
Oxidative Stress Promotes Corticosteroid Insensitivity in Asthma and COPD




Abstract
:1. Introduction
2. Corticosteroid Insensitivity in Asthma and COPD Pathophysiology
2.1. Airway Inflammation
2.2. Airway Structure and Function
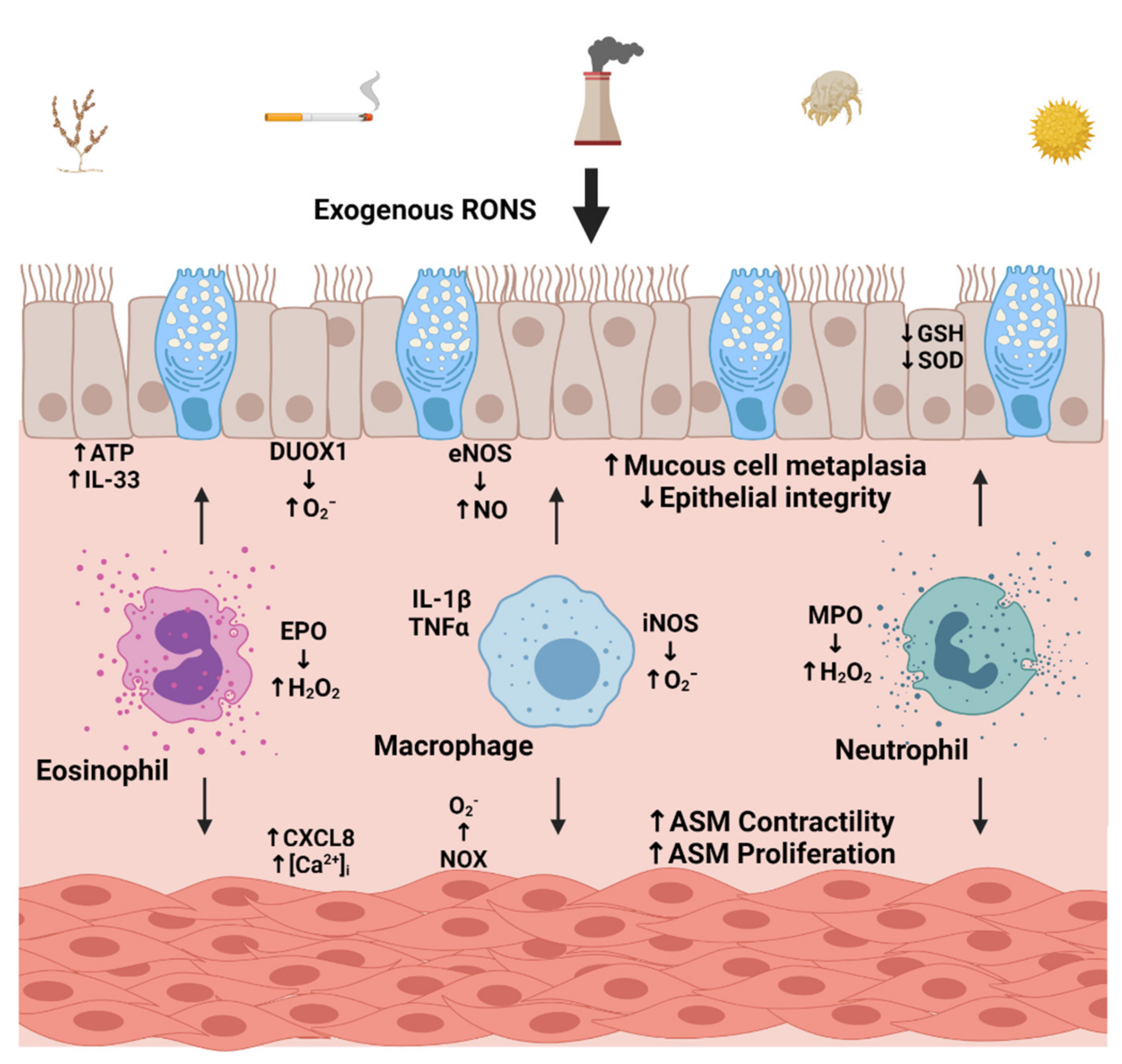
3. Factors Contributing to Oxidative Stress
3.1. Environmental Sources
3.2. Cellular Sources of Oxidative Stress
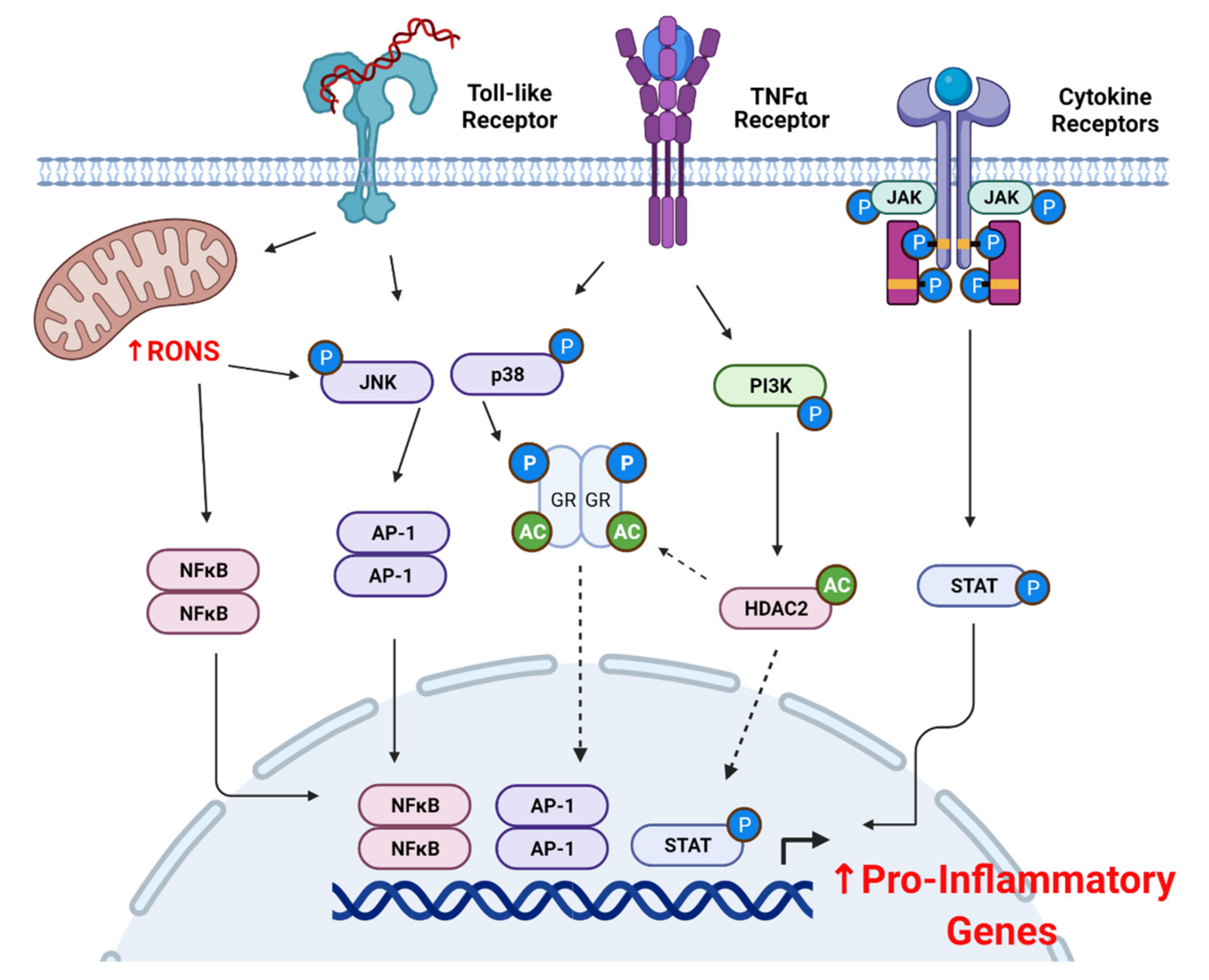
4. Oxidative Stress Promotes Corticosteroid Insensitivity
4.1. Disruption of Glucocorticoid Receptor (GR) Signaling
4.2. Oxidative Stress and Pro-Inflammatory Signaling
5. Targeting Oxidative Stress to Improve Corticosteroid Sensitivity
5.1. Key Antioxidant Systems
5.2. Endogenous Antioxidant Response and Corticosteroid Sensitivity
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
| Compound | Disease Model | Experimental Model | Primary Endpoint (s) | Dose | Reference |
|---|---|---|---|---|---|
| Antagomir-21 and LY294002 | Asthma | Mouse |
|
| [185] |
| MCC950 (NLRP3 Inhibitor) | Asthma | Mouse |
|
| [222] |
| γ-tocotrienol | Asthma | Mouse |
|
| [275] |
| Resveratrol (polyphenol) | Asthma | Mouse |
|
| [280] |
| Curcumin | COPD | Human monocytes (U937) |
|
| [283] |
| N-acetylcysteine (NAC) | Asthma | Mouse |
|
| [284] |
| Carbocysteine | COPD | Alveolar epithelial cells |
|
| [287] |
| Sulforaphane (Nrf2 Agonist) | Asthma | Mouse |
|
| [292] |
| Asthma/COPD | Mouse |
|
| [293] | |
| GYY4137 (H2S donor) | COPD | Human monocytes (U937) |
|
| [304] |
| Andrographolide | COPD | Human PBMCs and monocytes (U937) |
|
| [305] |
| Antagomir-9 | Asthma | Mouse pulmonary macrophages and human sputum cells |
|
| [306] |
| GW856553 (p38 inhibitor) | COPD | Human PBMCs |
|
| [307] |
References
- Jesenak, M.; Zelieskova, M.; Babusikova, E. Oxidative Stress and Bronchial Asthma in Children-Causes or Consequences? Front. Pediatr. 2017, 5, 162. [Google Scholar] [CrossRef]
- Kinnula, V.L.; Crapo, J.D. Superoxide dismutases in the lung and human lung diseases. Am. J. Respir. Crit. Care Med. 2003, 167, 1600–1619. [Google Scholar] [CrossRef] [PubMed]
- Zuo, L.; He, F.; Sergakis, G.G.; Koozehchian, M.S.; Stimpfl, J.N.; Rong, Y.; Diaz, P.T.; Best, T.M. Interrelated role of cigarette smoking, oxidative stress, and immune response in COPD and corresponding treatments. Am. J. Physiol. Lung Cell Mol. Physiol. 2014, 307, L205–L218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiegman, C.H.; Michaeloudes, C.; Haji, G.; Narang, P.; Clarke, C.J.; Russell, K.E.; Bao, W.; Pavlidis, S.; Barnes, P.J.; Kanerva, J.; et al. Oxidative stress-induced mitochondrial dysfunction drives inflammation and airway smooth muscle remodeling in patients with chronic obstructive pulmonary disease. J. Allergy Clin. Immunol. 2015, 136, 769–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mates, J.M.; Segura, J.A.; Alonso, F.J.; Marquez, J. Intracellular redox status and oxidative stress: Implications for cell proliferation, apoptosis, and carcinogenesis. Arch. Toxicol. 2008, 82, 273–299. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.G.; Campbell, H.R.; Iijima, H.; Gautrin, D.; Malo, J.L.; Eidelman, D.H.; Hamid, Q.; Maghni, K. Chlorine-induced injury to the airways in mice. Am. J. Respir. Crit. Care Med. 2003, 168, 568–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, W.; Wei, S.; Liu, G.; Yu, Z.; Estell, K.; Yadav, A.K.; Schwiebert, L.M.; Matalon, S. Postexposure administration of a {beta}2-agonist decreases chlorine-induced airway hyperreactivity in mice. Am. J. Respir. Cell Mol. Biol. 2011, 45, 88–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balakrishna, S.; Song, W.; Achanta, S.; Doran, S.F.; Liu, B.; Kaelberer, M.M.; Yu, Z.; Sui, A.; Cheung, M.; Leishman, E.; et al. TRPV4 inhibition counteracts edema and inflammation and improves pulmonary function and oxygen saturation in chemically induced acute lung injury. Am. J. Physiol. Lung Cell Mol. Physiol. 2014, 307, L158–L172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, M.K.; Kim, S.H.; Lee, K.Y.; Kim, T.B.; Moon, K.A.; Park, C.S.; Bae, Y.J.; Zhu, Z.; Moon, H.B.; Cho, Y.S. The tyrosine phosphatase, SHP-1, is involved in bronchial mucin production during oxidative stress. Biochem. Biophys. Res. Commun. 2010, 393, 137–143. [Google Scholar] [CrossRef]
- Uchida, M.; Anderson, E.L.; Squillace, D.L.; Patil, N.; Maniak, P.J.; Iijima, K.; Kita, H.; O’Grady, S.M. Oxidative stress serves as a key checkpoint for IL-33 release by airway epithelium. Allergy 2017, 72, 1521–1531. [Google Scholar] [CrossRef]
- Brown, D.M.; Hutchison, L.; Donaldson, K.; MacKenzie, S.J.; Dick, C.A.; Stone, V. The effect of oxidative stress on macrophages and lung epithelial cells: The role of phosphodiesterases 1 and 4. Toxicol. Lett. 2007, 168, 1–6. [Google Scholar] [CrossRef]
- Choo-Wing, R.; Syed, M.A.; Harijith, A.; Bowen, B.; Pryhuber, G.; Janer, C.; Andersson, S.; Homer, R.J.; Bhandari, V. Hyperoxia and interferon-gamma-induced injury in developing lungs occur via cyclooxygenase-2 and the endoplasmic reticulum stress-dependent pathway. Am. J. Respir. Cell Mol. Biol. 2013, 48, 749–757. [Google Scholar] [CrossRef] [Green Version]
