
Intermittent Hypoxia-Hyperoxia and Oxidative Stress in Developing Human Airway Smooth Muscle


Abstract
:1. Introduction
1.1. Perinatal Oxygen Transition and the Developing Lung
1.2. Intermittent Hypoxia-Hyperoxia
1.3. The Paradox of Oxygen and Oxidative Damage in the Neonatal Lung
1.4. Balancing Oxygen, ROS, and Antioxidants in the Human Fetal ASM

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| O2 | Model | Effects on Lung | Ref. |
|---|---|---|---|
| Hypoxia | Neonatal rats | Decreased alveolarization Differential regulation of genes involved in the pathogenesis of postnatal lung remodeling | [46] |
| Fetal lung explants from rats | Increased branching Increased epithelial proliferation, differentiation Decreased matrix metalloproteinases | [3] | |
| Hyperoxia | Prenatal LPS and postnatal hyperoxia in mice | Decreased alveolarization; Diffuse fibrosis Altered lung function (decreased compliance, increased resistance) | [47] |
| Neonatal rats | Increased lung vascular and airway smooth muscle contraction (hyperresponsiveness), potentially through ROS and endothelin pathways | [48] | |
| Human fASM exposed to increasing concentrations of O2 | Increased [Ca2+]i response to vasoconstrictor agonists; Increased proliferation, decreased apoptosis; Increased mitochondrial fragmentation | [35] | |
| Mouse lung epithelial cells | Increased senescence Increased glycolysis Increased proliferation Increased DNA damage | [49] | |
| Mouse lung epithelial cells and neonatal mice | Decreased mitochondrial respiration Increased mitochondrial networks Alveolar simplification in neonates | [50] | |
| Neonatal rats exposed to hyperoxia +/− vitamin A | Inhaled vitamin A during hyperoxia exposure increased markers of alveolar maturation, decreased lung damage from hyperoxia, and increased surfactant levels | [51] | |
| Neonatal mice exposed to moderate or severe hyperoxia | Moderate hyperoxia increased ROS formation/oxidative stress more than severe hyperoxia | [52] | |
| Hyperoxia-injury in neonatal rats +/− caffeine | Increased total glutathione and hydrogen peroxide, oxidative damage, and antioxidant stress response, effects of which were reduced by caffeine administration | [53] | |
| Neonatal hyperoxia mouse model with or without caveolin-1 scaffolding domain peptide | Moderate hyperoxia increased airway reactivity and decreased compliance Hyperoxia increased airway thickness and ASM mass Caveolin-1 scaffolding domain peptide mitigated effects of hyperoxia on airway | [45] | |
| Pulmonary epithelial II cells exposed to hyperoxia +/− NAC | Decreased cell viability, increased cell death Increased ROS, NO, inflammatory cytokines Free radical scavengers reversed these effects | [54] | |
| Human fASM exposed to hyperoxia +/− senolytics | Increased senescence-associated markers, cell cycle checkpoint markers, and DNA damage markers, leading to secretion of inflammatory markers | [55] | |
| Neonatal rats exposed to hyperoxia +/− caffeine | Hyperoxia decreased lung development, which was improved by the administration of caffeine | [56] | |
| Neonatal rats | Increased oxidative damage in the lung through increased malondialdehyde and myeloperoxidase Decreased antioxidant superoxide dismutase Increased infiltration of inflammatory cells and proinflammatory cytokines, effects which were partially reversed by placental growth factor inhibition (which was shown to inhibit NFκB signaling) | [57] | |
