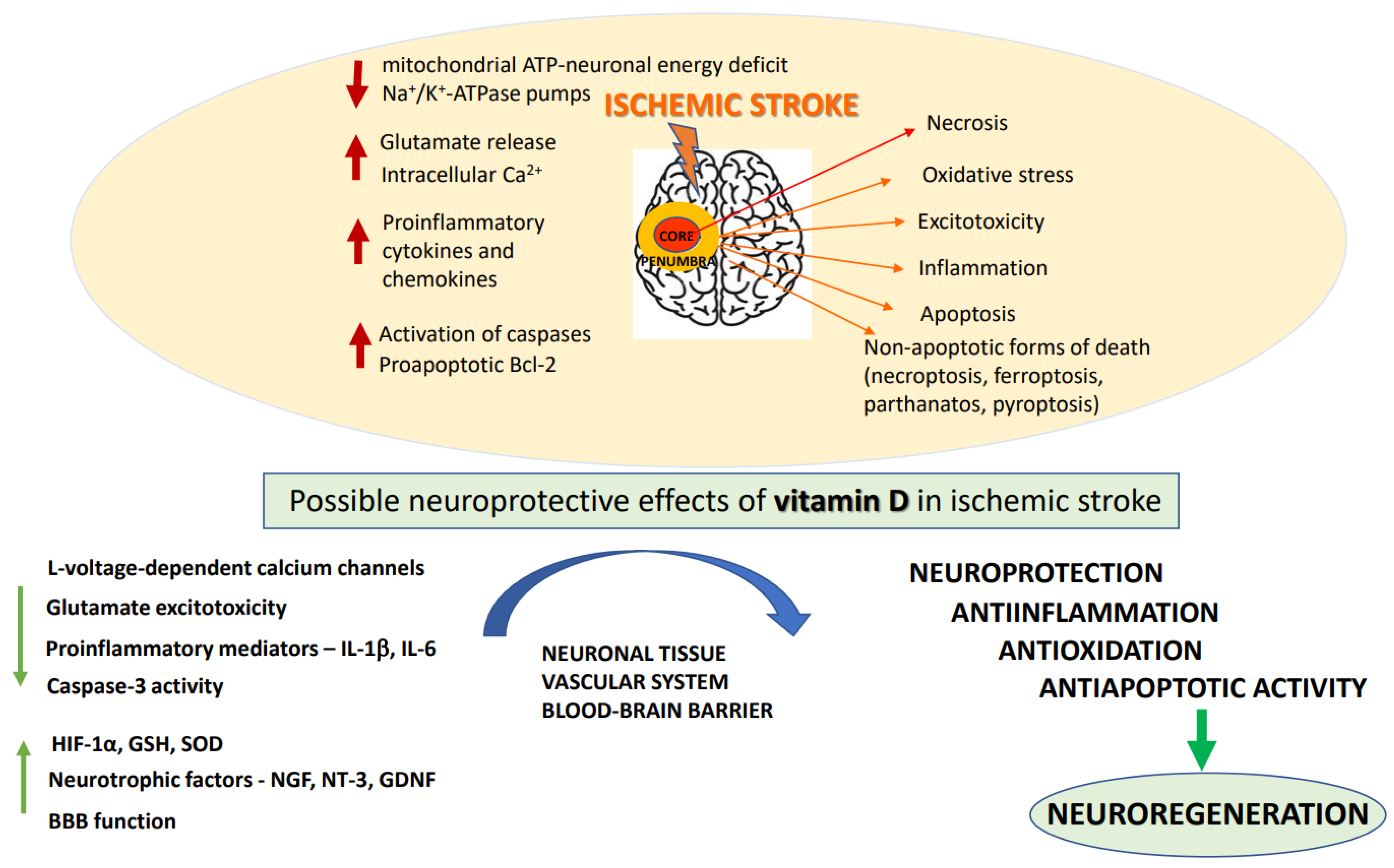
2. Pathomechanisms of Ischemic Stroke
2.1. Pathomorphological Features of Ischemic Stroke
2.2. Biochemical Basis of Ischemic Stroke and Neuroprotective Strategies of Its Treatment
3. The Basics of Vitamin D
3.1. Sources, Biosynthesis and Metabolism
3.2. Genomic and Non-Genomic Mechanisms of Vitamin D Action
3.3. Vitamin D Analogues
4. The Effects of Vitamin D3 in the CNS
5. The Effects of Calcitriol on Ischemia-Related Neuronal Injury—A Preclinical Evidence
5.1. In Vitro Experimental Studies
5.2. In Vivo Experimental Studies

{kind=link}
| Model | Animal Treatment | Effects of Vitamin D | Reference |
|---|---|---|---|
| Hypoxia ischemia (HI) rat model | Hypothermia treatment + NAC (50 mg/kg) + 1,25-(OH)2D3 (0.1 μg/kg/)/daily for 2 weeks | ↑motor skills ↓anxiety ↑spatial learning | [103] |
| Rat model of perinatal asphyxia in 7-day-old pups | 1,25(OH)2D3 (2 μg/kg, i.p., single dose)/30 min after the insult or for 6 consecutive days | ↓brain damage | [98] |
MCAO/R rat model of I/R injury | 1,25(OH)2D3 (1 μg/kg i.p.)/day/8 days before ischemia. DHA (250 mg/mL, from tail vein/30 min before MCAO/R | ↓MDA ↑GSH, SOD activity in cortex and corpus striatum in 1,25(OH)2D3 + DHA group | [104] |
| MCA ligation model in rat | 1,25(OH)2D3, 1 μg/kg/day, i.p., 4 or 8 days | ↓the amount of infarction in the cortex, ↑GDNF levels | [105] |
| MCAO/R model in C57BL6 mice | 1,25(OH)2D3 (100 ng/kg, i.p./day/5 day prior to stroke | ↓infarct volume ↓pro-inflammatory mediators IL-6, IL-1β, IL-23a, TGF-β and NADPH oxidase-2 | [106] |
| Spinal cord I/R injury in rabbit | 1,25(OH)2D3 (0.5 μg/kg, i.p./7 days before I/R) | ↓MDA, myeloperoxidase, xanthine oxidase activities, caspase-3 level, ↑catalase level, histopathological, ultrastructural, and neurological scores | [107] |
| BCAO model in Mongolian gerbils | 1,25(OH)2D3 1 μg/kg, i.p./day/7 days prior to ischemia. | ↓MMP-9 ↓lipid peroxidation ↓superoxide anion production ↑VDR expression | [108] |
| GCI model in rat | 1,25(OH)2D3 (1 μg/kg, i.p.)/30 min, 12 and 24 h after the GCI insult and PD98059 (5 μg, through the tail vein)/30 min prior to the insult | ↓brain edema, ↑neurological function, ↑ERK 1/2 pathway activation, ↓neuronal apoptosis | [109] |
| GCI model in rat | 1,25(OH)2D3 (1 μg/kg, i.p.)/30 min, 12 and 24 h after the GCI insult and PD98059 (5 μg, through the tail vein)/30 min prior to the insult | ↑the spatial learning and memory ↑neurological function ↓brain edema, ↓morphological defects in the CA1 area of the hippocampus ↓apoptosis ↑ VDR expression ↑ERK 1/2 pathway activation—PD98059 reversed the anti-apoptotic effect of 1,25(OH)2D3 | [110] |
| MCAO model in rat | 1,25(OH)2D3, 7 days prior to stroke induction | ↓lesion volume, ischemic neurobehavioral deficits, regulation of the glutamate receptor expression and CYP46A1 genes | [111] |
| MCAO model in rat | 1,25(OH)2D3 i.p., (a single dose of 2 μg/kg, immediately following ischemia) and subchronically (2 μg/kg on 6 consecutive days). | ↓infarct volumes 7 days following reperfusion, ↑NR3A and CREB activity in the hippocampal neurons, protection of the brain from I/R injury through the NR3A-MEK/ERK1/2-CREB pathway | [112] |
| Focal cortical ischemia (photothrombosis model) in rat | Lesioned rats were injected i.p. one hour after injury with either 1 μg 1,25(OH)2D3/kg or 7 μg 17β-estradiol/kg or a combination of both steroids | ↓HSP-27 within the infracted cerebral cortex | [114] |
| tMCAO model in rat | Progesterone (8 mg/kg), 1,25(OH)2D3 (1 μg/kg body weight/day) alone or in a combination, 5 min. i.p. prior to reperfusion followed by daily s.c. injections for 6 days. | ↓motor deficits, infarct reduction, ↑BDNF, TrkB and p-ERK1/2 expression, ↓apoptosis (↑Bcl-2, ↓caspase-3) ↓IL-6 and p-NF-κB ↑HO-1 | [100] |
| Focal cortical ischemia (photothrombosis model) in rat | Postlesional treatment with 1,25(OH)2D3 (4 μg/kg i.p.) | ↑glial HO-1 ↓GFAP | [115] |
| Focal cortical ischemia (photothrombosis model) in rat | 1,25(OH)2D3 (4 μg/kg i.p.) | no significant differences between 1,25(OH)2D3-treated and solvent-treated lesioned rats in neuronal COX-2 expression | [116] |
| MCAO model in female rat | Vit. D deficiency (VDD) diet for 8 weeks before MCAO; 10 μg/kg 1,25(OH)2D3, 4 h after MCAO and every 24 h thereafter for 5 days | VDD diet effects: ↑cortical and striatal infarct volumes, ↑severe poststroke behavioral impairment ↓IGF-I in plasma and the ischemic hemisphere, ↓IL-1α, IL-1β, IL-2, IL-4, IFN-γ, and IL-10 expression in ischemic brain tissue, ↑IL-6 Acute 1,25(OH)2D3 treatment did not improve infarct volume or behavioral performance | [117] |
| Hypoxia/reoxygenation (H/R) model in bEnd.3 cells | 1,25(OH)2D3 (5–200 nmol/L)/24 h before H/R, continued throughout the H/R period | ↑BBB function, zonula occludin-1, claudin-5, and occludin, ↓NF-κB ↓MMP-9 | [118] |
| MCAO model in rat | 1,25(OH)2D3 i.p. one group—12 μg/kg immediately after the ischemia period (60 min) second group—2 μg/kg after MCAO and over the next 5 days | ↓brain infarction volume, brain edema formation ↑BBB function ↑antioxidant enzyme activities ↓cell apoptosis ↑BDNF immunoreactivity | [119] |
| MCAO rat model | Vit. D3, 1000 IU/kg/day through gavage/14 days | ↓the size of cerebral infarction, ↑cerebral perfusion in the ischemic area ↑levels of vascular growth-related factor ↑micro-vessel density after cerebral infarction and ↑the proliferation of vascular endothelial cells in the ischemic cortex ↑Shh signaling in the ischemic cortex | [120] |
| MCAO rat model | Vit. D3 100 ng/kg i.v., vit. D3 nanoemulsion i.v. and intranasal (a mean size range of 49.29 ± 10.28 nm, equivalent to 100 ng/kg vit. D3 | ↑BBB permeation, deposition, and efficacy of vit. D3-nanoemulsion through the intranasal route in comparison i.v. vit. D3 or vit. D3 nanoemulsion | [121] |
| MCAO model in C57BL6 mice | 1,25(OH)2D3 (100 ng/kg, i.p./day/5 days before MCAO | ↓the volume of cerebral infarction ↓IL-6, IL-1β, IL-23a, TGF-β, Gp91phox | [106] |
| MCAO combined with CUMS in mice | Vit. D3 (6–50 μg/kg), icv/4 weeks | ↓motor dysfunction and depression-like behaviors ↑VDR expression and BDNF | [15] |
| tMCAO model in rat | Calcitriol 1 µg/kg, i.p., 7 consecutive days before experimental induction of stroke | ↓infarction volume ↓neurological deficits in brain, ↓MDA and NO levels ↑TAC level, ↑HO-1 and Nrf2 protein and mRNA | [124] |
5.3. The Effects of Calcitriol on Inflammatory Response in Ischemic Brain
6. Vitamin D3 Deficiency as a Risk Factor for HIE and Stroke Severity and Outcomes—Clinical Studies
| Objective | Participants | Vitamin D Administration/ Determination | Effect | Reference |
|---|---|---|---|---|
| vit. D metabolism in neonatal HIE and involvement cytokines related to Th17 function (*) | 50 HIE infants | serum samples from a multicenter randomized controlled trial of hypothermia 33 °C for 48 h after HIE birth vs. normothermia | ↓25(OH)D after birth in 70% of infants ↓IL-17E in all HIE neonates | [80] |