- Deng, X.; Zhang, F.; Rui, W.; Long, F.; Wang, L.; Feng, Z.; Chen, D.; Ding, W. PM2.5-induced oxidative stress triggers autophagy in human lung epithelial A549 cells. Toxicol. Vitr. 2013, 27, 1762–1770. [Google Scholar] [CrossRef] [PubMed]
- Eapen, M.S.; Lu, W.; Hackett, T.L.; Singhera, G.K.; Mahmood, M.Q.; Hardikar, A.; Ward, C.; Walters, E.H.; Sohal, S.S. Increased myofibroblasts in the small airways, and relationship to remodelling and functional changes in smokers and COPD patients: Potential role of epithelial-mesenchymal transition. ERJ Open Res. 2021, 7, 00876–2020. [Google Scholar] [CrossRef] [PubMed]
- Higham, A.; Quinn, A.M.; Cancado, J.E.D.; Singh, D. The pathology of small airways disease in COPD: Historical aspects and future directions. Respir. Res. 2019, 20, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bourdin, A.; Neveu, D.; Vachier, I.; Paganin, F.; Godard, P.; Chanez, P. Specificity of basement membrane thickening in severe asthma. J. Allergy Clin. Immunol. 2007, 119, 1367–1374. [Google Scholar] [CrossRef]
- Yaghoubi, M.; Adibi, A.; Safari, A.; FitzGerald, J.M.; Sadatsafavi, M. The Projected Economic and Health Burden of Uncontrolled Asthma in the United States. Am. J. Respir. Crit. Care Med. 2019, 200, 1102–1112. [Google Scholar] [CrossRef]
- Adeloye, D.; Chua, S.; Lee, C.; Basquill, C.; Papana, A.; Theodoratou, E.; Nair, H.; Gasevic, D.; Sridhar, D.; Campbell, H.; et al. Global and regional estimates of COPD prevalence: Systematic review and meta-analysis. J. Glob. Health 2015, 5, 020415. [Google Scholar] [CrossRef]
- Vogelmeier, C.F.; Criner, G.J.; Martinez, F.J.; Anzueto, A.; Barnes, P.J.; Bourbeau, J.; Celli, B.R.; Chen, R.; Decramer, M.; Fabbri, L.M.; et al. Global Strategy for the Diagnosis, Management, and Prevention of Chronic Obstructive Lung Disease 2017 Report. GOLD Executive Summary. Am. J. Respir. Crit. Care Med. 2017, 195, 557–582. [Google Scholar] [CrossRef]
- Papadopoulos, N.G.; Christodoulou, I.; Rohde, G.; Agache, I.; Almqvist, C.; Bruno, A.; Bonini, S.; Bont, L.; Bossios, A.; Bousquet, J.; et al. Viruses and bacteria in acute asthma exacerbations—A GA(2) LEN-DARE systematic review. Allergy 2011, 66, 458–468. [Google Scholar] [CrossRef]
- Rehman, A.U.; Shah, S.; Abbas, G.; Harun, S.N.; Shakeel, S.; Hussain, R.; Hassali, M.A.A.; Rasool, M.F. Assessment of risk factors responsible for rapid deterioration of lung function over a period of one year in patients with chronic obstructive pulmonary disease. Sci. Rep. 2021, 11, 13578. [Google Scholar] [CrossRef] [PubMed]
- Chung, K.F.; Wenzel, S.E.; Brozek, J.L.; Bush, A.; Castro, M.; Sterk, P.J.; Adcock, I.M.; Bateman, E.D.; Bel, E.H.; Bleecker, E.R.; et al. International ERS/ATS guidelines on definition, evaluation and treatment of severe asthma. Eur. Respir. J. 2014, 43, 343–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jarjour, N.N.; Erzurum, S.C.; Bleecker, E.R.; Calhoun, W.J.; Castro, M.; Comhair, S.A.; Chung, K.F.; Curran-Everett, D.; Dweik, R.A.; Fain, S.B.; et al. Severe asthma: Lessons learned from the National Heart, Lung, and Blood Institute Severe Asthma Research Program. Am. J. Respir. Crit. Care Med. 2012, 185, 356–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramadan, A.A.; Gaffin, J.M.; Israel, E.; Phipatanakul, W. Asthma and Corticosteroid Responses in Childhood and Adult Asthma. Clin. Chest Med. 2019, 40, 163–177. [Google Scholar] [CrossRef]
- Holgate, S.T. Pathophysiology of asthma: What has our current understanding taught us about new therapeutic approaches? J. Allergy Clin. Immunol. 2011, 128, 495–505. [Google Scholar] [CrossRef]
- Raissy, H.H.; Kelly, H.W.; Harkins, M.; Szefler, S.J. Inhaled corticosteroids in lung diseases. Am. J. Respir. Crit. Care Med. 2013, 187, 798–803. [Google Scholar] [CrossRef] [Green Version]
- Wenzel, S.E. Asthma phenotypes: The evolution from clinical to molecular approaches. Nat. Med. 2012, 18, 716–725. [Google Scholar] [CrossRef]
- Sullivan, S.D.; Rasouliyan, L.; Russo, P.A.; Kamath, T.; Chipps, B.E.; Group, T.S. Extent, patterns, and burden of uncontrolled disease in severe or difficult-to-treat asthma. Allergy 2007, 62, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Fitzpatrick, A.M.; Baena-Cagnani, C.E.; Bacharier, L.B. Severe asthma in childhood: Recent advances in phenotyping and pathogenesis. Curr. Opin. Allergy Clin. Immunol. 2012, 12, 193–201. [Google Scholar] [CrossRef] [Green Version]
- Patel, J.G.; Nagar, S.P.; Dalal, A.A. Indirect costs in chronic obstructive pulmonary disease: A review of the economic burden on employers and individuals in the United States. Int. J. Chronic Obstr. Pulm. Dis. 2014, 9, 289–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanania, N.A.; Chapman, K.R.; Kesten, S. Adverse effects of inhaled corticosteroids. Am. J. Med. 1995, 98, 196–208. [Google Scholar] [CrossRef]