| Neonatal mouse model of hyperoxia exposure +/− superoxide dismutase | Hyperoxia increased antioxidant genes via Nrf2 Hyperoxia decreased superoxide dismutase Overexpression of superoxide dismutase rescued hyperoxia effects on the lung | [58] | |
| Neonatal mouse model of hyperoxic lung injury +/− Nrf2 | Mice deficient in Nrf2 had more severe hyperoxic effects including mortality, decreased alveolarization, edema, inflammation, and DNA damage, and tissue oxidation | [59] | |
| Intermittent | Neonatal mice | Increased airway resistance and decreased compliance | [16] |
| Primary murine lung endothelial cell cultures | Intermittent hypoxia-hyperoxia had less of an effect on increasing pro-inflammatory cytokine compared to constant hyperoxia Intermittent hypoxia-hyperoxia increased peroxynitate | [15] | |
| Lung mitochondria of rats | Intermittent decreased in vitro lipid peroxidation, increased GSH/GSSG ratio, and decreased GSSG content seen in constant hypoxia | [60] | |
| Neonatal rats +/− superoxide dismutase mimetic, superoxide anion, and peroxynitrate scavenger | Intermittent altered biomarkers of angiogenesis and alveolarization that may be targetable by treatment with antioxidants to decrease lung inflammation | [61] | |
| Neonatal rats | Decreased lung function Decreased alveolarization Decreased total antioxidant capacity | [62] | |
| Neonatal mice | Decreased HIF1α and markers of angiogenesis | [63] | |
| Neonatal mice | Decreased alveolarization Decreased pulmonary total/oxidized glutathione ratio Increased carbonyl content Intermittent hypoxia-hyperoxia exacerbates oxidative stress in lung development | [64] |
2. Materials and Methods
2.1. Human Airway Smooth Muscle
2.2. Treatments and Exposures
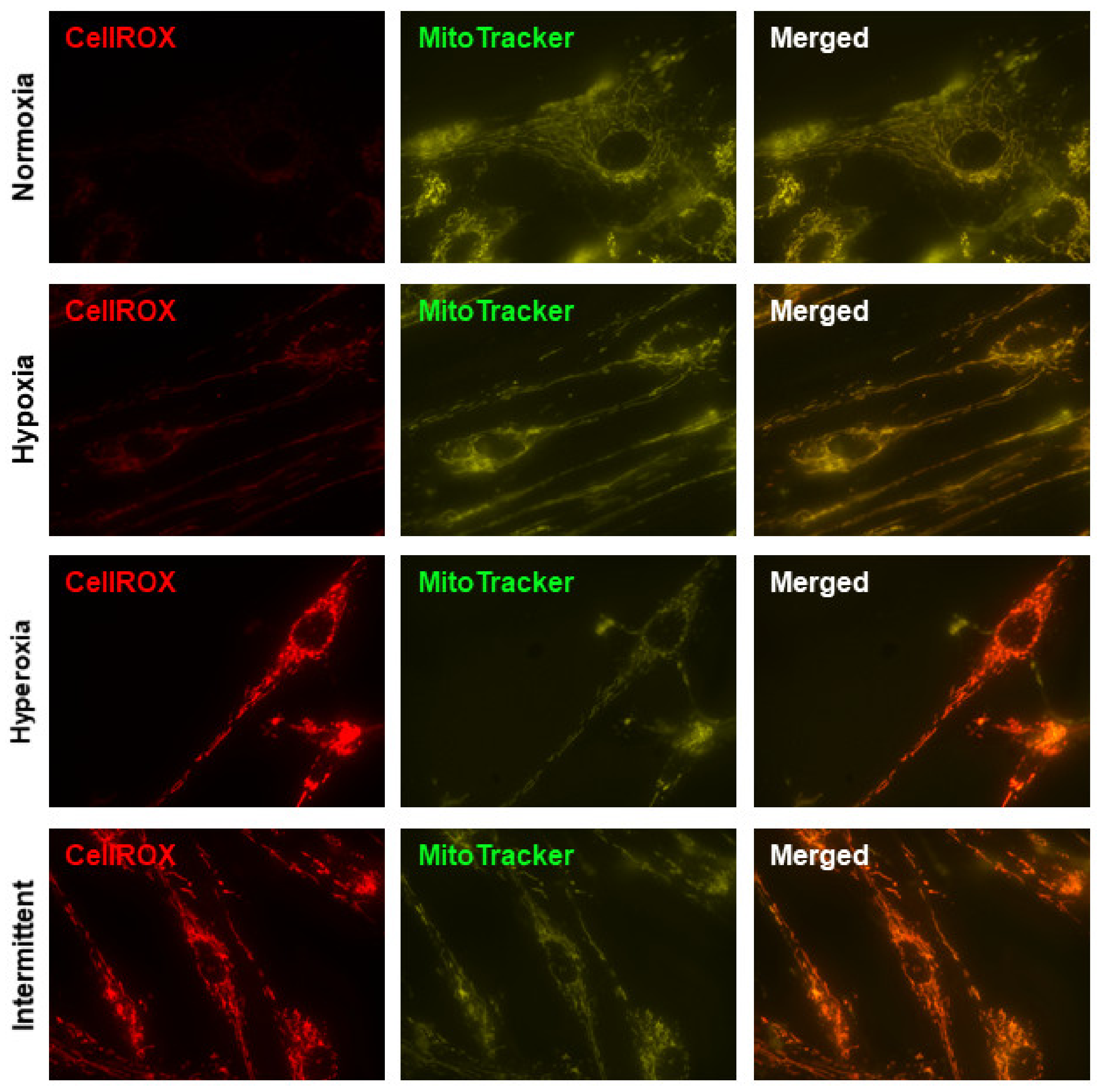
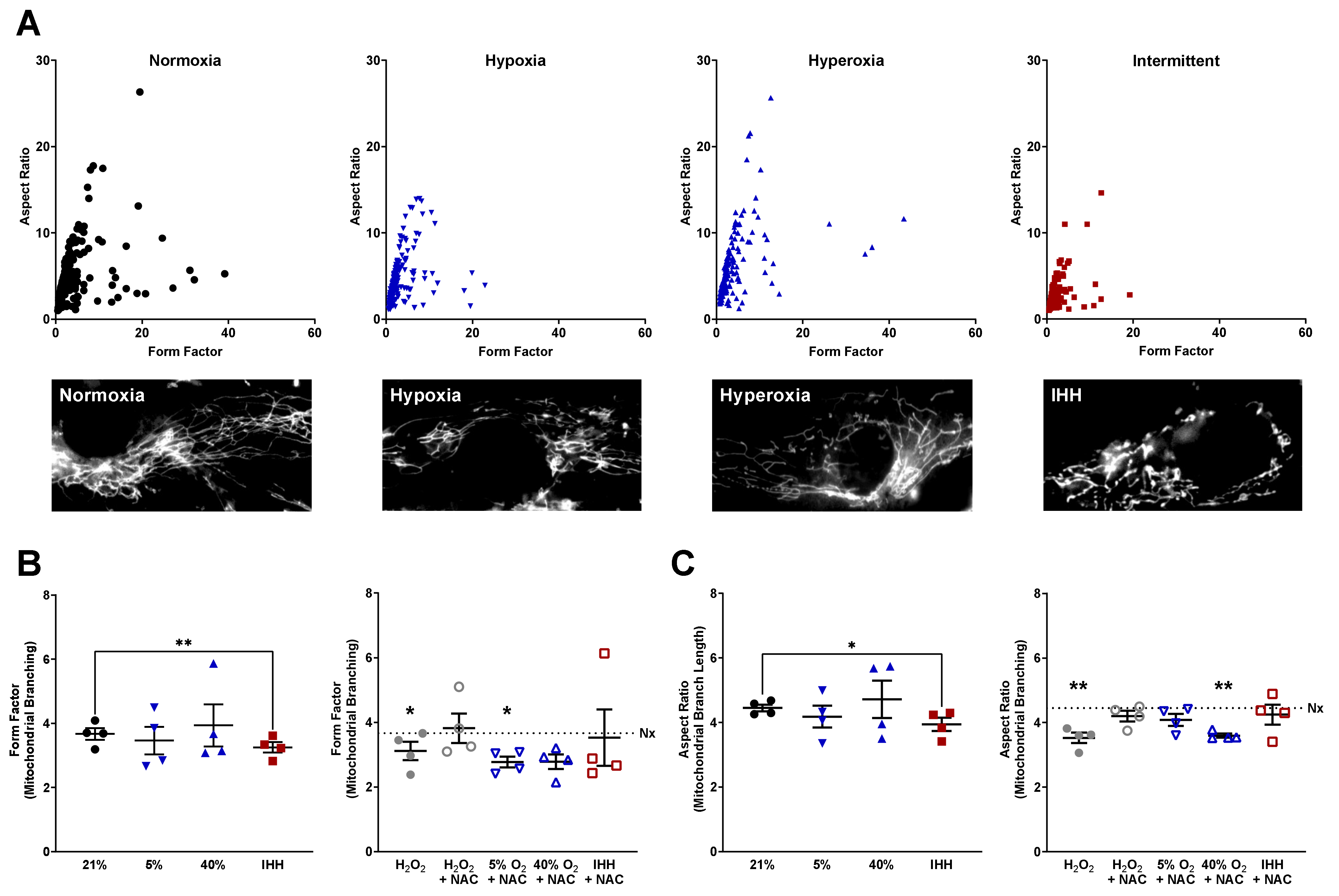
2.3. CellROX and MitoTracker Staining, Microscopy, and Mitochondrial Morphology
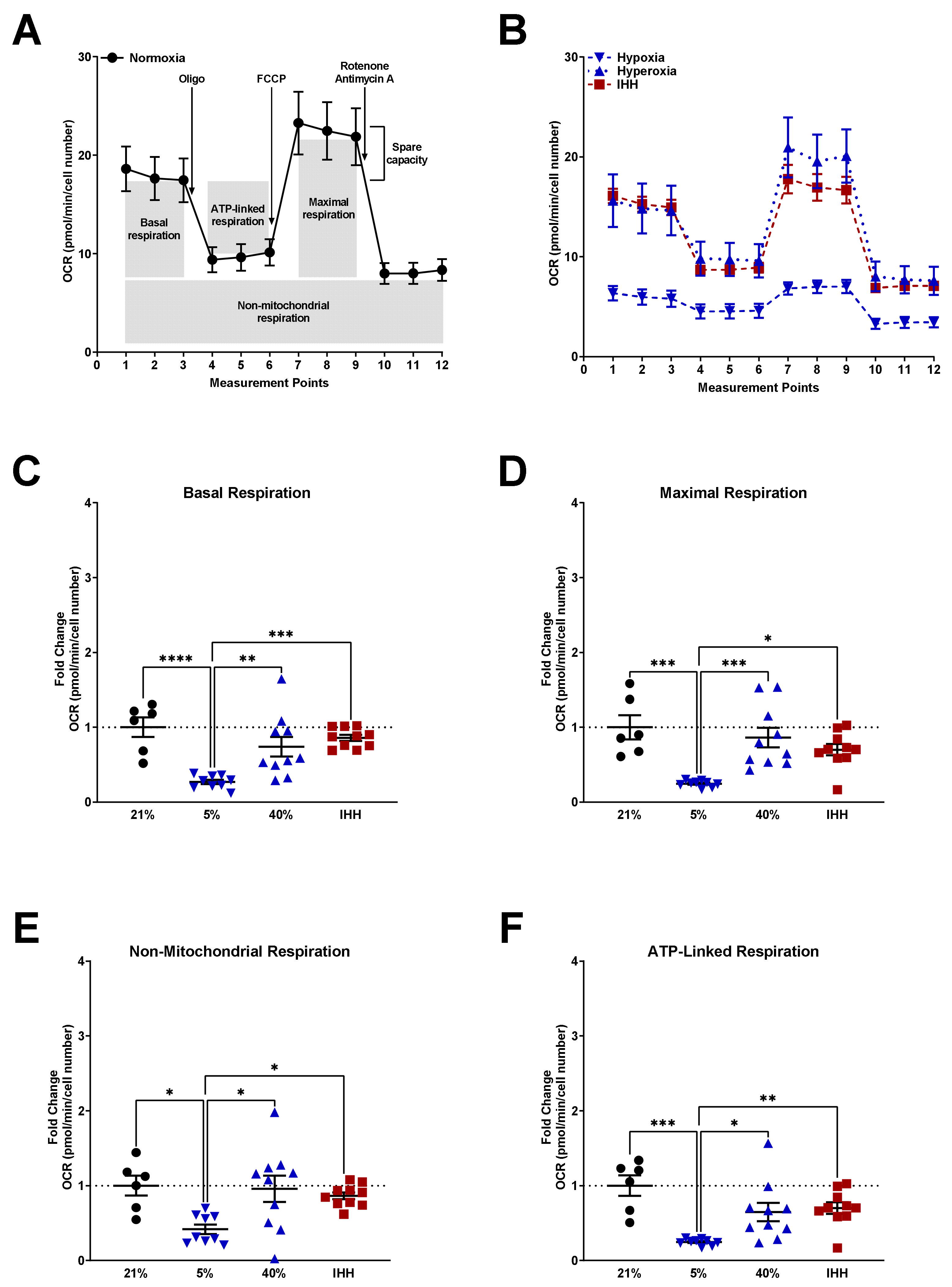
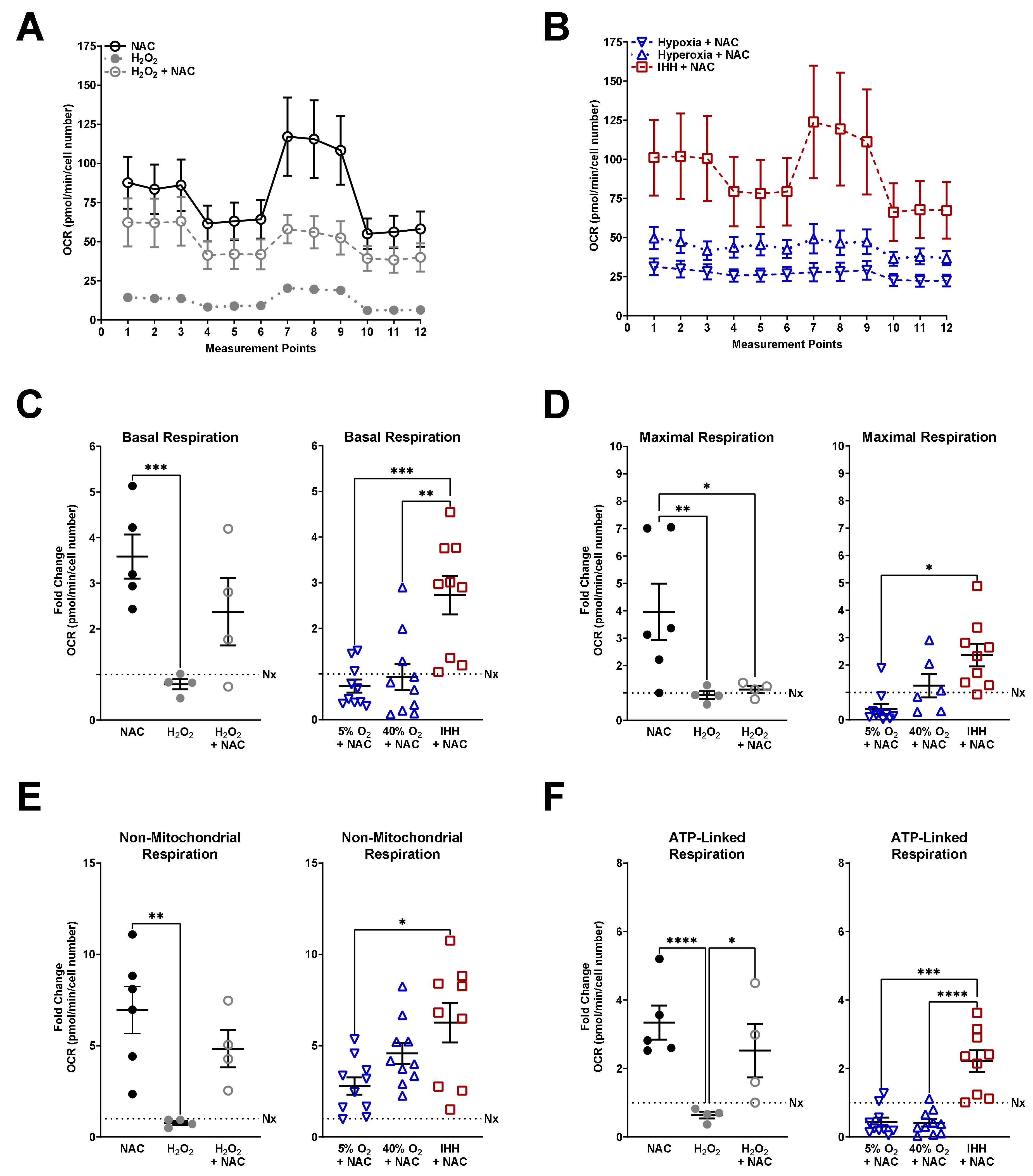
2.4. Seahorse Bioanalyzer Mitochondrial Stress Test
2.5. Statistics
3. Results
3.1. Hyperoxia and IHH Increase Oxidative Stress in fASM
3.2. Mitochondrial Morphology Altered by O2 Exposure and NAC
3.3. Mitochondrial Respiration Altered by O2 Exposure and NAC
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Henschen, M.; Stocks, J.; Brookes, I.; Frey, U. New aspects of airway mechanics in pre-term infants. Eur. Respir. J. 2006, 27, 913–920. [Google Scholar] [CrossRef] [Green Version]
- Ramchandani, R.; Shen, X.; Elmsley, C.L.; Ambrosius, W.T.; Gunst, S.J.; Tepper, R.S. Differences in airway structure in immature and mature rabbits. J. Appl. Physiol. 2000, 89, 1310–1316. [Google Scholar] [CrossRef]
- Gebb, S.A.; Jones, P.L. Hypoxia and lung branching morphogenesis. Adv. Exp. Med. Biol. 2003, 543, 117–125. [Google Scholar] [CrossRef]
- Haworth, S.G.; Hislop, A.A. Lung development-the effects of chronic hypoxia. Semin. Neonatol. 2003, 8, 1–8. [Google Scholar] [CrossRef]
- Chen, M.L.; Guo, L.; Smith, L.E.; Dammann, C.E.; Dammann, O. High or low oxygen saturation and severe retinopathy of prematurity: A meta-analysis. Pediatrics 2010, 125, e1483–e1492. [Google Scholar] [CrossRef] [Green Version]
- Bashambu, M.T.; Bhola, M.; Walsh, M. Evidence for oxygen use in preterm infants. Acta. Paediatr. 2012, 101, 29–33. [Google Scholar] [CrossRef]