| vit. D as an adjuvant therapy for management of neonatal HIE | 60 HIE grade II neonates | vit. D3 (1000 IU, oral)/day/2 weeks and human recombinant erythropoietin (2500 IU/kg/s.c.)/day/5 days and magnesium sulphate 250 mg/kg/i.m. or i.v. half an hour of birth, and subsequently 125 mg/kg/24 and 48 h of life | before therapy: ↓serum 25(OH)D ↑serum S100-B after vit. D: ↓serum S100-B level | [134] |
| associations between serum 25(OH)D level and the ischemic infarct volume and long-term outcome | 96 AIS patients retrospective study | serum 25(OH)D level; calculation the volume of cerebral infarction | low 25(OH)D associated with higher infarct volumes and worse outcome | [136] |
| associations between serum 25(OH)D level and the functional outcome | 818 AIS patients | serum 25(OH)D level | ↑serum 25(OH)D in patients with good outcomes | [137] |
| associations between VDD and inflammatory markers, and short-term outcome | 168 AIS patients and 118 controls | serum 25(OH)D, IL-6, TNF-α, hsCRP level | in AIS patients: ↓25(OH)D, ↑frequency of VDD, ↑inflammatory markers (IL-6, hsCRP) | [138] |
| associations between 25(OH)D serum level and cardiovascular disease (CVD) or all-cause mortality | 387 patients with ischemic stroke | serum 25(OH)D | negative correlation between 25(OH)D and infarct volume | [139] |
| association between 25(OH)D serum level and severity of stroke | 986 stroke patients (629 males, 357 females) | serum 25(OH)D, apolipoprotein A-I, apolipoprotein B, ApoA-I/ApoB, cholesterol, fibrinogen, blood glucose, high-density lipoprotein, low-density lipoprotein cholesterol, triglyceride | female gender and higher blood fibrinogen level as the risk factors of VDD and higher severity of stroke | [141] |
| association between serum 25(OH)D and stroke severity | 90 ischemic stroke patients, 39 controls | serum 25(OH)D, DBP and VDR gene expression in leukocytes | negative correlation 25(OH)D levels and the severity of ischemic stroke no changes in DBP and VDR | [142] |
| associations of 25(OH)D with risk of mortality | 240 consecutive patients admitted within the 24 h after the onset of IS | serum 25(OH)D | severe VDD strong negative predictor for survival after IS | [143] |
| association of serum 25(OH)D with prevalent and incident stroke—population-based study | 9680 participants | serum 25(OH)D | severe VDD associated with a higher stroke risk | [145] |
| association between 25(OH)D and functional outcomes in stroke patients | 120 ischemic and hemorrhagic stroke patients participating the neurological rehabilitation program | serum 25(OH)D, motor functional status, cognitive status | correlation between 25(OH)D and cognitive impairment ↑25(OH)D associated with greater functional gain | [146] |
| vit. D supplementation and rehabilitation outcomes in patients having hemiplegia—a randomized, double-blind, placebo-controlled study (*) | 132 ischemic stroke patients hospitalized for 3-month hemiplegia rehabilitation | vit. D 300,000 IU or saline (i.m.), BRS, FAC, BBS-tests at the beginning and at the end of the rehabilitation program. | Significantly vit. D improved the BBS parameter | [147] |
| vit. D supplementation on stroke rehabilitation efficacy | 76 patients receiving inpatient stroke rehabilitation treatment | weekly vit. D (50,000 IU, orally) for 4–12 weeks, BRS and FAC scores before and after rehabilitation | higher changes in FAC and BRS scores in patients receiving vit. D. | [148] |
| vit. D supplementation on rehabilitation after acute stroke (*) | 100 patients after acute stroke multicentre, randomized, double-blind study | vit. D3 (2000 IU/day) or a placebo Barthel index scores | no significant differences between the groups | [149] |
| meta-analysis of association between vit. D status and the risk of stroke | 19 studies ischemic and hemorrhagic stroke | circulating vit. D/vit. D intake | low vit. D level associated with ischemic stroke | [150] |
| associations of 25(OH)D and risk of poor functional outcome in nondiabetic stroke | 266 nondiabetic Chinese stroke patients | serum 25(OH)D | VDD associated with an increased risk of poor functional outcome | [151] |
| vit. D, hypertension and ischemic stroke—Observational and genetic study | 11,6655 Danish individuals genotyped for genetic variants in DHCR7 and CYP2R1 | serum 25(OH)D blood pressure, hypertension and ischemic stroke | DHCR7 and CYP2R1 allele score associated with lower 25(OH)D and higher blood pressure and hypertension | [153] |
| association of 25(OH)D with incident stroke REGARDS study | 610 participants with incident stroke and 937 stroke-free individuals | serum 25(OH)D | low 25(OH)D associated with higher risk of stroke irresprective of black or white race | [154] |
| associations of 25(OH)D level and SNP status with incident stroke | 12,158 participants in ARIC study | serum 25(OH)D level; DBP SNPs: rs7041, rs4588 | low 25(OH)D level associated with higher stroke risk; possible associacion with DBP SNPs | [155] |
| association between vit. D metabolites, cognitive function and brain atrophy in elderly individuals | 390 community-dwelling elderly individuals with normal neurological status and without history of stroke and dementia | serum 25(OH)D3, 25(OH)D2 and 24,25(OH)2D3 | worse memory in individuals with low 25(OH)D and 24,25(OH)2D3 | [157] |
| association between vit. D and the development of PSD | 89 patients with acute ischemic stroke and 100 healthy controls | serum 25(OH)D | lower 25(OH)D level in non-PSD and PSD patients vs. healthy controls lower 25(OH)D level in PSD vs. non-PSD | [161] |
| association between vit. D and PSA | 226 first acute ischemic stroke patients and 100 healthy subjects | serum 25(OH)D | low serum 25(OH)D level in PSA and non-PSA patients vs. healthy subjects significant association between PSA and low 25(OH)D | [162] |
| association between vit. D supplementation and rehabilitation after stroke—randomized double blind, parallel, monocentric clinical trial (*) | 40 patients undergoing intensive neuro-rehabilitation treatment after stroke | cholecalciferol 2000 IU, oral/day/12 weeks, | no significant effect of vit. D supplementation on the beneficial effects of rehabilitation | [16] |
| association between 25(OH)D serum level with initial stroke severity and infarct volume | 235 patients who were admitted within 24 h of acute ischemic stroke onset | serum 25(OH)D the volume of cerebral infarction | low 25(OH)D level in acute ischemic stroke as early predictor of larger infarct volume and neurological deficits | [144] |
| associations of vit. D with stroke recurrence in a 3-month follow-up study | 349 Chinese patients with first-ever ischemic stroke | serum 25(OH)D | low serum 25(OH)D in patients with recurrent stroke | [140] |
| association between vit. D and rehabilitation in stroke patients | 100 stroke patients | serum 25(OH)D | rehabilitation efficacy positively correlated with 25(OH)D level and negatively | [163] |
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbrevations
| CNS | central nervous system |
| BBB | blood brain barrier |
| I/R | ischemia/reperfusion |