- Loke, Y.K.; Gilbert, D.; Thavarajah, M.; Blanco, P.; Wilson, A.M. Bone mineral density and fracture risk with long-term use of inhaled corticosteroids in patients with asthma: Systematic review and meta-analysis. BMJ Open 2015, 5, e008554. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, P.W.; Ghushchyan, V.H.; Globe, G.; Schatz, M. Oral corticosteroid exposure and adverse effects in asthmatic patients. J. Allergy Clin. Immunol. 2018, 141, 110–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singanayagam, A.; Johnston, S.L. Long-term impact of inhaled corticosteroid use in asthma and chronic obstructive pulmonary disease (COPD): Review of mechanisms that underlie risks. J. Allergy Clin. Immunol. 2020, 146, 1292–1294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fahy, J.V. Type 2 inflammation in asthma--present in most, absent in many. Nat. Rev. Immunol. 2015, 15, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J. Glucocorticosteroids: Current and future directions. Br. J. Pharmacol. 2011, 163, 29–43. [Google Scholar] [CrossRef] [Green Version]
- Mei, D.; Tan, W.S.D.; Wong, W.S.F. Pharmacological strategies to regain steroid sensitivity in severe asthma and COPD. Curr. Opin. Pharmacol. 2019, 46, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Fitzpatrick, A.M.; Stephenson, S.T.; Hadley, G.R.; Burwell, L.; Penugonda, M.; Simon, D.M.; Hansen, J.; Jones, D.P.; Brown, L.A. Thiol redox disturbances in children with severe asthma are associated with posttranslational modification of the transcription factor nuclear factor (erythroid-derived 2)-like 2. J. Allergy Clin. Immunol. 2011, 127, 1604–1611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fitzpatrick, A.M.; Teague, W.G.; Burwell, L.; Brown, M.S.; Brown, L.A.; Program, N.N.S.A.R. Glutathione oxidation is associated with airway macrophage functional impairment in children with severe asthma. Pediatr. Res. 2011, 69, 154–159. [Google Scholar] [CrossRef] [Green Version]
- Zhu, T.; Li, S.; Wang, J.; Liu, C.; Gao, L.; Zeng, Y.; Mao, R.; Cui, B.; Ji, H.; Chen, Z. Induced sputum metabolomic profiles and oxidative stress are associated with chronic obstructive pulmonary disease (COPD) severity: Potential use for predictive, preventive, and personalized medicine. EPMA J. 2020, 11, 645–659. [Google Scholar] [CrossRef]
- Vestbo, J.; Hansen, E.F. Airway hyperresponsiveness and COPD mortality. Thorax 2001, 56 (Suppl. 2), ii11–ii14. [Google Scholar]
- Ray, A.; Camiolo, M.; Fitzpatrick, A.; Gauthier, M.; Wenzel, S.E. Are We Meeting the Promise of Endotypes and Precision Medicine in Asthma? Physiol. Rev. 2020, 100, 983–1017. [Google Scholar] [CrossRef]
- Phipatanakul, W.; Mauger, D.T.; Sorkness, R.L.; Gaffin, J.M.; Holguin, F.; Woodruff, P.G.; Ly, N.P.; Bacharier, L.B.; Bhakta, N.R.; Moore, W.C.; et al. Effects of Age and Disease Severity on Systemic Corticosteroid Responses in Asthma. Am. J. Respir. Crit. Care Med. 2017, 195, 1439–1448. [Google Scholar] [CrossRef]
- Woodruff, P.G.; Modrek, B.; Choy, D.F.; Jia, G.; Abbas, A.R.; Ellwanger, A.; Koth, L.L.; Arron, J.R.; Fahy, J.V. T-helper type 2-driven inflammation defines major subphenotypes of asthma. Am. J. Respir. Crit. Care Med. 2009, 180, 388–395. [Google Scholar] [CrossRef] [PubMed]
- Kubo, M. Innate and adaptive type 2 immunity in lung allergic inflammation. Immunol. Rev. 2017, 278, 162–172. [Google Scholar] [CrossRef]
- Wisniewski, J.A.; Muehling, L.M.; Eccles, J.D.; Capaldo, B.J.; Agrawal, R.; Shirley, D.A.; Patrie, J.T.; Workman, L.J.; Schuyler, A.J.; Lawrence, M.G.; et al. TH1 signatures are present in the lower airways of children with severe asthma, regardless of allergic status. J. Allergy Clin. Immunol. 2018, 141, 2048–2060.e2013. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Zhu, Z.; Zuo, X.; Pan, H.; Gu, Y.; Yuan, Y.; Wang, G.; Wang, S.; Zheng, R.; Liu, Z.; et al. The role of NTHi colonization and infection in the pathogenesis of neutrophilic asthma. Respir. Res. 2020, 21, 170. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Li, R.; Sun, Y. Increased IFN-gamma-producing Th17/Th1 cells and their association with lung function and current smoking status in patients with chronic obstructive pulmonary disease. BMC Pulm. Med. 2019, 19, 137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raundhal, M.; Morse, C.; Khare, A.; Oriss, T.B.; Milosevic, J.; Trudeau, J.; Huff, R.; Pilewski, J.; Holguin, F.; Kolls, J.; et al. High IFN-gamma and low SLPI mark severe asthma in mice and humans. J. Clin. Investig. 2015, 125, 3037–3050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, W.C.; Hastie, A.T.; Li, X.; Li, H.; Busse, W.W.; Jarjour, N.N.; Wenzel, S.E.; Peters, S.P.; Meyers, D.A.; Bleecker, E.R.; et al. Sputum neutrophil counts are associated with more severe asthma phenotypes using cluster analysis. J. Allergy Clin. Immunol. 2014, 133, 1557–1563.e1555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christenson, S.A.; van den Berge, M.; Faiz, A.; Inkamp, K.; Bhakta, N.; Bonser, L.R.; Zlock, L.T.; Barjaktarevic, I.Z.; Barr, R.G.; Bleecker, E.R.; et al. An airway epithelial IL-17A response signature identifies a steroid-unresponsive COPD patient subgroup. J. Clin. Investig. 2019, 129, 169–181. [Google Scholar] [CrossRef]
- Yanagisawa, H.; Hashimoto, M.; Minagawa, S.; Takasaka, N.; Ma, R.; Moermans, C.; Ito, S.; Araya, J.; Budelsky, A.; Goodsell, A.; et al. Role of IL-17A in murine models of COPD airway disease. Am. J. Physiol. Lung Cell Mol. Physiol. 2017, 312, L122–L130. [Google Scholar] [CrossRef]
- Ouyang, S.; Liu, C.; Xiao, J.; Chen, X.; Lui, A.C.; Li, X. Targeting IL-17A/glucocorticoid synergy to CSF3 expression in neutrophilic airway diseases. JCI Insight 2020, 5, e132836. [Google Scholar] [CrossRef]