- Cherian, S.; Morris, I.; Evans, J.; Kotecha, S. Oxygen therapy in preterm infants. Paediatr. Respir. Rev. 2014, 15, 135–141. [Google Scholar] [CrossRef]
- Halvorsen, T.; Skadberg, B.T.; Eide, G.E.; Roksund, O.; Aksnes, L.; Oymar, K. Characteristics of asthma and airway hyper-responsiveness after premature birth. Pediatr. Allergy Immunol. 2005, 16, 487–494. [Google Scholar] [CrossRef]
- Been, J.V.; Lugtenberg, M.J.; Smets, E.; van Schayck, C.P.; Kramer, B.W.; Mommers, M.; Sheikh, A. Preterm birth and childhood wheezing disorders: A systematic review and meta-analysis. PLoS Med. 2014, 11, e1001596. [Google Scholar] [CrossRef]
- Doyle, L.W.; Anderson, P.J. Pulmonary and neurological follow-up of extremely preterm infants. Neonatology 2010, 97, 388–394. [Google Scholar] [CrossRef]
- Holditch-Davis, D.; Merrill, P.; Schwartz, T.; Scher, M. Predictors of wheezing in prematurely born children. J. Obstet. Gynecol. Neonatal. Nurs. 2008, 37, 262–273. [Google Scholar] [CrossRef] [Green Version]
- Martin, R.J.; Prakash, Y.S.; Hibbs, A.M. Why do former preterm infants wheeze? J. Pediatr. 2013, 162, 443–444. [Google Scholar] [CrossRef] [Green Version]
- Goyal, N.K.; Fiks, A.G.; Lorch, S.A. Association of late-preterm birth with asthma in young children: Practice-based study. Pediatrics 2011, 128, e830–e838. [Google Scholar] [CrossRef] [Green Version]
- Hoo, A.F.; Dezateux, C.; Henschen, M.; Costeloe, K.; Stocks, J. Development of airway function in infancy after preterm delivery. J. Pediatr. 2002, 141, 652–658. [Google Scholar] [CrossRef] [PubMed]
- Wohlrab, P.; Johann Danhofer, M.; Schaubmayr, W.; Tiboldi, A.; Krenn, K.; Markstaller, K.; Ullrich, R.; Ulrich Klein, K.; Tretter, V. Oxygen conditions oscillating between hypoxia and hyperoxia induce different effects in the pulmonary endothelium compared to constant oxygen conditions. Physiol. Rep. 2021, 9, e14590. [Google Scholar] [CrossRef] [PubMed]
- Dylag, A.M.; Mayer, C.A.; Raffay, T.M.; Martin, R.J.; Jafri, A.; MacFarlane, P.M. Long-term effects of recurrent intermittent hypoxia and hyperoxia on respiratory system mechanics in neonatal mice. Pediatr. Res. 2017, 81, 565–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kroncke, K.D. Nitrosative stress and transcription. Biol. Chem. 2003, 384, 1365–1377. [Google Scholar] [CrossRef]
- Yang, D.; Elner, S.G.; Bian, Z.M.; Till, G.O.; Petty, H.R.; Elner, V.M. Pro-inflammatory cytokines increase reactive oxygen species through mitochondria and NADPH oxidase in cultured RPE cells. Exp. Eye Res. 2007, 85, 462–472. [Google Scholar] [CrossRef] [Green Version]
- Lo, Y.Y.; Wong, J.M.; Cruz, T.F. Reactive oxygen species mediate cytokine activation of c-Jun NH2-terminal kinases. J. Biol. Chem. 1996, 271, 15703–15707. [Google Scholar] [CrossRef] [Green Version]
- Cai, H.; Li, Z.; Dikalov, S.; Holland, S.M.; Hwang, J.; Jo, H.; Dudley, S.C., Jr.; Harrison, D.G. NAD(P)H oxidase-derived hydrogen peroxide mediates endothelial nitric oxide production in response to angiotensin II. J. Biol. Chem. 2002, 277, 48311–48317. [Google Scholar] [CrossRef] [Green Version]