| VDR | vitamin D receptor |
| VDR-GP | VDR genomic pocket |
| VDR-AR | VDR alternative pocket |
| VDR-RXR | VDR- retinoic acid X receptor |
| VDREs | vitamin D responsive elements |
| PI3K | phosphatidylinositol 3 kinase |
| Akt | protein kinase B |
| GSK-3β | glycogen synthase kinase-3 beta |
| BDNF | brain derived neurotrophic factor |
| L-VGCC | L-type voltage-gated calcium channel |
| NT3 | neurotrophin 3 |
| GDNF | glial-derived neurotrophic factor |
| NGF | nerve growth factor |
| MCAO/R | middle cerebral artery occlusion/reperfusion |
| GSH | Glutathione |
| SOD | superoxide dismutase |
| MCA | middle cerebral artery |
| HIE | hypoxic-ischemic encephalopathy |
| ROS | reactive oxygen species |
| VDD | vitamin D deficient |
References
- Aho, K.; Harmsen, P.; Hatano, S.; Marquardsen, J.; Smirnov, V.E.; Strasser, T. Cerebrovascular Disease in the Community: Results of a WHO Collaborative Study. Bull. World Health Organ. 1980, 58, 113–130. [Google Scholar] [PubMed]
- Sacco, R.L.; Kasner, S.E.; Broderick, J.P.; Caplan, L.R.; Connors, J.J.; Culebras, A.; Elkind, M.S.V.; George, M.G.; Hamdan, A.D.; Higashida, R.T.; et al. An Updated Definition of Stroke for the 21st Century: A Statement for Healthcare Professionals from the American Heart Association/American Stroke Association. Stroke 2013, 44, 2064–2089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Naghavi, M.; Allen, C.; Barber, R.M.; Bhutta, Z.A.; Carter, A.; Casey, D.C.; Charlson, F.J.; Chen, A.Z.; Coates, M.M.; et al. Global, Regional, and National Life Expectancy, All-Cause Mortality, and Cause-Specific Mortality for 249 Causes of Death, 1980–2015: A Systematic Analysis for the Global Burden of Disease Study 2015. Lancet 2016, 388, 1459–1544. [Google Scholar] [CrossRef] [Green Version]
- Swanepoel, A.C.; Pretorius, E. Prevention and Follow-up in Thromboembolic Ischemic Stroke: Do We Need to Think out of the Box? Thromb. Res. 2015, 136, 1067–1073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berger, J.R. COVID-19 and the Nervous System. J. Neurovirol. 2020, 26, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Higgins, V.; Sohaei, D.; Diamandis, E.P.; Prassas, I. COVID-19: From an Acute to Chronic Disease? Potential Long-Term Health Consequences. Crit. Rev. Clin. Lab. Sci. 2021, 58, 297–310. [Google Scholar] [CrossRef]
- Montalvan, V.; Lee, J.; Bueso, T.; De Toledo, J.; Rivas, K. Neurological Manifestations of COVID-19 and Other Coronavirus Infections: A Systematic Review. Clin. Neurol. Neurosurg. 2020, 194, 105921. [Google Scholar] [CrossRef]
- Nannoni, S.; de Groot, R.; Bell, S.; Markus, H.S. Stroke in COVID-19: A Systematic Review and Meta-Analysis. Int. J. Stroke 2021, 16, 137–149. [Google Scholar] [CrossRef]
- Sagris, D.; Papanikolaou, A.; Kvernland, A.; Korompoki, E.; Frontera, J.A.; Troxel, A.B.; Gavriatopoulou, M.; Milionis, H.; Lip, G.Y.H.; Michel, P.; et al. COVID-19 and Ischemic Stroke. Eur. J. Neurol. 2021, 28, 3826–3836. [Google Scholar] [CrossRef]
- Silva Andrade, B.; Siqueira, S.; de Assis Soares, W.R.; de Souza Rangel, F.; Santos, N.O.; dos Santos Freitas, A.; Ribeiro da Silveira, P.; Tiwari, S.; Alzahrani, K.J.; Góes-Neto, A.; et al. Long-COVID and Post-COVID Health Complications: An Up-to-Date Review on Clinical Conditions and Their Possible Molecular Mechanisms. Viruses 2021, 13, 700. [Google Scholar] [CrossRef]
- Albers, G.W.; Goldstein, L.B.; Hess, D.C.; Wechsler, L.R.; Furie, K.L.; Gorelick, P.B.; Hurn, P.; Liebeskind, D.S.; Nogueira, R.G.; Saver, J.L. Stroke Treatment Academic Industry Roundtable (STAIR) Recommendations for Maximizing the Use of Intravenous Thrombolytics and Expanding Treatment Options with Intra-Arterial and Neuroprotective Therapies. Stroke 2011, 42, 2645–2650. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Gooch, H.; Petty, A.; McGrath, J.J.; Eyles, D. Vitamin D and the Brain: Genomic and Non-Genomic Actions. Mol. Cell. Endocrinol. 2017, 453, 131–143. [Google Scholar] [CrossRef] [PubMed]
- Abbott, N.J.; Rönnbäck, L.; Hansson, E. Astrocyte-Endothelial Interactions at the Blood-Brain Barrier. Nat. Rev. Neurosci. 2006, 7, 41–53. [Google Scholar] [CrossRef]
- Iadecola, C. The Neurovascular Unit Coming of Age: A Journey through Neurovascular Coupling in Health and Disease. Neuron 2017, 96, 17–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Liang, L. Vitamin D3/Vitamin D Receptor Signaling Mitigates Symptoms of Post-Stroke Depression in Mice by Upregulating Hippocampal BDNF Expression. Neurosci. Res. 2021, 170, 306–313. [Google Scholar] [CrossRef] [PubMed]
- Torrisi, M.; Bonanno, L.; Formica, C.; Arcadi, F.A.; Cardile, D.; Cimino, V.; Bramanti, P.; Morini, E. The Role of Rehabilitation and Vitamin D Supplementation on Motor and Psychological Outcomes in Poststroke Patients. Medicine 2021, 100, e27747. [Google Scholar] [CrossRef] [PubMed]
- Marek, K.; Cicho, N. The Role of Vitamin D in Stroke Prevention and the Effects of Its Supplementation for Post-Stroke Rehabilitation. Nutrients 2022, 14, 2761. [Google Scholar] [CrossRef] [PubMed]
- Detante, O.; Jaillard, A.; Moisan, A.; Barbieux, M.; Favre, I.M.; Garambois, K.; Hommel, M.; Remy, C. Biotherapies in Stroke. Rev. Neurol. 2014, 170, 779–798. [Google Scholar] [CrossRef] [PubMed]
- Hossmann, K.-A. Viability Thresholds and the Penumbra of Focal Ischemia. Ann. Neurol. 1994, 36, 557–565. [Google Scholar] [CrossRef]
- Sakai, S.; Shichita, T. Inflammation and Neural Repair after Ischemic Brain Injury. Neurochem. Int. 2019, 130, 104316. [Google Scholar] [CrossRef]
- Doyle, K.; Simon, R.; Stenzel-poore, M. Mechanisms of Ischemic Brain Damage—Review Article. Neuropharmacology 2008, 55, 310–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sekerdag, E.; Solaroglu, I.; Gursoy-Ozdemir, Y. Cell Death Mechanisms in Stroke and Novel Molecular and Cellular Treatment Options. Curr. Neuropharmacol. 2018, 16, 1396–1415. [Google Scholar] [CrossRef] [PubMed]
- Radak, D.; Katsiki, N.; Resanovic, I.; Jovanovic, A.; Sudar-Milovanovic, E.; Zafirovic, S.; Mousad, S.A.; Isenovic, E.R. Apoptosis and Acute Brain Ischemia in Ischemic Stroke. Curr. Vasc. Pharmacol. 2016, 15, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Rossi, D.J.; Oshima, T.; Attwell, D. Glutamate Release in Severe Brain Ischaemia Is Mainly by Reversed Uptake. Nature 2000, 403, 316–321. [Google Scholar] [CrossRef]
- Krzyzanowska, W.; Pomierny, B.; Filip, M.; Pera, J. Glutamate Transporters in Brain Ischemia: To Modulate or Not? Acta Pharmacol. Sin. 2014, 35, 444–462. [Google Scholar] [CrossRef] [Green Version]
- Choi, D.W. Excitotoxicity: Still Hammering the Ischemic Brain in 2020. Front. Neurosci. 2020, 14, 579953. [Google Scholar] [CrossRef]
- Bruno, V.; Battaglia, G.; Copani, A.; D’Onofrio, M.; Di Iorio, P.; De Blasi, A.; Melchiorri, D.; Flor, P.J.; Nicoletti, F. Metabotropic Glutamate Receptor Subtypes as Targets for Neuroprotective Drugs. J. Cereb. Blood Flow Metab. 2001, 21, 1013–1033. [Google Scholar] [CrossRef] [Green Version]