- Holgate, S.T. The sentinel role of the airway epithelium in asthma pathogenesis. Immunol. Rev. 2011, 242, 205–219. [Google Scholar] [CrossRef] [PubMed]
- Curran, D.R.; Cohn, L. Advances in mucous cell metaplasia: A plug for mucus as a therapeutic focus in chronic airway disease. Am. J. Respir. Cell Mol. Biol. 2010, 42, 268–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlier, F.M.; de Fays, C.; Pilette, C. Epithelial Barrier Dysfunction in Chronic Respiratory Diseases. Front. Physiol. 2021, 12, 691227. [Google Scholar] [CrossRef]
- Drake, L.Y.; Kita, H. IL-33: Biological properties, functions, and roles in airway disease. Immunol. Rev. 2017, 278, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Rada, B.; Gardina, P.; Myers, T.G.; Leto, T.L. Reactive oxygen species mediate inflammatory cytokine release and EGFR-dependent mucin secretion in airway epithelial cells exposed to Pseudomonas pyocyanin. Mucosal Immunol. 2011, 4, 158–171. [Google Scholar] [CrossRef] [PubMed]
- Heijink, I.; van Oosterhout, A.; Kliphuis, N.; Jonker, M.; Hoffmann, R.; Telenga, E.; Klooster, K.; Slebos, D.J.; ten Hacken, N.; Postma, D.; et al. Oxidant-induced corticosteroid unresponsiveness in human bronchial epithelial cells. Thorax 2014, 69, 5–13. [Google Scholar] [CrossRef] [Green Version]
- Klassen, C.; Karabinskaya, A.; Dejager, L.; Vettorazzi, S.; Van Moorleghem, J.; Luhder, F.; Meijsing, S.H.; Tuckermann, J.P.; Bohnenberger, H.; Libert, C.; et al. Airway Epithelial Cells Are Crucial Targets of Glucocorticoids in a Mouse Model of Allergic Asthma. J. Immunol. 2017, 199, 48–61. [Google Scholar] [CrossRef] [Green Version]
- Sekiyama, A.; Gon, Y.; Terakado, M.; Takeshita, I.; Kozu, Y.; Maruoka, S.; Matsumoto, K.; Hashimoto, S. Glucocorticoids enhance airway epithelial barrier integrity. Int. Immunopharmacol. 2012, 12, 350–357. [Google Scholar] [CrossRef] [PubMed]
- Milara, J.; Morell, A.; de Diego, A.; Artigues, E.; Morcillo, E.; Cortijo, J. Mucin 1 deficiency mediates corticosteroid insensitivity in asthma. Allergy 2019, 74, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Pepe, C.; Foley, S.; Shannon, J.; Lemiere, C.; Olivenstein, R.; Ernst, P.; Ludwig, M.S.; Martin, J.G.; Hamid, Q. Differences in airway remodeling between subjects with severe and moderate asthma. J. Allergy Clin. Immunol. 2005, 116, 544–549. [Google Scholar] [CrossRef]
- Smith, B.M.; Zhao, N.; Olivenstein, R.; Lemiere, C.; Hamid, Q.; Martin, J.G. Asthma and fixed airflow obstruction: Long-term trajectories suggest distinct endotypes. Clin. Exp. Allergy 2021, 51, 39–48. [Google Scholar] [CrossRef]
- Hogg, J.C.; Chu, F.; Utokaparch, S.; Woods, R.; Elliott, W.M.; Buzatu, L.; Cherniack, R.M.; Rogers, R.M.; Sciurba, F.C.; Coxson, H.O.; et al. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N. Engl. J. Med. 2004, 350, 2645–2653. [Google Scholar] [CrossRef] [PubMed]
- Bento, A.M.; Hershenson, M.B. Airway remodeling: Potential contributions of subepithelial fibrosis and airway smooth muscle hypertrophy/hyperplasia to airway narrowing in asthma. Allergy Asthma Proc. 1998, 19, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Jeffery, P.K. Remodeling in asthma and chronic obstructive lung disease. Am. J. Respir. Crit. Care Med. 2001, 164, S28–S38. [Google Scholar] [CrossRef]
- Prakash, Y.S. Emerging concepts in smooth muscle contributions to airway structure and function: Implications for health and disease. Am. J. Physiol. Lung Cell Mol. Physiol. 2016, 311, L1113–L1140. [Google Scholar] [CrossRef]
- James, A.L.; Bai, T.R.; Mauad, T.; Abramson, M.J.; Dolhnikoff, M.; McKay, K.O.; Maxwell, P.S.; Elliot, J.G.; Green, F.H. Airway smooth muscle thickness in asthma is related to severity but not duration of asthma. Eur. Respir. J. 2009, 34, 1040–1045. [Google Scholar] [CrossRef] [PubMed]
- Munakata, M. Airway remodeling and airway smooth muscle in asthma. Allergol. Int. 2006, 55, 235–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camoretti-Mercado, B.; Lockey, R.F. Airway smooth muscle pathophysiology in asthma. J. Allergy Clin. Immunol. 2021, 147, 1983–1995. [Google Scholar] [CrossRef] [PubMed]
- Berry, M.; Morgan, A.; Shaw, D.E.; Parker, D.; Green, R.; Brightling, C.; Bradding, P.; Wardlaw, A.J.; Pavord, I.D. Pathological features and inhaled corticosteroid response of eosinophilic and non-eosinophilic asthma. Thorax 2007, 62, 1043–1049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo, C.Y.; Michaeloudes, C.; Bhavsar, P.K.; Huang, C.D.; Wang, C.H.; Kuo, H.P.; Chung, K.F. Increased phenotypic differentiation and reduced corticosteroid sensitivity of fibrocytes in severe asthma. J. Allergy Clin. Immunol. 2015, 135, 1186–1195.e6. [Google Scholar] [CrossRef] [PubMed]
- Bossley, C.J.; Saglani, S.; Kavanagh, C.; Payne, D.N.; Wilson, N.; Tsartsali, L.; Rosenthal, M.; Balfour-Lynn, I.M.; Nicholson, A.G.; Bush, A. Corticosteroid responsiveness and clinical characteristics in childhood difficult asthma. Eur. Respir. J. 2009, 34, 1052–1059. [Google Scholar] [CrossRef]