- Waypa, G.B.; Marks, J.D.; Mack, M.M.; Boriboun, C.; Mungai, P.T.; Schumacker, P.T. Mitochondrial reactive oxygen species trigger calcium increases during hypoxia in pulmonary arterial myocytes. Circ. Res. 2002, 91, 719–726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, F.S.; Wang, C.J.; Chen, Y.J.; Chang, P.R.; Huang, Y.T.; Sun, Y.C.; Huang, H.C.; Yang, Y.J.; Yang, K.D. Ras induction of superoxide activates ERK-dependent angiogenic transcription factor HIF-1alpha and VEGF-A expression in shock wave-stimulated osteoblasts. J. Biol. Chem. 2004, 279, 10331–10337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Connor, K.M.; Subbaram, S.; Regan, K.J.; Nelson, K.K.; Mazurkiewicz, J.E.; Bartholomew, P.J.; Aplin, A.E.; Tai, Y.T.; Aguirre-Ghiso, J.; Flores, S.C.; et al. Mitochondrial H2O2 regulates the angiogenic phenotype via PTEN oxidation. J. Biol. Chem. 2005, 280, 16916–16924. [Google Scholar] [CrossRef] [Green Version]
- Ray, P.D.; Huang, B.W.; Tsuji, Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell. Signal. 2012, 24, 981–990. [Google Scholar] [CrossRef] [Green Version]
- Trachootham, D.; Lu, W.; Ogasawara, M.A.; Nilsa, R.D.; Huang, P. Redox regulation of cell survival. Antioxid. Redox Signal. 2008, 10, 1343–1374. [Google Scholar] [CrossRef] [Green Version]
- Dennery, P.A. Role of redox in fetal development and neonatal diseases. Antioxid. Redox Signal. 2004, 6, 147–153. [Google Scholar] [CrossRef]
- Ofman, G.; Tipple, T.E. Thiol-Redox Regulation in Lung Development and Vascular Remodeling. Antioxid. Redox Signal. 2019, 31, 858–873. [Google Scholar] [CrossRef]
- Upham, B.L.; Trosko, J.E. Oxidative-dependent integration of signal transduction with intercellular gap junctional communication in the control of gene expression. Antioxid. Redox Signal. 2009, 11, 297–307. [Google Scholar] [CrossRef] [Green Version]
- Wright, C.J.; Dennery, P.A. Manipulation of gene expression by oxygen: A primer from bedside to bench. Pediatr. Res. 2009, 66, 3–10. [Google Scholar] [CrossRef] [Green Version]
- Das, K.C. Thioredoxin system in premature and newborn biology. Antioxid. Redox Signal. 2004, 6, 177–184. [Google Scholar] [CrossRef]
- Asikainen, T.M.; White, C.W. Pulmonary antioxidant defenses in the preterm newborn with respiratory distress and bronchopulmonary dysplasia in evolution: Implications for antioxidant therapy. Antioxid. Redox Signal. 2004, 6, 155–167. [Google Scholar] [CrossRef]
- Murrell, G.A.; Francis, M.J.; Bromley, L. Modulation of fibroblast proliferation by oxygen free radicals. Biochem. J. 1990, 265, 659–665. [Google Scholar] [CrossRef] [Green Version]
- Kroemer, G.; Zamzami, N.; Susin, S.A. Mitochondrial control of apoptosis. Immunol. Today 1997, 18, 44–51. [Google Scholar] [CrossRef]
- Prakash, Y.S.; Pabelick, C.M.; Sieck, G.C. Mitochondrial Dysfunction in Airway Disease. Chest 2017, 152, 618–626. [Google Scholar] [CrossRef] [PubMed]
- Hartman, W.R.; Smelter, D.F.; Sathish, V.; Karass, M.; Kim, S.; Aravamudan, B.; Thompson, M.A.; Amrani, Y.; Pandya, H.C.; Martin, R.J.; et al. Oxygen dose responsiveness of human fetal airway smooth muscle cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 303, L711–L719. [Google Scholar] [CrossRef] [Green Version]
- Vogel, E.R.; Britt, R.D., Jr.; Faksh, A.; Kuipers, I.; Pandya, H.; Prakash, Y.S.; Martin, R.J.; Pabelick, C.M. Moderate hyperoxia induces extracellular matrix remodeling by human fetal airway smooth muscle cells. Pediatr. Res. 2017, 81, 376–383. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Jafri, A.; Martin, R.J.; Nnanabu, J.; Farver, C.; Prakash, Y.S.; MacFarlane, P.M. Severity of neonatal hyperoxia determines structural and functional changes in developing mouse airway. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 307, L295–L301. [Google Scholar] [CrossRef] [Green Version]