- Ogden, K.K.; Traynelis, S.F. New Advances in NMDA Receptor Pharmacology. Trends Pharmacol. Sci. 2011, 32, 726–733. [Google Scholar] [CrossRef] [Green Version]
- Hardingham, G.E.; Fukunaga, Y.; Bading, H. Extrasynaptic NMDARs Oppose Synaptic NMDARs by Triggering CREB Shut-off and Cell Death Pathways. Nat. Neurosci. 2002, 5, 405–414. [Google Scholar] [CrossRef]
- Parsons, M.P.; Raymond, L.A. Extrasynaptic NMDA Receptor Involvement in Central Nervous System Disorders. Neuron 2014, 82, 279–293. [Google Scholar] [CrossRef]
- Yan, J.; Peter Bengtson, C.; Buchthal, B.; Hagenston, A.M.; Bading, H. Coupling of NMDA Receptors and TRPM4 Guides Discovery of Unconventional Neuroprotectants. Science 2020, 370, eaay3302. [Google Scholar] [CrossRef] [PubMed]
- Szydlowska, K.; Tymianski, M. Calcium, Ischemia and Excitotoxicity. Cell Calcium 2010, 47, 122–129. [Google Scholar] [CrossRef]
- Crack, P.J.; Taylor, J.M. Reactive Oxygen Species and the Modulation of Stroke. Free Radic. Biol. Med. 2005, 38, 1433–1444. [Google Scholar] [CrossRef] [PubMed]
- Gürsoy-Özdemir, Y.; Can, A.; Dalkara, T. Reperfusion-Induced Oxidative/Nitrativie Injury to Neurovascular Unit after Focal Cerebral Ischemia. Stroke 2004, 35, 1449–1453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Datta, A.; Sarmah, D.; Mounica, L.; Kaur, H.; Kesharwani, R.; Verma, G.; Veeresh, P.; Kotian, V.; Kalia, K.; Borah, A.; et al. Cell Death Pathways in Ischemic Stroke and Targeted Pharmacotherapy. Transl. Stroke Res. 2020, 11, 1185–1202. [Google Scholar] [CrossRef]
- Tuo, Q.Z.; Zhang, S.T.; Lei, P. Mechanisms of Neuronal Cell Death in Ischemic Stroke and Their Therapeutic Implications. Med. Res. Rev. 2022, 42, 259–305. [Google Scholar] [CrossRef]
- Zhou, Y.; Liao, J.; Mei, Z.; Liu, X.; Ge, J. Insight into Crosstalk between Ferroptosis and Necroptosis: Novel Therapeutics in Ischemic Stroke. Oxid. Med. Cell. Longev. 2021, 2021, 9991001. [Google Scholar] [CrossRef]
- Kono, H.; Rock, K.L. How Dying Cells Alert the Immune System to Danger. Nat. Rev. Immunol. 2008, 8, 279–289. [Google Scholar] [CrossRef]
- Bohacek, I.; Cordeau, P.; Lalancette-Hébert, M.; Gorup, D.; Weng, Y.C.; Gajovic, S.; Kriz, J. Toll-like Receptor 2 Deficiency Leads to Delayed Exacerbation of Ischemic Injury. J. Neuroinflamm. 2012, 9, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Linnik, M.D.; Zobrist, R.H.; Hatfield, M.D. Evidence Supporting a Role for Programmed Cell Death in Focal Cerebral Ischemia in Rats. Stroke 1993, 24, 2002–2008. [Google Scholar] [CrossRef]
- Alonso De Leciñana, M.; Díez-Tejedor, E.; Gutierrez, M.; Guerrero, S.; Carceller, F.; Roda, J.M. New Goals in Ischemic Stroke Therapy: The Experimental Approach—Harmonizing Science with Practice. Cerebrovasc. Dis. 2005, 20, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Uzdensky, A.B. Apoptosis Regulation in the Penumbra after Ischemic Stroke: Expression of pro- and Antiapoptotic Proteins. Apoptosis 2019, 24, 687–702. [Google Scholar] [CrossRef] [PubMed]
- Charriaut-Marlangue, C.; Margaill, I.; Represa, A.; Popovici, T.; Plotkine, M.; Ben-Ari, Y. Apoptosis and Necrosis after Reversible Focal Ischemia: An in Situ DNA Fragmentation Analysis. J. Cereb. Blood Flow Metab. 1996, 16, 186–194. [Google Scholar] [CrossRef] [PubMed]
- MacManus, J.P.; Hill, I.E.; Huang, Z.-G.; Rasquinha, I.; Xue, D.; Buchan, A.M. DNA Damage Consistent with Apoptosis in Transient Focal Ischaemic Neocortex. Neuroreport 1994, 5, 493–496. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, G.; Granger, D.N. Cell Adhesion Molecules and Ischemic Stroke. Neurol. Res. 2008, 30, 783–793. [Google Scholar] [CrossRef]
- Kassis, H.; Shehadah, A.; Chopp, M.; Zhang, Z.G. Epigenetics in Stroke Recovery. Genes 2017, 8, 89. [Google Scholar] [CrossRef] [Green Version]
- Hu, Z.; Zhong, B.; Tan, J.; Chen, C.; Lei, Q.; Zeng, L. The Emerging Role of Epigenetics in Cerebral Ischemia. Mol. Neurobiol. 2017, 54, 1887–1905. [Google Scholar] [CrossRef]
- Bendik, I.; Friedel, A.; Roos, F.F.; Weber, P.; Eggersdorfer, M. Vitamin D: A Critical and Essential Micronutrient for Human Health. Front. Physiol. 2014, 5, 248. [Google Scholar] [CrossRef]
- Norman, A.W. From Vitamin D to Hormone D: Fundamentals of the Vitamin D Endocrine System Essential for Good Health. Am. J. Clin. Nutr. 2008, 88, 491S–499S. [Google Scholar] [CrossRef] [Green Version]
- Bikle, D.D. Vitamin D Metabolism, Mechanism of Action, and Clinical Applications. Chem. Biol. 2014, 21, 319–329. [Google Scholar] [CrossRef]
- Gil, Á.; Plaza-Diaz, J.; Mesa, M.D. Vitamin D: Classic and Novel Actions. Ann. Nutr. Metab. 2018, 72, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Carlberg, C. Nutrigenomics of Vitamin D. Nutrients 2019, 11, 676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norlin, M. Effects of Vitamin D in the Nervous System: Special Focus on Interaction with Steroid Hormone Signalling and a Possible Role in the Treatment of Brain Cancer. J. Neuroendocrinol. 2020, 32, e12799. [Google Scholar] [CrossRef] [PubMed]
- Bivona, G.; Gambino, C.M.; Iacolino, G.; Ciaccio, M. Vitamin D and the Nervous System. Neurol. Res. 2019, 41, 827–835. [Google Scholar] [CrossRef] [PubMed]
- Slominski, A.T.; Janjetovic, Z.; Fuller, B.E.; Zmijewski, M.A.; Tuckey, R.C.; Nguyen, M.N.; Sweatman, T.; Li, W.; Zjawiony, J.; Miller, D.; et al. Products of Vitamin D3 or 7-Dehydrocholesterol Metabolism by Cytochrome P450scc Show Anti-Leukemia Effects, Having Low or Absent Calcemic Activity. PLoS ONE 2010, 5, 7–10. [Google Scholar] [CrossRef]
- Slominski, A.T.; Kim, T.; Hobrath, J.V.; Oak, A.S.W.; Edith, K.Y.; Tieu, E.W.; Li, W.; Tuckey, R.C.; Jetten, A.M. Endogenously Produced Nonclassical Vitamin D Hydroxy-Metabolites Act as “Biased” Agonists on Vdr and Inverse Agonists on Rorα and Rorγ. J. Steroid Biochem. Mol. Biol. 2017, 173, 42–56. [Google Scholar] [CrossRef] [Green Version]
- Christakos, S.; Dhawan, P.; Verstuyf, A.; Verlinden, L.; Carmeliet, G. Vitamin D: Metabolism, Molecular Mechanism of Action, and Pleiotropic Effects. Physiol. Rev. 2015, 96, 365–408. [Google Scholar] [CrossRef] [Green Version]
- Bikle, D.D.; Schwartz, J. Vitamin D Binding Protein, Total and Free Vitamin D Levels in Different Physiological and Pathophysiological Conditions. Front. Endocrinol. 2019, 10, 317. [Google Scholar] [CrossRef] [Green Version]
- Wimalawansa, S.J. Vitamin D deficiency: Effects on oxidative stress, epigenetics, gene regulation, and aging. Biology 2019, 8, 30. [Google Scholar] [CrossRef] [Green Version]
- Garcion, E.; Wion-Barbot, N.; Montero-Menei, C.N.; Berger, F.; Wion, D. New Clues about Vitamin D Functions in the Nervous System. Trends Endocrinol. Metab. 2002, 13, 100–105. [Google Scholar] [CrossRef]
- Zmijewski, M.A.; Carlberg, C. Vitamin D Receptor(s): In the Nucleus but Also at Membranes? Exp. Dermatol. 2020, 29, 876–884. [Google Scholar] [CrossRef]
- Haussler, M.R.; Jurutka, P.W.; Mizwicki, M.; Norman, A.W. Vitamin D Receptor (VDR)-Mediated Actions of 1α,25(OH) 2 Vitamin D 3: Genomic and Non-Genomic Mechanisms. Best Pract. Res. Clin. Endocrinol. Metab. 2011, 25, 543–559. [Google Scholar] [CrossRef]