- Comhair, S.A.; Erzurum, S.C. Redox control of asthma: Molecular mechanisms and therapeutic opportunities. Antioxid. Redox Signal. 2010, 12, 93–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zinellu, E.; Zinellu, A.; Fois, A.G.; Pau, M.C.; Scano, V.; Piras, B.; Carru, C.; Pirina, P. Oxidative Stress Biomarkers in Chronic Obstructive Pulmonary Disease Exacerbations: A Systematic Review. Antioxidants 2021, 10, 710. [Google Scholar] [CrossRef]
- Couillard, S.; Shrimanker, R.; Chaudhuri, R.; Mansur, A.H.; McGarvey, L.P.; Heaney, L.G.; Fowler, S.J.; Bradding, P.; Pavord, I.D.; Hinks, T.S.C.; et al. FeNO Non-Suppression Identifies Corticosteroid-Resistant Type-2 Signaling in Severe Asthma. Am. J. Respir. Crit. Care Med. 2021. Published electronically. [Google Scholar] [CrossRef]
- Schwartz, J.; Timonen, K.L.; Pekkanen, J. Respiratory effects of environmental tobacco smoke in a panel study of asthmatic and symptomatic children. Am. J. Respir. Crit. Care Med. 2000, 161, 802–806. [Google Scholar] [CrossRef] [Green Version]
- Flayer, C.H.; Ge, M.Q.; Hwang, J.W.; Kokalari, B.; Redai, I.G.; Jiang, Z.; Haczku, A. Ozone Inhalation Attenuated the Effects of Budesonide on Aspergillus fumigatus-Induced Airway Inflammation and Hyperreactivity in Mice. Front. Immunol. 2019, 10, 2173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zmirou, D.; Gauvin, S.; Pin, I.; Momas, I.; Sahraoui, F.; Just, J.; Le Moullec, Y.; Bremont, F.; Cassadou, S.; Reungoat, P.; et al. Traffic related air pollution and incidence of childhood asthma: Results of the Vesta case-control study. J. Epidemiol. Community Health 2004, 58, 18–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilhelm, M.; Meng, Y.Y.; Rull, R.P.; English, P.; Balmes, J.; Ritz, B. Environmental public health tracking of childhood asthma using California health interview survey, traffic, and outdoor air pollution data. Environ. Health Perspect. 2008, 116, 1254–1260. [Google Scholar] [CrossRef] [Green Version]
- De Homdedeu, M.; Cruz, M.; Sanchez-Diez, S.; Ojanguren, I.; Romero-Mesones, C.; Vanoirbeek, J.; Velde, G.V.; Muñoz, X. The immunomodulatory effects of diesel exhaust particles in asthma. Environ. Pollut. 2020, 263, 114600. [Google Scholar] [CrossRef]
- He, X.; Zhang, L.; Xiong, A.; Ran, Q.; Wang, J.; Wu, D.; Niu, B.; Liu, S.; Li, G. PM2.5 aggravates NQO1-induced mucus hyper-secretion through release of neutrophil extracellular traps in an asthma model. Ecotoxicol. Environ. Saf. 2021, 218, 112272. [Google Scholar] [CrossRef] [PubMed]
- McGovern, T.K.; Chen, M.; Allard, B.; Larsson, K.; Martin, J.G.; Adner, M. Neutrophilic oxidative stress mediates organic dust-induced pulmonary inflammation and airway hyperresponsiveness. Am. J. Physiol. Lung Cell Mol. Physiol. 2016, 310, L155–L165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prakash, Y.S.; Pabelick, C.M.; Sieck, G.C. Mitochondrial Dysfunction in Airway Disease. Chest 2017, 152, 618–626. [Google Scholar] [CrossRef] [PubMed]
- Schumacker, P.T.; Gillespie, M.N.; Nakahira, K.; Choi, A.M.; Crouser, E.D.; Piantadosi, C.A.; Bhattacharya, J. Mitochondria in lung biology and pathology: More than just a powerhouse. Am. J. Physiol. Lung Cell Mol. Physiol. 2014, 306, L962–L974. [Google Scholar] [CrossRef] [Green Version]
- Thannickal, V.J.; Fanburg, B.L. Reactive oxygen species in cell signaling. Am. J. Physiol. Lung Cell Mol. Physiol. 2000, 279, L1005–L1028. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.Y. The innate immune response in house dust mite-induced allergic inflammation. Allergy Asthma Immunol. Res. 2013, 5, 68–74. [Google Scholar] [CrossRef] [Green Version]
- Tham, R.; Vicendese, D.; Dharmage, S.C.; Hyndman, R.J.; Newbigin, E.; Lewis, E.; O’Sullivan, M.; Lowe, A.J.; Taylor, P.; Bardin, P.; et al. Associations between outdoor fungal spores and childhood and adolescent asthma hospitalizations. J. Allergy Clin. Immunol. 2017, 139, 1140–1147.e1144. [Google Scholar] [CrossRef] [Green Version]
- Roberts, G.; Fontanella, S.; Selby, A.; Howard, R.; Filippi, S.; Hedlin, G.; Nordlund, B.; Howarth, P.; Hashimoto, S.; Brinkman, P.; et al. Connectivity patterns between multiple allergen specific IgE antibodies and their association with severe asthma. J. Allergy Clin. Immunol. 2020, 146, 821–830. [Google Scholar] [CrossRef]
- Fraczek, M.G.; Chishimba, L.; Niven, R.M.; Bromley, M.; Simpson, A.; Smyth, L.; Denning, D.W.; Bowyer, P. Corticosteroid treatment is associated with increased filamentous fungal burden in allergic fungal disease. J. Allergy Clin. Immunol. 2018, 142, 407–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, T.K.; Loh, X.Y.; Peh, H.Y.; Tan, W.N.F.; Tan, W.S.D.; Li, N.; Tay, I.J.J.; Wong, W.S.F.; Engelward, B.P. House dust mite-induced asthma causes oxidative damage and DNA double-strand breaks in the lungs. J. Allergy Clin. Immunol. 2016, 138, 84–96.e81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mannino, D.M.; Homa, D.M.; Redd, S.C. Involuntary smoking and asthma severity in children: Data from the Third National Health and Nutrition Examination Survey. Chest 2002, 122, 409–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chalmers, G.W.; Macleod, K.J.; Little, S.A.; Thomson, L.J.; McSharry, C.P.; Thomson, N.C. Influence of cigarette smoking on inhaled corticosteroid treatment in mild asthma. Thorax 2002, 57, 226–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazarus, S.C.; Chinchilli, V.M.; Rollings, N.J.; Boushey, H.A.; Cherniack, R.; Craig, T.J.; Deykin, A.; DiMango, E.; Fish, J.E.; Ford, J.G.; et al. Smoking affects response to inhaled corticosteroids or leukotriene receptor antagonists in asthma. Am. J. Respir. Crit. Care Med. 2007, 175, 783–790. [Google Scholar] [CrossRef]