- Bhandari, V. Molecular mechanisms of hyperoxia-induced acute lung injury. Front. Biosci. 2008, 13, 6653–6661. [Google Scholar] [CrossRef] [Green Version]
- Hernansanz-Agustin, P.; Izquierdo-Alvarez, A.; Sanchez-Gomez, F.J.; Ramos, E.; Villa-Pina, T.; Lamas, S.; Bogdanova, A.; Martinez-Ruiz, A. Acute hypoxia produces a superoxide burst in cells. Free Radic. Biol. Med. 2014, 71, 146–156. [Google Scholar] [CrossRef] [PubMed]
- Hamanaka, R.B.; Chandel, N.S. Mitochondrial reactive oxygen species regulate hypoxic signaling. Curr. Opin. Cell Biol. 2009, 21, 894–899. [Google Scholar] [CrossRef] [Green Version]
- Guzy, R.D.; Schumacker, P.T. Oxygen sensing by mitochondria at complex III: The paradox of increased reactive oxygen species during hypoxia. Exp. Physiol. 2006, 91, 807–819. [Google Scholar] [CrossRef] [PubMed]
- Quintero, M.; Colombo, S.L.; Godfrey, A.; Moncada, S. Mitochondria as signaling organelles in the vascular endothelium. Proc. Natl. Acad. Sci. USA 2006, 103, 5379–5384. [Google Scholar] [CrossRef] [Green Version]
- Schroedl, C.; McClintock, D.S.; Budinger, G.R.; Chandel, N.S. Hypoxic but not anoxic stabilization of HIF-1alpha requires mitochondrial reactive oxygen species. Am. J. Physiol. Lung Cell. Mol. Physiol. 2002, 283, L922–L931. [Google Scholar] [CrossRef] [Green Version]
- Gitto, E.; Reiter, R.J.; Karbownik, M.; Xian-Tan, D.; Barberi, I. Respiratory distress syndrome in the newborn: Role of oxidative stress. Intensive Care Med. 2001, 27, 1116–1123. [Google Scholar] [CrossRef]
- Vogel, E.R.; Manlove, L.J.; Kuipers, I.; Thompson, M.A.; Fang, Y.H.; Freeman, M.R.; Britt, R.D., Jr.; Faksh, A.; Yang, B.; Prakash, Y.S.; et al. Caveolin-1 scaffolding domain peptide prevents hyperoxia-induced airway remodeling in a neonatal mouse model. Am. J. Physiol. Lung Cell. Mol. Physiol. 2019, 317, L99–L108. [Google Scholar] [CrossRef] [PubMed]
- Truog, W.E.; Xu, D.; Ekekezie, I.I.; Mabry, S.; Rezaiekhaligh, M.; Svojanovsky, S.; Soares, M.J. Chronic hypoxia and rat lung development: Analysis by morphometry and directed microarray. Pediatr. Res. 2008, 64, 56–62. [Google Scholar] [CrossRef] [Green Version]
- Velten, M.; Heyob, K.M.; Rogers, L.K.; Welty, S.E. Deficits in lung alveolarization and function after systemic maternal inflammation and neonatal hyperoxia exposure. J. Appl. Physiol. 2010, 108, 1347–1356. [Google Scholar] [CrossRef]
- Belik, J.; Jankov, R.P.; Pan, J.; Tanswell, A.K. Chronic O2 exposure enhances vascular and airway smooth muscle contraction in the newborn but not adult rat. J. Appl. Physiol. 2003, 94, 2303–2312. [Google Scholar] [CrossRef] [Green Version]
- Scaffa, A.M.; Peterson, A.L.; Carr, J.F.; Garcia, D.; Yao, H.; Dennery, P.A. Hyperoxia causes senescence and increases glycolysis in cultured lung epithelial cells. Physiol. Rep. 2021, 9, e14839. [Google Scholar] [CrossRef] [PubMed]
- Garcia, D.; Carr, J.F.; Chan, F.; Peterson, A.L.; Ellis, K.A.; Scaffa, A.; Ghio, A.J.; Yao, H.; Dennery, P.A. Short exposure to hyperoxia causes cultured lung epithelial cell mitochondrial dysregulation and alveolar simplification in mice. Pediatr. Res. 2020, 90, 58–65. [Google Scholar] [CrossRef]