- Norman, A.W. Minireview: Vitamin D Receptor: New Assignments for an Already Busy Receptor. Endocrinology 2006, 147, 5542–5548. [Google Scholar] [CrossRef] [Green Version]
- Shaffer, P.L.; Gewirth, D.T. Structural Basis of VDR-DNA Interactions on Direct Repeat Response Elements. EMBO J. 2002, 21, 2242–2252. [Google Scholar] [CrossRef] [Green Version]
- Pertile, R.A.N.; Cui, X.; Eyles, D.W. Vitamin D Signaling and the Differentiation of Developing Dopamine Systems. Neuroscience 2016, 333, 193–203. [Google Scholar] [CrossRef]
- Alroy, I.; Towers, T.L.; Freedman, L.P. Transcriptional Repression of the Interleukin-2 Gene by Vitamin D3: Direct Inhibition of NFATp/AP-1 Complex Formation by a Nuclear Hormone Receptor. Mol. Cell. Biol. 1995, 15, 5789–5799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurlek, A.; Pittelkow, M.R.; Kumar, R. Modulation of Growth Factor/Cytokine Synthesis and Signaling by 1α,25-Dihydroxyvitamin D3: Implications in Cell Growth and Differentiation. Endocr. Rev. 2002, 23, 763–786. [Google Scholar] [CrossRef] [PubMed]
- Snegarova, V.; Naydenova, D. Vitamin D: A Review of Its Effects on Epigenetics and Gene Regulation. Folia Med. 2020, 62, 662–669. [Google Scholar] [CrossRef] [PubMed]
- Ricca, C.; Aillon, A.; Bergandi, L.; Alotto, D.; Castagnoli, C.; Silvagno, F. Vitamin D Receptor Is Necessary for Mitochondrial Function and Cell Health. Int. J. Mol. Sci. 2018, 19, 1672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Tang, Z.; Slominski, A.T.; Li, W.; Żmijewski, M.A.; Liu, Y.; Chen, J. Vitamin D and Its Analogs as Anticancer and Anti-Inflammatory Agents. Eur. J. Med. Chem. 2020, 207, 112738. [Google Scholar] [CrossRef]
- Leyssens, C.; Verlinden, L.; Verstuyf, A. The Future of Vitamin D Analogs. Front. Physiol. 2014, 5, 122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maestro, M.A.; Molnár, F.; Carlberg, C. Vitamin D and Its Synthetic Analogs. J. Med. Chem. 2019, 62, 6854–6875. [Google Scholar] [CrossRef] [PubMed]
- Regulska, M.; Leśkiewicz, M.; Budziszewska, B.; Kutner, A.; Jantas, D.; Basta-Kaim, A.; Kubera, M.; Jaworska-Feil, L.; Lasoń, W. Inhibitory Effects of 1,25-Dihydroxyvitamin D3 and Its Low-Calcemic Analogues on Staurosporine-Induced Apoptosis. Pharmacol. Rep. 2007, 59, 393–401. [Google Scholar] [PubMed]
- Regulska, M.; Leśkiewicz, M.; Budziszewska, B.; Kutner, A.; Basta-Kaim, A.; Kubera, M.; Jaworska-Feil, L.; Lasoń, W. Involvement of P13-K in Neuroprotective Effects of the 1,25-Dihydroxyvitamin D3 Analogue—PRI-2191. Pharmacol. Rep. 2006, 58, 900–907. [Google Scholar]
- Pierucci, F.; Garcia-Gil, M.; Frati, A.; Bini, F.; Martinesi, M.; Vannini, E.; Mainardi, M.; Luzzati, F.; Peretto, P.; Caleo, M.; et al. Vitamin D3 Protects against Aβ Peptide Cytotoxicity in Differentiated Human Neuroblastoma SH-SY5Y Cells: A Role for S1P1/P38MAPK/ATF4 Axis. Neuropharmacology 2017, 116, 328–342. [Google Scholar] [CrossRef]
- Grimm, M.O.W.; Thiel, A.; Lauer, A.A.; Winkler, J.; Lehmann, J.; Regner, L.; Nelke, C.; Janitschke, D.; Benoist, C.; Streidenberger, O.; et al. Vitamin D and Its Analogues Decrease Amyloid-β (Aβ) Formation and Increase Aβ-Degradation. Int. J. Mol. Sci. 2017, 18, 2764. [Google Scholar] [CrossRef] [Green Version]
- Eyles, D.W.; Smith, S.; Kinobe, R.; Hewison, M.; McGrath, J.J. Distribution of the Vitamin D Receptor and 1α-Hydroxylase in Human Brain. J. Chem. Neuroanat. 2005, 29, 21–30. [Google Scholar] [CrossRef]
- Cui, X.; Gooch, H.; Groves, N.J.; Sah, P.; Burne, T.H.; Eyles, D.W.; McGrath, J.J. Vitamin D and the Brain: Key Questions for Future Research. J. Steroid Biochem. Mol. Biol. 2015, 148, 305–309. [Google Scholar] [CrossRef] [Green Version]
- Lang, F.; Ma, K.; Leibrock, C.B. 1,25(OH)2D3 in Brain Function and Neuropsychiatric Disease. Neurosignals 2019, 27, 40–49. [Google Scholar] [CrossRef]
- Lowe, D.W.; Hollis, B.W.; Wagner, C.L.; Bass, T.; Kaufman, D.A.; Horgan, M.J.; Givelichian, L.M.; Sankaran, K.; Yager, J.Y.; Katikaneni, L.D.; et al. Vitamin D Insufficiency in Neonatal Hypoxic–ischemic Encephalopathy. Pediatr. Res. 2017, 82, 55–62. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.B.; Lin, A.M.Y.; Chiu, T.H. Systemic Vitamin D3 Attenuated Oxidative Injuries in the Locus Coeruleus of Rat Brain. Ann. NY Acad. Sci. 2003, 993, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Landel, V.; Stephan, D.; Cui, X.; Eyles, D.; Feron, F. Differential Expression of Vitamin D-Associated Enzymes and Receptors in Brain Cell Subtypes. J. Steroid Biochem. Mol. Biol. 2018, 177, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Neveu, I.; Naveilhan, P.; Jehan, F.; Baudet, C.; Wion, D.; De Luca, H.F.; Brachet, P. 1,25-Dihydroxyvitamin D3 Regulates the Synthesis of Nerve Growth Factor in Primary Cultures of Glial Cells. Mol. Brain Res. 1994, 24, 70–76. [Google Scholar] [CrossRef]
- Neveu, I.; Naveilhan, P.; Baudet, C.; Brachet, P.; Metsis, M. 1,25-Dihydroxyvitamin D3 Regulates NT-3, NT-4 but Not BDNF MRNA in Astrocytes. Neuroreport 1994, 6, 124–126. [Google Scholar] [CrossRef]
- Pertile, R.A.N.; Cui, X.; Hammond, L.; Eyles, D.W. Vitamin D Regulation of GDNF/Ret Signaling in Dopaminergic Neurons. FASEB J. 2018, 32, 819–828. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.-I.; Chang, Y.; Kao, N.; Lee, W.; Cross, T.; Lin, S. 1,25(OH)2D3 Alleviates Aβ(25-35)-Induced Tau Hyperphosphorylation, Excessive Reactive Oxygen Species, and Apoptosis Through Interplay with Glial Cell Line-Derived Neurotrophic Factor Signaling in SH-SY5Y Cells. Int. J. Mol. Sci. 2020, 21, 4215. [Google Scholar] [CrossRef]
- McGrath, J.J.; Féron, F.P.; Burne, T.H.J.; MacKay-Sim, A.; Eyles, D.W. Vitamin D3—Implications for Brain Development. J. Steroid Biochem. Mol. Biol. 2004, 89–90, 557–560. [Google Scholar] [CrossRef]
- Ilie, P.C.; Stefanescu, S.; Smith, L. The Role of Vitamin D in the Prevention of Coronavirus Disease 2019 Infection and Mortality. Aging Clin. Exp. Res. 2020, 32, 1195–1198. [Google Scholar] [CrossRef]
- Eyles, D.; Almeras, L.; Benech, P.; Patatian, A.; Mackay-Sim, A.; McGrath, J.; Féron, F. Developmental Vitamin D Deficiency Alters the Expression of Genes Encoding Mitochondrial, Cytoskeletal and Synaptic Proteins in the Adult Rat Brain. J. Steroid Biochem. Mol. Biol. 2007, 103, 538–545. [Google Scholar] [CrossRef]
- Levenson, C.W.; Figueirôa, S.M. Gestational Vitamin D Deficiency: Long-Term Effects on the Brain. Nutr. Rev. 2008, 66, 726–729. [Google Scholar] [CrossRef]
- Monteggia, L.M.; Björkholm, C. BDNF—A Key Transducer of Antidepressant Effects. Neuropharmacology 2016, 102, 72–79. [Google Scholar] [CrossRef]
- Duman, R.S.; Deyama, S.; Fogaça, M.V. Role of BDNF in the Pathophysiology and Treatment of Depression: Activity-dependent Effects Distinguish Rapid-acting Antidepressants. Eur. J. Neurosci. 2021, 53, 126–139. [Google Scholar] [CrossRef] [PubMed]
- Kouba, B.R.; Camargo, A.; Gil-Mohapel, J.; Rodrigues, A.L.S. Molecular Basis Underlying the Therapeutic Potential of Vitamin D for the Treatment of Depression and Anxiety. Int. J. Mol. Sci. 2022, 23, 7077. [Google Scholar] [CrossRef] [PubMed]