- Winickoff, J.P.; Friebely, J.; Tanski, S.E.; Sherrod, C.; Matt, G.E.; Hovell, M.F.; McMillen, R.C. Beliefs about the health effects of "thirdhand" smoke and home smoking bans. Pediatrics 2009, 123, e74–e79. [Google Scholar] [CrossRef] [Green Version]
- Heijink, I.H.; Brandenburg, S.M.; Postma, D.S.; van Oosterhout, A.J. Cigarette smoke impairs airway epithelial barrier function and cell-cell contact recovery. Eur. Respir. J. 2012, 39, 419–428. [Google Scholar] [CrossRef] [Green Version]
- Hackett, T.L.; Singhera, G.K.; Shaheen, F.; Hayden, P.; Jackson, G.R.; Hegele, R.G.; Van Eeden, S.; Bai, T.R.; Dorscheid, D.R.; Knight, D.A. Intrinsic phenotypic differences of asthmatic epithelium and its inflammatory responses to respiratory syncytial virus and air pollution. Am. J. Respir. Cell Mol. Biol. 2011, 45, 1090–1100. [Google Scholar] [CrossRef]
- Valavanidis, A.; Vlachogianni, T.; Fiotakis, K. Tobacco smoke: Involvement of reactive oxygen species and stable free radicals in mechanisms of oxidative damage, carcinogenesis and synergistic effects with other respirable particles. Int. J. Environ. Res. Public Health 2009, 6, 445–462. [Google Scholar] [CrossRef]
- van der Toorn, M.; Rezayat, D.; Kauffman, H.F.; Bakker, S.J.; Gans, R.O.; Koeter, G.H.; Choi, A.M.; van Oosterhout, A.J.; Slebos, D.J. Lipid-soluble components in cigarette smoke induce mitochondrial production of reactive oxygen species in lung epithelial cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2009, 297, L109–L114. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Z.; Zhu, L. Update on molecular mechanisms of corticosteroid resistance in chronic obstructive pulmonary disease. Pulm. Pharmacol. Ther. 2016, 37, 1–8. [Google Scholar] [CrossRef]
- Brandt, E.B.; Myers, J.M.; Ryan, P.H.; Hershey, G.K. Air pollution and allergic diseases. Curr. Opin. Pediatr. 2015, 27, 724–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Che, L.; Jin, Y.; Zhang, C.; Lai, T.; Zhou, H.; Xia, L.; Tian, B.; Zhao, Y.; Liu, J.; Wu, Y.; et al. Ozone-induced IL-17A and neutrophilic airway inflammation is orchestrated by the caspase-1-IL-1 cascade. Sci. Rep. 2016, 6, 18680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathews, J.A.; Krishnamoorthy, N.; Kasahara, D.I.; Hutchinson, J.; Cho, Y.; Brand, J.D.; Williams, A.S.; Wurmbrand, A.P.; Ribeiro, L.; Cuttitta, F.; et al. Augmented Responses to Ozone in Obese Mice Require IL-17A and Gastrin-Releasing Peptide. Am. J. Respir. Cell Mol. Biol. 2018, 58, 341–351. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.B.; Brandt, E.B.; Xiao, C.; Gibson, A.M.; Le Cras, T.D.; Brown, L.A.; Fitzpatrick, A.M.; Khurana Hershey, G.K. Diesel exhaust particles induce cysteine oxidation and s-glutathionylation in house dust mite induced murine asthma. PLoS ONE 2013, 8, e60632. [Google Scholar] [CrossRef] [Green Version]
- Brandt, E.B.; Kovacic, M.B.; Lee, G.B.; Gibson, A.M.; Acciani, T.H.; Le Cras, T.D.; Ryan, P.H.; Budelsky, A.L.; Khurana Hershey, G.K. Diesel exhaust particle induction of IL-17A contributes to severe asthma. J. Allergy Clin. Immunol. 2013, 132, 1194–1204.e1192. [Google Scholar] [CrossRef] [Green Version]
- Acciani, T.H.; Brandt, E.B.; Khurana Hershey, G.K.; Le Cras, T.D. Diesel exhaust particle exposure increases severity of allergic asthma in young mice. Clin. Exp. Allergy 2013, 43, 1406–1418. [Google Scholar] [CrossRef]
- Brandt, E.B.; Khurana Hershey, G.K. A combination of dexamethasone and anti-IL-17A treatment can alleviate diesel exhaust particle-induced steroid insensitive asthma. J. Allergy Clin. Immunol. 2016, 138, 924–928.e922. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Shi, Z.; Yin, Y.; Wang, Y.; Mu, N.; Li, C.; Ma, H.; Wang, Q. Particulate matter 2.5 triggers airway inflammation and bronchial hyperresponsiveness in mice by activating the SIRT2-p65 pathway. Front. Med. 2021. Published electronically. [Google Scholar] [CrossRef]
- Castaneda, A.R.; Vogel, C.F.A.; Bein, K.J.; Hughes, H.K.; Smiley-Jewell, S.; Pinkerton, K.E. Ambient particulate matter enhances the pulmonary allergic immune response to house dust mite in a BALB/c mouse model by augmenting Th2- and Th17-immune responses. Physiol. Rep. 2018, 6, e13827. [Google Scholar] [CrossRef]
- Tiotiu, A.; Kermani, N.Z.; Badi, Y.; Pavlidis, S.; Hansbro, P.M.; Guo, Y.K.; Chung, K.F.; Adcock, I.M.; U-BIOPRED Consortium Project Team. Sputum macrophage diversity and activation in asthma: Role of severity and inflammatory phenotype. Allergy 2021, 76, 775–788. [Google Scholar] [CrossRef] [PubMed]
- De Groot, L.E.S.; van der Veen, T.A.; Martinez, F.O.; Hamann, J.; Lutter, R.; Melgert, B.N. Oxidative stress and macrophages: Driving forces behind exacerbations of asthma and chronic obstructive pulmonary disease? Am. J. Physiol. Lung Cell Mol. Physiol. 2019, 316, L369–L384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slauch, J.M. How does the oxidative burst of macrophages kill bacteria? Still an open question. Mol. Microbiol. 2011, 80, 580–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balhara, J.; Gounni, A.S. The alveolar macrophages in asthma: A double-edged sword. Mucosal Immunol. 2012, 5, 605–609. [Google Scholar] [CrossRef]