- Gelfand, C.A.; Sakurai, R.; Wang, Y.; Liu, Y.; Segal, R.; Rehan, V.K. Inhaled vitamin A is more effective than intramuscular dosing in mitigating hyperoxia-induced lung injury in a neonatal rat model of bronchopulmonary dysplasia. Am. J. Physiol. Lung Cell. Mol. Physiol. 2020, 319, L576–L584. [Google Scholar] [CrossRef]
- Kindermann, A.; Binder, L.; Baier, J.; Gundel, B.; Simm, A.; Haase, R.; Bartling, B. Severe but not moderate hyperoxia of newborn mice causes an emphysematous lung phenotype in adulthood without persisting oxidative stress and inflammation. BMC Pulm. Med. 2019, 19, 245. [Google Scholar] [CrossRef]
- Endesfelder, S.; Strauss, E.; Scheuer, T.; Schmitz, T.; Buhrer, C. Antioxidative effects of caffeine in a hyperoxia-based rat model of bronchopulmonary dysplasia. Respir. Res. 2019, 20, 88. [Google Scholar] [CrossRef] [Green Version]
- Zou, D.; Li, J.; Fan, Q.; Zheng, X.; Deng, J.; Wang, S. Reactive oxygen and nitrogen species induce cell apoptosis via a mitochondria-dependent pathway in hyperoxia lung injury. J. Cell. Biochem. 2019, 120, 4837–4850. [Google Scholar] [CrossRef]
- Parikh, P.; Britt, R.D., Jr.; Manlove, L.J.; Wicher, S.A.; Roesler, A.; Ravix, J.; Teske, J.; Thompson, M.A.; Sieck, G.C.; Kirkland, J.L.; et al. Hyperoxia-induced Cellular Senescence in Fetal Airway Smooth Muscle Cells. Am. J. Respir. Cell Mol. Biol. 2019, 61, 51–60. [Google Scholar] [CrossRef]
- Jing, X.; Huang, Y.W.; Jarzembowski, J.; Shi, Y.; Konduri, G.G.; Teng, R.J. Caffeine ameliorates hyperoxia-induced lung injury by protecting GCH1 function in neonatal rat pups. Pediatr. Res. 2017, 82, 483–489. [Google Scholar] [CrossRef]
- Zhang, L.; Zhao, S.; Yuan, L.; Wu, H.; Jiang, H.; Luo, G.; Zhao, S. Knockdown of placental growth factor (PLGF) mitigates hyperoxia-induced acute lung injury in neonatal rats: Suppressive effects on NFkappaB signaling pathway. Int. Immunopharm. 2016, 38, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Poonyagariyagorn, H.K.; Metzger, S.; Dikeman, D.; Mercado, A.L.; Malinina, A.; Calvi, C.; McGrath-Morrow, S.; Neptune, E.R. Superoxide dismutase 3 dysregulation in a murine model of neonatal lung injury. Am. J. Respir. Cell Mol. Biol. 2014, 51, 380–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, H.Y.; van Houten, B.; Wang, X.; Miller-DeGraff, L.; Fostel, J.; Gladwell, W.; Perrow, L.; Panduri, V.; Kobzik, L.; Yamamoto, M.; et al. Targeted deletion of nrf2 impairs lung development and oxidant injury in neonatal mice. Antioxid. Redox Signal. 2012, 17, 1066–1082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonchar, O.; Mankovska, I. Moderate hypoxia/hyperoxia attenuates acute hypoxia-induced oxidative damage and improves antioxidant defense in lung mitochondria. Acta Physiol. Hung. 2012, 99, 436–446. [Google Scholar] [CrossRef]
- Chang, M.; Bany-Mohammed, F.; Kenney, M.C.; Beharry, K.D. Effects of a superoxide dismutase mimetic on biomarkers of lung angiogenesis and alveolarization during hyperoxia with intermittent hypoxia. Am. J. Transl. Res. 2013, 5, 594–607. [Google Scholar] [PubMed]
- Wang, J.; Zhang, A.; Li, Y.; Xu, J.; Huang, F.; Zhao, M.; Wu, B.; He, S. Effect of intermittent hypoxia or hyperoxia on lung development in preterm rat neonates during constant oxygen therapy. J. Cell. Biochem. 2019, 120, 17545–17554. [Google Scholar] [CrossRef]
- Elberson, V.D.; Nielsen, L.C.; Wang, H.; Kumar, H.S. Effects of intermittent hypoxia and hyperoxia on angiogenesis and lung development in newborn mice. J. Neonatal. Perinatal. Med. 2015, 8, 313–322. [Google Scholar] [CrossRef]