- Marazziti, D.; Parra, E.; Palermo, S.; Barberi, F.M.; Buccianelli, B.; Ricciardulli, S.; Cappelli, A.; Mucci, F.; Dell’Osso, L. Vitamin D: A Pleiotropic Hormone with Possible Psychotropic Activities. Curr. Med. Chem. 2021, 28, 3843–3864. [Google Scholar] [CrossRef]
- Brewer, L.D.; Thibault, V.; Chen, K.C.; Langub, M.C.; Landfield, P.W.; Porter, N.M. Vitamin D Hormone Confers Neuroprotection in Parallel with Downregulation of L-Type Calcium Channel Expression in Hippocampal Neurons. J. Neurosci. 2001, 21, 98–108. [Google Scholar] [CrossRef] [Green Version]
- Ibi, M.; Sawada, H.; Nakanishi, M.; Kume, T.; Katsuki, H.; Kaneko, S.; Shimohama, S.; Akaike, A. Protective Effects of 1α,25-(OH)2D3 against the Neurotoxicity of Glutamate and Reactive Oxygen Species in Mesencephalic Culture. Neuropharmacology 2001, 40, 761–771. [Google Scholar] [CrossRef]
- Taniura, H.; Ito, M.; Sanada, N.; Kuramoto, N.; Ohno, Y.; Nakamichi, N.; Yoneda, Y. Chronic Vitamin D3 Treatment Protects against Neurotoxicity by Glutamate in Association with Upregulation of Vitamin D Receptor MRNA Expression in Cultured Rat Cortical Neurons. J. Neurosci. Res. 2006, 83, 1179–1189. [Google Scholar] [CrossRef]
- Kajta, M.; Makarewicz, D.; Ziemińska, E.; Jantas, D.; Domin, H.; Lasoń, W.; Kutner, A.; Łazarewicz, J.W. Neuroprotection by Co-Treatment and Post-Treating with Calcitriol Following the Ischemic and Excitotoxic Insult in Vivo and in Vitro. Neurochem. Int. 2009, 55, 265–274. [Google Scholar] [CrossRef]
- Atif, F.; Sayeed, I.; Ishrat, T.; Stein, D.G. Progesterone with Vitamin D Affords Better Neuroprotection against Excitotoxicity in Cultured Cortical Neurons than Progesterone Alone. Mol. Med. 2009, 15, 328–336. [Google Scholar] [CrossRef]
- Atif, F.; Yousuf, S.; Sayeed, I.; Ishrat, T.; Hua, F.; Stein, D.G. Combination Treatment with Progesterone and Vitamin D Hormone Is More Effective than Monotherapy in Ischemic Stroke: The Role of BDNF/TrkB/Erk1/2 Signaling in Neuroprotection. Neuropharmacology 2013, 67, 78–87. [Google Scholar] [CrossRef] [Green Version]
- Loginova, M.; Mishchenko, T.; Savyuk, M.; Guseva, S.; Gavrish, M.; Krivonosov, M.; Ivanchenko, M.; Fedotova, J.; Vedunova, M. Double-Edged Sword of Vitamin D3 Effects on Primary Neuronal Cultures in Hypoxic States. Int. J. Mol. Sci. 2021, 22, 5417. [Google Scholar] [CrossRef] [PubMed]
- Stessman, L.E.; Peeples, E.S. Vitamin D and Its Role in Neonatal Hypoxic-Ischemic Brain Injury. Neonatology 2018, 113, 305–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lowe, D.W.; Fraser, J.L.; Rollins, L.G.; Bentzley, J.; Nie, X.; Martin, R.; Singh, I.; Jenkins, D. Vitamin D Improves Functional Outcomes in Neonatal Hypoxic Ischemic Male Rats Treated with N-Acetylcysteine and Hypothermia. Neuropharmacology 2017, 123, 186–200. [Google Scholar] [CrossRef] [PubMed]
- Ekici, F.; Ozyurt, B.; Erdogan, H. The Combination of Vitamin D3 and Dehydroascorbic Acid Administration Attenuates Brain Damage in Focal Ischemia. Neurol. Sci. 2009, 30, 207–212. [Google Scholar] [CrossRef]
- Wang, Y.; Chiang, Y.H.; Su, T.P.; Hayashi, T.; Morales, M.; Hoffer, B.J.; Lin, S.Z. Vitamin D3 Attenuates Cortical Infarction Induced by Middle Cerebral Arterial Ligation in Rats. Neuropharmacology 2000, 39, 873–880. [Google Scholar] [CrossRef]
- Evans, M.A.; Kim, H.A.; Ling, Y.H.; Uong, S.; Vinh, A.; De Silva, T.M.; Arumugam, T.V.; Clarkson, A.N.; Zosky, G.R.; Drummond, G.R.; et al. Vitamin D3 Supplementation Reduces Subsequent Brain Injury and Inflammation Associated with Ischemic Stroke. NeuroMol. Med. 2018, 20, 147–159. [Google Scholar] [CrossRef] [Green Version]
- Gürer, B.; Karakoç, A.; Bektaşoğlu, P.K.; Kertmen, H.; Kanat, M.A.; Arıkök, A.T.; Ergüder, B.İ.; Sargon, M.F.; Öztürk, Ö.Ç.; Çelikoğlu, E. Comparative Effects of Vitamin D and Methylprednisolone against Ischemia/Reperfusion Injury of Rabbit Spinal Cords. Eur. J. Pharmacol. 2017, 813, 50–60. [Google Scholar] [CrossRef]
- Velimirović, M.; Dožudić, G.J.; Selaković, V.; Stojković, T.; Puškaš, N.; Zaletel, I.; Živković, M.; Dragutinović, V.; Nikolić, T.; Jelenković, A.; et al. Effects of Vitamin D3 on the NADPH Oxidase and Matrix Metalloproteinase 9 in an Animal Model of Global Cerebral Ischemia. Oxid. Med. Cell. Longev. 2018, 2018, 3273654. [Google Scholar] [CrossRef] [Green Version]
- Yuan, J.; Guo, X.; Liu, Z.; Zhao, X.; Feng, Y.; Song, S.; Cui, C.; Jiang, P. Vitamin D Receptor Activation Influences the ERK Pathway and Protects against Neurological Deficits and Neuronal Death. Int. J. Mol. Med. 2018, 41, 364–372. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.; Yuan, J.; Wang, J.; Cui, C.; Jiang, P. Calcitriol Alleviates Global Cerebral Ischemia-Induced Cognitive Impairment by Reducing Apoptosis Regulated by VDR/ERK Signaling Pathway in Rat Hippocampus. Brain Res. 2019, 1724, 146430. [Google Scholar] [CrossRef]
- Khassafi, N.; Zahraei, Z.; Vahidinia, Z.; Karimian, M.; Azami Tameh, A. Calcitriol Pretreatment Attenuates Glutamate Neurotoxicity by Regulating NMDAR and CYP46A1 Gene Expression in Rats Subjected to Transient Middle Cerebral Artery Occlusion. J. Neuropathol. Exp. Neurol. 2022, 81, 252–259. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Xue, R.; Gu, J.; Xiao, Y.; Zhong, H.; Pan, X.; Ran, R. Neuroprotective Effect of Calcitriol on Ischemic/Reperfusion Injury through the NR3A/CREB Pathways in the Rat Hippocampus. Mol. Med. Rep. 2013, 8, 1708–1714. [Google Scholar] [CrossRef] [Green Version]
- Losem-Heinrichs, E.; Görg, B.; Schleicher, A.; Redecker, C.; Witte, O.W.; Zilles, K.; Bidmon, H.J. A Combined Treatment with 1α,25-Dihydroxy-Vitamin D3 and 17β-Estradiol Reduces the Expression of Heat Shock Protein-32 (HSP-32) Following Cerebral Cortical Ischemia. J. Steroid Biochem. Mol. Biol. 2004, 89–90, 371–374. [Google Scholar] [CrossRef] [PubMed]
- Losem-Heinrichs, E.; Görg, B.; Redecker, C.; Schleicher, A.; Witte, O.W.; Zilles, K.; Bidmon, H.J. 1α,25-Dihydroxy-Vitamin D3 in Combination with 17β-Estradiol Lowers the Cortical Expression of Heat Shock Protein-27 Following Experimentally Induced Focal Cortical Ischemia in Rats. Arch. Biochem. Biophys. 2005, 439, 70–79. [Google Scholar] [CrossRef] [PubMed]
- Oermann, E.; Bidmon, H.J.; Witte, O.W.; Zilles, K. Effects of 1α,25 Dihydroxyvitamin D 3 on the Expression of HO-1 and GFAP in Glial Cells of the Photothrombotically Lesioned Cerebral Cortex. J. Chem. Neuroanat. 2004, 28, 225–238. [Google Scholar] [CrossRef] [PubMed]
- Oermann, E.; Bidmon, H.J.; Witte, O.W.; Zilles, K. 1α,25-Dihydroxyvitamin D3 Treatment Does Not Alter Neuronal Cyclooxygenase-2 Expression in the Cerebral Cortex after Stroke. Anat. Embryol. 2006, 211, 129–137. [Google Scholar] [CrossRef]
- Balden, R.; Selvamani, A.; Sohrabji, F. Vitamin D Deficiency Exacerbates Experimental Stroke Injury and Dysregulates Ischemia-Induced Inflammation in Adult Rats. Endocrinology 2012, 153, 2420–2435. [Google Scholar] [CrossRef] [Green Version]
- Won, S.; Sayeed, I.; Peterson, B.L.; Wali, B.; Kahn, J.S.; Stein, D.G. Vitamin D Prevents Hypoxia/Reoxygenation-Induced Blood-Brain Barrier Disruption via Vitamin D Receptor-Mediated NF-KB Signaling Pathways. PLoS ONE 2015, 10, e0122821. [Google Scholar] [CrossRef] [Green Version]