- Miki, H.; Pei, H.; Gracias, D.T.; Linden, J.; Croft, M. Clearance of apoptotic cells by lung alveolar macrophages prevents development of house dust mite-induced asthmatic lung inflammation. J. Allergy Clin. Immunol. 2021, 147, 1087–1092. [Google Scholar] [CrossRef] [PubMed]
- Iles, K.E.; Forman, H.J. Macrophage signaling and respiratory burst. Immunol. Res. 2002, 26, 95–105. [Google Scholar] [CrossRef]
- Brune, B.; Dehne, N.; Grossmann, N.; Jung, M.; Namgaladze, D.; Schmid, T.; von Knethen, A.; Weigert, A. Redox control of inflammation in macrophages. Antioxid. Redox Signal. 2013, 19, 595–637. [Google Scholar] [CrossRef] [Green Version]
- Belchamber, K.B.R.; Singh, R.; Batista, C.M.; Whyte, M.K.; Dockrell, D.H.; Kilty, I.; Robinson, M.J.; Wedzicha, J.A.; Barnes, P.J.; Donnelly, L.E.; et al. Defective bacterial phagocytosis is associated with dysfunctional mitochondria in COPD macrophages. Eur. Respir. J. 2019, 54, 1802244. [Google Scholar] [CrossRef]
- Higham, A.; Booth, G.; Lea, S.; Southworth, T.; Plumb, J.; Singh, D. The effects of corticosteroids on COPD lung macrophages: A pooled analysis. Respir. Res. 2015, 16, 98. [Google Scholar] [CrossRef] [Green Version]
- Staples, K.J.; Hinks, T.S.; Ward, J.A.; Gunn, V.; Smith, C.; Djukanovic, R. Phenotypic characterization of lung macrophages in asthmatic patients: Overexpression of CCL17. J. Allergy Clin. Immunol. 2012, 130, 1404–1412.e1407. [Google Scholar] [CrossRef] [Green Version]
- Morshed, M.; Yousefi, S.; Stockle, C.; Simon, H.U.; Simon, D. Thymic stromal lymphopoietin stimulates the formation of eosinophil extracellular traps. Allergy 2012, 67, 1127–1137. [Google Scholar] [CrossRef]
- Aldridge, R.E.; Chan, T.; van Dalen, C.J.; Senthilmohan, R.; Winn, M.; Venge, P.; Town, G.I.; Kettle, A.J. Eosinophil peroxidase produces hypobromous acid in the airways of stable asthmatics. Free Radic. Biol. Med. 2002, 33, 847–856. [Google Scholar] [CrossRef]
- Silveira, J.S.; Antunes, G.L.; Kaiber, D.B.; da Costa, M.S.; Marques, E.P.; Ferreira, F.S.; Gassen, R.B.; Breda, R.V.; Wyse, A.T.S.; Pitrez, P.; et al. Reactive oxygen species are involved in eosinophil extracellular traps release and in airway inflammation in asthma. J. Cell. Physiol. 2019, 234, 23633–23646. [Google Scholar] [CrossRef] [PubMed]
- Reis, A.C.; Alessandri, A.L.; Athayde, R.M.; Perez, D.A.; Vago, J.P.; Avila, T.V.; Ferreira, T.P.; de Arantes, A.C.; Coutinho Dde, S.; Rachid, M.A.; et al. Induction of eosinophil apoptosis by hydrogen peroxide promotes the resolution of allergic inflammation. Cell Death Dis. 2015, 6, e1632. [Google Scholar] [CrossRef] [PubMed]
- Sousa, A.R.; Marshall, R.P.; Warnock, L.C.; Bolton, S.; Hastie, A.; Symon, F.; Hargadon, B.; Marshall, H.; Richardson, M.; Brightling, C.E.; et al. Responsiveness to oral prednisolone in severe asthma is related to the degree of eosinophilic airway inflammation. Clin. Exp. Allergy 2017, 47, 890–899. [Google Scholar] [CrossRef] [Green Version]
- Demarche, S.F.; Schleich, F.N.; Henket, M.A.; Paulus, V.A.; van Hees, T.J.; Louis, R.E. Effectiveness of inhaled corticosteroids in real life on clinical outcomes, sputum cells and systemic inflammation in asthmatics: A retrospective cohort study in a secondary care centre. BMJ Open 2017, 7, e018186. [Google Scholar] [CrossRef]
- Nabe, T. Steroid-Resistant Asthma and Neutrophils. Biol. Pharm. Bull. 2020, 43, 31–35. [Google Scholar] [CrossRef] [Green Version]
- Cox, G. Glucocorticoid treatment inhibits apoptosis in human neutrophils. Separation of survival and activation outcomes. J. Immunol. 1995, 154, 4719–4725. [Google Scholar]
- Fahy, J.V. Eosinophilic and neutrophilic inflammation in asthma: Insights from clinical studies. Proc. Am. Thorac Soc. 2009, 6, 256–259. [Google Scholar] [CrossRef]
- Bruijnzeel, P.L.; Uddin, M.; Koenderman, L. Targeting neutrophilic inflammation in severe neutrophilic asthma: Can we target the disease-relevant neutrophil phenotype? J. Leukoc. Biol. 2015, 98, 549–556. [Google Scholar] [CrossRef]
- Mann, B.S.; Chung, K.F. Blood neutrophil activation markers in severe asthma: Lack of inhibition by prednisolone therapy. Respir. Res. 2006, 7, 59. [Google Scholar] [CrossRef] [Green Version]
- Grunwell, J.R.; Stephenson, S.T.; Tirouvanziam, R.; Brown, L.A.S.; Brown, M.R.; Fitzpatrick, A.M. Children with Neutrophil-Predominant Severe Asthma Have Proinflammatory Neutrophils With Enhanced Survival and Impaired Clearance. J. Allergy Clin. Immunol. Pract 2019, 7, 516–525.e516. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, W.; Wang, N.; Tall, A.R.; Tabas, I. Mitochondrial Oxidative Stress Promotes Atherosclerosis and Neutrophil Extracellular Traps in Aged Mice. Arter. Thromb. Vasc. Biol. 2017, 37, e99–e107. [Google Scholar] [CrossRef] [Green Version]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef]
- Grabcanovic-Musija, F.; Obermayer, A.; Stoiber, W.; Krautgartner, W.D.; Steinbacher, P.; Winterberg, N.; Bathke, A.C.; Klappacher, M.; Studnicka, M. Neutrophil extracellular trap (NET) formation characterises stable and exacerbated COPD and correlates with airflow limitation. Respir. Res. 2015, 16, 59. [Google Scholar] [CrossRef] [Green Version]