- Ratner, V.; Slinko, S.; Utkina-Sosunova, I.; Starkov, A.; Polin, R.A.; Ten, V.S. Hypoxic stress exacerbates hyperoxia-induced lung injury in a neonatal mouse model of bronchopulmonary dysplasia. Neonatology 2009, 95, 299–305. [Google Scholar] [CrossRef] [PubMed]
- Pandya, H.C.; Innes, J.; Hodge, R.; Bustani, P.; Silverman, M.; Kotecha, S. Spontaneous contraction of pseudoglandular-stage human airspaces is associated with the presence of smooth muscle-alpha-actin and smooth muscle-specific myosin heavy chain in recently differentiated fetal human airway smooth muscle. Biol. Neonate 2006, 89, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Britt, R.D., Jr.; Faksh, A.; Vogel, E.R.; Thompson, M.A.; Chu, V.; Pandya, H.C.; Amrani, Y.; Martin, R.J.; Pabelick, C.M.; Prakash, Y.S. Vitamin D attenuates cytokine-induced remodeling in human fetal airway smooth muscle cells. J. Cell. Physiol. 2015, 230, 1189–1198. [Google Scholar] [CrossRef] [PubMed]
- Bartman, C.M.; Matveyenko, A.; Pabelick, C.; Prakash, Y.S. Cellular clocks in hyperoxia effects on [Ca(2+)]i regulation in developing human airway smooth muscle. Am. J. Physiol. Lung Cell Mol. Physiol. 2021, 320, L451–L466. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Krebiehl, G.; Ruckerbauer, S.; Burbulla, L.F.; Kieper, N.; Maurer, B.; Waak, J.; Wolburg, H.; Gizatullina, Z.; Gellerich, F.N.; Woitalla, D.; et al. Reduced basal autophagy and impaired mitochondrial dynamics due to loss of Parkinson’s disease-associated protein DJ-1. PLoS ONE 2010, 5, e9367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aravamudan, B.; Thompson, M.A.; Pabelick, C.M.; Prakash, Y.S. Mitochondria in lung diseases. Expert Rev. Respir. Med. 2013, 7, 631–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cloonan, S.M.; Choi, A.M. Mitochondria in lung disease. J. Clin. Investig. 2016, 126, 809–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mishra, P.; Chan, D.C. Mitochondrial dynamics and inheritance during cell division, development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 634–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trian, T.; Benard, G.; Begueret, H.; Rossignol, R.; Girodet, P.O.; Ghosh, D.; Ousova, O.; Vernejoux, J.M.; Marthan, R.; Tunon-de-Lara, J.M.; et al. Bronchial smooth muscle remodeling involves calcium-dependent enhanced mitochondrial biogenesis in asthma. J. Exp. Med. 2007, 204, 3173–3181. [Google Scholar] [CrossRef] [Green Version]
- Aravamudan, B.; Thompson, M.; Sieck, G.C.; Vassallo, R.; Pabelick, C.M.; Prakash, Y.S. Functional Effects of Cigarette Smoke-Induced Changes in Airway Smooth Muscle Mitochondrial Morphology. J. Cell Physiol. 2017, 232, 1053–1068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faksh, A.; Britt, R.D., Jr.; Vogel, E.R.; Kuipers, I.; Thompson, M.A.; Sieck, G.C.; Pabelick, C.M.; Martin, R.J.; Prakash, Y.S. Effects of antenatal lipopolysaccharide and postnatal hyperoxia on airway reactivity and remodeling in a neonatal mouse model. Pediatr. Res. 2016, 79, 391–400. [Google Scholar] [CrossRef]
- MacFarlane, P.M.; Mayer, C.A.; Jafri, A.; Pabelick, C.M.; Prakash, Y.S.; Martin, R.J. CPAP protects against hyperoxia-induced increase in airway reactivity in neonatal mice. Pediatr. Res. 2020, 90, 52–57. [Google Scholar] [CrossRef]