- Sadeghian, N.; Shadman, J.; Moradi, A.; ghasem Golmohammadi, M.; Panahpour, H. Calcitriol Protects the Blood-Brain Barrier Integrity against Ischemic Stroke and Reduces Vasogenic Brain Edema via Antioxidant and Antiapoptotic Actions in Rats. Brain Res. Bull. 2019, 150, 281–289. [Google Scholar] [CrossRef]
- Bao, G.Q.; Yu, J.Y. Vitamin D3 Promotes Cerebral Angiogenesis after Cerebral Infarction in Rats by Activating Shh Signaling Pathway. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 7069–7077. [Google Scholar]
- Kumar, M.; Nishad, D.K.; Kumar, A.; Bhatnagar, A.; Karwasra, R.; Khanna, K.; Sharma, D.; Dua, K.; Mudaliyar, V.; Sharma, N. Enhancement in Brain Uptake of Vitamin D3 Nanoemulsion for Treatment of Cerebral Ischemia: Formulation, Gamma Scintigraphy and Efficacy Study in Transient Middle Cerebral Artery Occlusion Rat Models. J. Microencapsul. 2020, 37, 492–501. [Google Scholar] [CrossRef]
- Takenaka, T.; Inoue, T.; Ohno, Y.; Miyazaki, T.; Nishiyama, A.; Ishii, N.; Suzuki, H. Calcitriol Supplementation Improves Endothelium-Dependent Vasodilation in Rat Hypertensive Renal Injury. Kidney Blood Press. Res. 2014, 39, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Dawoud, H.; Malinski, T. Nanomedical Studies of the Restoration of Nitric Oxide/Peroxynitrite Balance in Dysfunctional Endothelium by 1,25-Dihydroxy Vitamin D3—Clinical Implications for Cardiovascular Diseases. Int. J. Nanomed. 2018, 13, 455–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vahidinia, Z.; Khassafi, N.; Tameh, A.A.; Karimian, M.; Zare-Dehghanani, Z.; Moradi, F.; Joghataei, M.T. Calcitriol Ameliorates Brain Injury in the Rat Model of Cerebral Ischemia-Reperfusion Through Nrf2/HO-1 Signalling Axis: An in Silico and in Vivo Study. J. Stroke Cerebrovasc. Dis. 2022, 31, 106331. [Google Scholar] [CrossRef]
- Yang, C.; Hawkins, K.E.; Doré, S.; Candelario-Jalil, E. Neuroinflammatory Mechanisms of Blood-Brain Barrier Damage in Ischemic Stroke. Am. J. Physiol. Cell Physiol. 2019, 316, C135–C153. [Google Scholar] [CrossRef]
- Galoppin, M.; Kari, S.; Soldati, S.; Pal, A.; Rival, M.; Engelhardt, B.; Astier, A.; Thouvenot, E. Full Spectrum of Vitamin D Immunomodulation in Multiple Sclerosis: Mechanisms and Therapeutic Implications. Brain Commun. 2022, 4, fcac171. [Google Scholar] [CrossRef] [PubMed]
- Kondo, T.; Kawai, T.; Akira, S. Dissecting Negative Regulation of Toll-like Receptor Signaling. Trends Immunol. 2012, 33, 449–458. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.A.; Perrelli, A.; Ragni, A.; Retta, F.; De Silva, T.M.; Sobey, C.G.; Retta, S.F. Vitamin D Deficiency and the Risk of Cerebrovascular Disease. Antioxidants 2020, 9, 327. [Google Scholar] [CrossRef] [PubMed]
- Ślusarczyk, J.; Piotrowski, M.; Szczepanowicz, K.; Regulska, M.; Leśkiewicz, M.; Warszyński, P.; Budziszewska, B.; Lasoń, W.; Basta-Kaim, A. Nanocapsules with Polyelectrolyte Shell as a Platform for 1,25-Dihydroxyvitamin D3 Neuroprotection: Study in Organotypic Hippocampal Slices. Neurotox. Res. 2016, 30, 581–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uberti, F.; Lattuada, D.; Morsanuto, V.; Nava, U.; Bolis, G.; Vacca, G.; Squarzanti, D.F.; Cisari, C.; Molinari, C. Vitamin D Protects Human Endothelial Cells from Oxidative Stress through the Autophagic and Survival Pathways. J. Clin. Endocrinol. Metab. 2014, 99, 1367–1374. [Google Scholar] [CrossRef] [Green Version]
- Uberti, F.; Morsanuto, V.; Bardelli, C.; Molinari, C. Protective Effects of 1α,25-Dihydroxyvitamin D3 on Cultured Neural Cells Exposed to Catalytic Iron. Physiol. Rep. 2016, 4, e12769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Atifi, M.; Dreyfus, M.; Berger, F.; Wion, D. Expression of CYP2R1 and VDR in Human Brain Pericytes: The Neurovascular Vitamin D Autocrine/Paracrine Model. Neuroreport 2015, 26, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Nissou, M.F.; Guttin, A.; Zenga, C.; Berger, F.; Issartel, J.P.; Wion, D. Additional Clues for a Protective Role of Vitamin D in Neurodegenerative Diseases: 1,25-Dihydroxyvitamin D3 Triggers an Anti-Inflammatory Response in Brain Pericytes. J. Alzheimer’s Dis. 2014, 42, 789–799. [Google Scholar] [CrossRef] [Green Version]
- Hagag, A.A.; El Frargy, M.S.; Abd El-Latif, A.E. Vitamin D as an Adjuvant Therapy in Neonatal Hypoxia: Is It Beneficial? Endocr. Metab. Immune Disord. Drug Targets 2018, 19, 341–348. [Google Scholar] [CrossRef]
- Uluduz, D.; Adil, M.A.; Rahim, B.; Rahman, H.A.; Gilani, W.I.; Gilani, S.I.; Qureshi, A.I. Abstract WP422: Vitamin D Deficiency and Osteoporosis in Stroke Survivors: An Analysis of National Health and Nutritional Examination Survey (NHANES) 2001–2006. Stroke 2013, 44, AWP422. [Google Scholar] [CrossRef]
- Turetsky, A.; Goddeau, R.P.; Henninger, N. Low Serum Vitamin D Is Independently Associated with Larger Lesion Volumes after Ischemic Stroke. J. Stroke Cerebrovasc. Dis. 2015, 24, 1555–1563. [Google Scholar] [CrossRef] [PubMed]
- Park, K.Y.; Chung, P.W.; Kim, Y.B.; Moon, H.S.; Suh, B.C.; Won, Y.S.; Kim, J.M.; Youn, Y.C.; Kwon, O.S. Serum Vitamin D Status as a Predictor of Prognosis in Patients with Acute Ischemic Stroke. Cerebrovasc. Dis. 2015, 40, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Alfieri, D.F.; Lehmann, M.F.; Oliveira, S.R.; Flauzino, T.; Delongui, F.; de Araújo, M.C.M.; Dichi, I.; Delfino, V.D.; Mezzaroba, L.; Simão, A.N.C.; et al. Vitamin D Deficiency Is Associated with Acute Ischemic Stroke, C-Reactive Protein, and Short-Term Outcome. Metab. Brain Dis. 2017, 32, 493–502. [Google Scholar] [CrossRef] [PubMed]
- Nie, Z.; Ji, X.C.; Wang, J.; Zhang, H.X. Serum Levels of 25-Hydroxyvitamin D Predicts Infarct Volume and Mortality in Ischemic Stroke Patients. J. Neuroimmunol. 2017, 313, 41–45. [Google Scholar] [CrossRef]
- Huang, H.; Zheng, T.; Wang, S.; Wei, L.; Wang, Q.; Sun, Z. Serum 25-Hydroxyvitamin D Predicts Early Recurrent Stroke in Ischemic Stroke Patients. Nutr. Metab. Cardiovasc. Dis. 2016, 26, 908–914. [Google Scholar] [CrossRef]
- Miao, H.; Zhu, H.; Luan, X.; Huang, G.; Chen, M.; Yuan, Z.; Wang, Z. Risk Factors of Vitamin D Deficiency in Chinese Ischemic Stroke Patients: A Cross-Sectional Study. Front. Aging Neurosci. 2021, 12, 613498. [Google Scholar] [CrossRef] [PubMed]
- Arslan, S.; Sener, E.F.; Gunay, N.E.; Demiryurek, S.; Gulderen, U.R.; Topaloglu, T.; Gunay, N.; Demiryurek, A.T. Serum Vitamin D, Vitamin D Binding Protein Levels and Leukocyte Vitamin D Receptor Gene Expression in Patients with Ischaemic Stroke. J. Pak. Med. Assoc. 2020, 70, 1340–1344. [Google Scholar] [CrossRef] [PubMed]