- Reyburn, B.; Martin, R.J.; Prakash, Y.S.; MacFarlane, P.M. Mechanisms of injury to the preterm lung and airway: Implications for long-term pulmonary outcome. Neonatology 2012, 101, 345–352. [Google Scholar] [CrossRef] [Green Version]
- Ruiz-Meana, M.; Fernandez-Sanz, C.; Garcia-Dorado, D. The SR-mitochondria interaction: A new player in cardiac pathophysiology. Cardiovasc. Res. 2010, 88, 30–39. [Google Scholar] [CrossRef] [Green Version]
- Roesler, A.M.; Ravix, J.; Bartman, C.M.; Patel, B.S.; Schiliro, M.; Roos, B.; Nesbitt, L.; Pabelick, C.M.; Martin, R.J.; MacFarlane, P.M.; et al. Calcium-Sensing Receptor Contributes to Hyperoxia Effects on Human Fetal Airway Smooth Muscle. Front. Physiol. 2021, 12, 585895. [Google Scholar] [CrossRef]
- Su, W.Y.; Folz, R.; Chen, J.S.; Crapo, J.D.; Chang, L.Y. Extracellular superoxide dismutase mRNA expressions in the human lung by in situ hybridization. Am. J. Respir. Cell Mol. Biol. 1997, 16, 162–170. [Google Scholar] [CrossRef]
- Vasconcelos, L.H.C.; Ferreira, S.R.D.; Silva, M.; Ferreira, P.B.; de Souza, I.L.L.; Cavalcante, F.A.; da Silva, B.A. Uncovering the Role of Oxidative Imbalance in the Development and Progression of Bronchial Asthma. Oxid. Med. Cell. Longev. 2021, 2021, 6692110. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bayir, H. Reactive oxygen species. Crit. Care Med. 2005, 33, S498–S501. [Google Scholar] [CrossRef]
- Pietarinen-Runtti, P.; Lakari, E.; Raivio, K.O.; Kinnula, V.L. Expression of antioxidant enzymes in human inflammatory cells. Am. J. Physiol. Cell Physiol. 2000, 278, C118–C125. [Google Scholar] [CrossRef] [PubMed]
- Schumacker, P.T. Lung cell hypoxia: Role of mitochondrial reactive oxygen species signaling in triggering responses. Proc. Am. Thorac Soc. 2011, 8, 477–484. [Google Scholar] [CrossRef]
- Ward, J.P.; Snetkov, V.A.; Aaronson, P.I. Calcium, mitochondria and oxygen sensing in the pulmonary circulation. Cell Calcium 2004, 36, 209–220. [Google Scholar] [CrossRef]
- Becklake, M.R.; Kauffmann, F. Gender differences in airway behaviour over the human life span. Thorax 1999, 54, 1119–1138. [Google Scholar] [CrossRef] [Green Version]
- Thurlbeck, W.M. Postnatal human lung growth. Thorax 1982, 37, 564–571. [Google Scholar] [CrossRef] [Green Version]
- Torday, J.S.; Nielsen, H.C. The sex difference in fetal lung surfactant production. Exp. Lung Res. 1987, 12, 1–19. [Google Scholar] [CrossRef]
- O’Reilly, M.; Hansbro, P.M.; Horvat, J.C.; Beckett, E.L.; Harding, R.; Sozo, F. Bronchiolar remodeling in adult mice following neonatal exposure to hyperoxia: Relation to growth. Anat. Rec. 2014, 297, 758–769. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bartman, C.M.; Awari, D.W.; Pabelick, C.M.; Prakash, Y.S. Intermittent Hypoxia-Hyperoxia and Oxidative Stress in Developing Human Airway Smooth Muscle. Antioxidants 2021, 10, 1400. https://doi.org/10.3390/antiox10091400
Bartman CM, Awari DW, Pabelick CM, Prakash YS. Intermittent Hypoxia-Hyperoxia and Oxidative Stress in Developing Human Airway Smooth Muscle. Antioxidants. 2021; 10(9):1400. https://doi.org/10.3390/antiox10091400
Chicago/Turabian StyleBartman, Colleen M., Daniel Wasim Awari, Christina M. Pabelick, and Y. S. Prakash. 2021. "Intermittent Hypoxia-Hyperoxia and Oxidative Stress in Developing Human Airway Smooth Muscle" Antioxidants 10, no. 9: 1400. https://doi.org/10.3390/antiox10091400