- Wajda, J.; Świat, M.; Owczarek, A.J.; Brzozowska, A.; Olszanecka-Glinianowicz, M.; Chudek, J. Severity of Vitamin D Deficiency Predicts Mortality in Ischemic Stroke Patients. Dis. Markers 2019, 2019, 3652894. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.Y.; Wang, Y.S.; Chen, Y.; Hu, Y.H.; Cui, W.; Shi, X.Y.; Jiang, W.; Zhang, J.M. Association of Serum 25(OH) D Levels with Infarct Volumes and Stroke Severity in Acute Ischemic Stroke. J. Nutr. Health Aging 2018, 22, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Berghout, B.P.; Fani, L.; Heshmatollah, A.; Koudstaal, P.J.; Ikram, M.A.; Zillikens, M.C.; Ikram, M.K. Vitamin D Status and Risk of Stroke: The Rotterdam Study. Stroke 2019, 50, 2293–2298. [Google Scholar] [CrossRef]
- Yalbuzdag, S.A.; Sarifakioglu, B.; Afsar, S.I.; Celik, C.; Can, A.; Yegin, T.; Senturk, B.; Guzelant, A.Y. Is 25(OH)D Associated with Cognitive Impairment and Functional Improvement in Stroke? A Retrospective Clinical Study. J. Stroke Cerebrovasc. Dis. 2015, 24, 1479–1486. [Google Scholar] [CrossRef]
- Sari, A.; Durmus, B.; Karaman, C.A.; Ogut, E.; Aktas, I. A Randomized, Double-Blind Study to Assess If Vitamin D Treatment Affects the Outcomes of Rehabilitation and Balance in Hemiplegic Patients. J. Phys. Ther. Sci. 2018, 30, 874–878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Utkan Karasu, A.; Kaymak Karataş, G. Effect of Vitamin d Supplementation on Lower Extremity Motor Function and Ambulation in Stroke Patients. Turk. J. Med. Sci. 2021, 51, 1413–1419. [Google Scholar] [CrossRef] [PubMed]
- Momosaki, R.; Abo, M.; Urashima, M. Vitamin D Supplementation and Post-Stroke Rehabilitation: A Randomized, Double-Blind, Placebo-Controlled Trial. Nutrients 2019, 11, 1295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, R.; Wang, M.; Huang, H.; Li, W.; Hu, Y.; Wu, T. Lower Vitamin D Status Is Associated with an Increased Risk of Ischemic Stroke: A Systematic Review and Meta-Analysis. Nutrients 2018, 10, 277. [Google Scholar] [CrossRef] [Green Version]
- Wei, Z.N.; Kuang, J.G. Vitamin D Deficiency in Relation to the Poor Functional Outcomes in Nondiabetic Patients with Ischemic Stroke. Biosci. Rep. 2018, 38, BSR20171509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yarlagadda, K.; Ma, N.; Doré, S. Vitamin D and Stroke: Effects on Incidence, Severity, and Outcome and the Potential Benefits of Supplementation. Front. Neurol. 2020, 11, 384. [Google Scholar] [CrossRef] [PubMed]
- Afzal, S.; Nordestgaard, B.G. Vitamin D, Hypertension, and Ischemic Stroke in 116 655 Individuals from the General Population. Hypertension 2017, 70, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Judd, S.E.; Morgan, C.J.; Panwar, B.; Howard, V.J.; Wadley, V.G.; Jenny, N.S.; Kissela, B.M.; Gutiérrez, O.M. Vitamin D Deficiency and Incident Stroke Risk in Community-Living Black and White Adults. Int. J. Stroke 2016, 11, 93–102. [Google Scholar] [CrossRef]
- Schneider, A.L.C.; Lutsey, P.L.; Selvin, E.; Mosley, T.H.; Sharrett, A.R.; Carson, K.A.; Post, W.S.; Pankow, J.S.; Folsom, A.R.; Gottesman, R.F.; et al. Vitamin D, Vitamin D Binding Protein Gene Polymorphisms, Race and Risk of Incident Stroke: The Atherosclerosis Risk in Communities (ARIC) Study. Eur. J. Neurol. 2015, 22, 1220–1227. [Google Scholar] [CrossRef] [Green Version]
- Manson, J.E.; Cook, N.R.; Lee, I.-M.; Christen, W.; Bassuk, S.S.; Mora, S.; Gibson, H.; Gordon, D.; Copeland, T.; D’Agostino, D.; et al. Vitamin D Supplements and Prevention of Cancer and Cardiovascular Disease. N. Engl. J. Med. 2019, 380, 33–44. [Google Scholar] [CrossRef]
- Zelzer, S.; Hofer, E.; Meinitzer, A.; Fritz-Petrin, E.; Simstich, S.; Goessler, W.; Schmidt, R.; Herrmann, M. Association of Vitamin D Metabolites with Cognitive Function and Brain Atrophy in Elderly Individuals -the Austrian Stroke Prevention Study. Aging 2021, 13, 9455–9467. [Google Scholar] [CrossRef]
- Hackett, M.L.; Pickles, K. Part I: Frequency of Depression after Stroke: An Updated Systematic Review and Meta-Analysis of Observational Studies. Int. J. Stroke 2014, 9, 1017–1025. [Google Scholar] [CrossRef]
- Villa, R.F.; Ferrari, F.; Moretti, A. Post-Stroke Depression: Mechanisms and Pharmacological Treatment. Pharmacol. Ther. 2018, 184, 131–144. [Google Scholar] [CrossRef]
- Ayerbe, L.; Ayis, S.; Wolfe, C.D.A.; Rudd, A.G. Natural History, Predictors and Outcomes of Depression after Stroke: Systematic Review and Meta-Analysis. Br. J. Psychiatry 2013, 202, 14–21. [Google Scholar] [CrossRef] [Green Version]
- Han, B.; Lyu, Y.; Sun, H.; Wei, Y.; He, J. Low Serum Levels of Vitamin D Are Associated with Post-Stroke Depression. Eur. J. Neurol. 2015, 22, 1269–1274. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Ren, W.; Cheng, J.; Zhu, B.; Jin, Q.; Wang, L.; Chen, C.; Zhu, L.; Chang, Y.; Gu, Y.; et al. Association Between Serum Levels of Vitamin D and the Risk of Post-Stroke Anxiety. Medicine 2016, 95, e3566. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Li, C.J.; He, J.; Zheng, X. Study on the Relationship between 25-Hydroxyvitamin D Level and Rehabilitation of Stroke Patients. Folia Neuropathol. 2022, 60, 114–121. [Google Scholar] [CrossRef] [PubMed]

| Model | Cell Treatment | Effects of Vitamin D | Reference |
|---|---|---|---|
| Glu and NMDA-induced excitotoxic damage in rat primary hippocampal neurons | 1,25(OH)2D3 (0.1–1000 nM) NMDA (100 μM), Glu (5 μM) | 1,25(OH)2D3 (1–100 nM) ↑cell viability ↓VGCC | [95] |
| Glu and dopaminergic toxins-induced rat mesencephalic cells damage | 1,25(OH)2D3 (1–100 nM) Glu (1 mM) H2O2 (30 μM) Calcium ionophore (A23187, 1 μM) MPP+ (30 μM) 6-OHDA (100 μM) | ↑cell viability ↓ROS | [96] |
| Glu-induced damage in primary rat cortical neurons | 1,25(OH)2D3 (10–100 nM)/3–9 DIV Glu (100 μM) | ↑cell viability ↑VDR mRNA ↑MAP-2 ↑GAP-43 ↑synapsin-1 | [97] |
| Primary neocortical, hippocampal and cerebellar cell cultures exposed to Glu | 1,25(OH)2D3 (50 and 100 nM)/30 min, 1, 3, 6 or 9 h after Glu (1 mM) | ↓excitotoxicity ↓caspase-3 activity | [98] |
| Glu-induced primary cortical neurons damage | Progesterone (0.1-80 μM) and 1,25(OH)2D3 (1–100 nM) individually or in different combinations Glu (0.5 μM) | ↓neuronal loss ↑p-ERK1/2 | [99] |
| OGD model in primary cortical neurons | Progesterone (0.1–80 μM) and 1,25(OH)2D3 (0.001–5 μM) individually or in different combinations during OGD and reoxygenation | ↓neuronal loss | [100] |
| Hypoxia model in C57BL/6J mice primary neuronal cells | cholecalciferol, 0.01–1 µM/14DIV, 20 min before hypoxia, during hypoxia and immediately after reoxygenation | cholecalciferol-1 µM ↓cell viability, ↓the neuron-glial functional structure cholecalciferol-0.01-0.1 µM ↑cell viability ↑the functional structure and activity of neuron–glial networks | [101] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lasoń, W.; Jantas, D.; Leśkiewicz, M.; Regulska, M.; Basta-Kaim, A. Vitamin D3 and Ischemic Stroke: A Narrative Review. Antioxidants 2022, 11, 2120. https://doi.org/10.3390/antiox11112120
Lasoń W, Jantas D, Leśkiewicz M, Regulska M, Basta-Kaim A. Vitamin D3 and Ischemic Stroke: A Narrative Review. Antioxidants. 2022; 11(11):2120. https://doi.org/10.3390/antiox11112120
Chicago/Turabian StyleLasoń, Władysław, Danuta Jantas, Monika Leśkiewicz, Magdalena Regulska, and Agnieszka Basta-Kaim. 2022. "Vitamin D3 and Ischemic Stroke: A Narrative Review" Antioxidants 11, no. 11: 2120. https://doi.org/10.3390/antiox11112120