- Pham, D.L.; Ban, G.Y.; Kim, S.H.; Shin, Y.S.; Ye, Y.M.; Chwae, Y.J.; Park, H.S. Neutrophil autophagy and extracellular DNA traps contribute to airway inflammation in severe asthma. Clin. Exp. Allergy 2017, 47, 57–70. [Google Scholar] [CrossRef]
- Gal, Z.; Gezsi, A.; Pallinger, E.; Visnovitz, T.; Nagy, A.; Kiss, A.; Sultesz, M.; Csoma, Z.; Tamasi, L.; Galffy, G.; et al. Plasma neutrophil extracellular trap level is modified by disease severity and inhaled corticosteroids in chronic inflammatory lung diseases. Sci. Rep. 2020, 10, 4320. [Google Scholar] [CrossRef] [Green Version]
- Saito, T.; Ichikawa, T.; Numakura, T.; Yamada, M.; Koarai, A.; Fujino, N.; Murakami, K.; Yamanaka, S.; Sasaki, Y.; Kyogoku, Y.; et al. PGC-1alpha regulates airway epithelial barrier dysfunction induced by house dust mite. Respir. Res. 2021, 22, 63. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Tan, X.; Liang, Y.; Hou, C.; Qu, D.; Li, M.; Huang, Q. Differential DAMP release was observed in the sputum of COPD, asthma and asthma-COPD overlap (ACO) patients. Sci. Rep. 2019, 9, 19241. [Google Scholar] [CrossRef] [Green Version]
- Srisomboon, Y.; Squillace, D.L.; Maniak, P.J.; Kita, H.; O’Grady, S.M. Fungal allergen-induced IL-33 secretion involves cholesterol-dependent, VDAC-1-mediated ATP release from the airway epithelium. J. Physiol. 2020, 598, 1829–1845. [Google Scholar] [CrossRef] [Green Version]
- Arzola Martinez, L.; Benavente, R.; Vega, G.; Rios, M.; Fonseca, W.; Rasky, A.J.; Morris, S.; Lukacs, N.W.; Villalon, M.J. Blocking ATP releasing channels prevents high extracellular ATP levels and airway hyperreactivity in an asthmatic mouse model. Am. J. Physiol. Lung Cell Mol. Physiol. 2021, 321, L466–L476. [Google Scholar] [CrossRef] [PubMed]
- Idzko, M.; Hammad, H.; van Nimwegen, M.; Kool, M.; Willart, M.A.; Muskens, F.; Hoogsteden, H.C.; Luttmann, W.; Ferrari, D.; di Virgilio, F.; et al. Extracellular ATP triggers and maintains asthmatic airway inflammation by activating dendritic cells. Nat. Med. 2007, 13, 913–919. [Google Scholar] [CrossRef]
- Pouwels, S.D.; Zijlstra, G.J.; van der Toorn, M.; Hesse, L.; Gras, R.; Ten Hacken, N.H.; Krysko, D.V.; Vandenabeele, P.; de Vries, M.; van Oosterhout, A.J.; et al. Cigarette smoke-induced necroptosis and DAMP release trigger neutrophilic airway inflammation in mice. Am. J. Physiol. Lung Cell Mol. Physiol. 2016, 310, L377–L386. [Google Scholar] [CrossRef] [Green Version]
- Zou, S.C.; Jiang, J.; Song, J. IL-33 induced inflammation exacerbated the development of chronic obstructive pulmonary disease through oxidative stress. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 1758–1764. [Google Scholar] [CrossRef]
- Dickinson, J.D.; Sweeter, J.M.; Warren, K.J.; Ahmad, I.M.; De Deken, X.; Zimmerman, M.C.; Brody, S.L. Autophagy regulates DUOX1 localization and superoxide production in airway epithelial cells during chronic IL-13 stimulation. Redox Biol. 2018, 14, 272–284. [Google Scholar] [CrossRef]
- Ravasi, S.; Citro, S.; Viviani, B.; Capra, V.; Rovati, G.E. CysLT1 receptor-induced human airway smooth muscle cells proliferation requires ROS generation, EGF receptor transactivation and ERK1/2 phosphorylation. Respir. Res. 2006, 7, 42. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.; Khalil, N. TGF-beta1 increases proliferation of airway smooth muscle cells by phosphorylation of map kinases. Respir. Res. 2006, 7, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, L.M.; Han, X.J.; Duan, T.T.; Xu, Y.D.; Wang, Y.; Ulloa, L.; Yang, Y.Q. Decreased S100A9 Expression Promoted Rat Airway Smooth Muscle Cell Proliferation by Stimulating ROS Generation and Inhibiting p38 MAPK. Can. Respir. J. 2016, 2016, 1462563. [Google Scholar] [CrossRef]
- Delmotte, P.; Yang, B.; Thompson, M.A.; Pabelick, C.M.; Prakash, Y.S.; Sieck, G.C. Inflammation alters regional mitochondrial Ca(2)+ in human airway smooth muscle cells. Am. J. Physiol. Cell Physiol. 2012, 303, C244–C256. [Google Scholar] [CrossRef] [Green Version]
- Hartman, W.R.; Smelter, D.F.; Sathish, V.; Karass, M.; Kim, S.; Aravamudan, B.; Thompson, M.A.; Amrani, Y.; Pandya, H.C.; Martin, R.J.; et al. Oxygen dose responsiveness of human fetal airway smooth muscle cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2012, 303, L711–L719. [Google Scholar] [CrossRef] [Green Version]
- Vandevyver, S.; Dejager, L.; Tuckermann, J.; Libert, C. New insights into the anti-inflammatory mechanisms of glucocorticoids: An emerging role for glucocorticoid-receptor-mediated transactivation. Endocrinology 2013, 154, 993–1007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerber, A.N.; Newton, R.; Sasse, S.K. Repression of transcription by the glucocorticoid receptor: A parsimonious model for the genomics era. J. Biol. Chem. 2021, 296, 100687. [Google Scholar] [CrossRef] [PubMed]
- Syed, A.P.; Greulich, F.; Ansari, S.A.; Uhlenhaut, N.H. Anti-inflammatory glucocorticoid action: Genomic insights and emerging concepts. Curr. Opin. Pharmacol. 2020, 53, 35–44. [Google Scholar] [CrossRef]
- Franco, L.M.; Gadkari, M.; Howe, K.N.; Sun, J.; Kardava, L.; Kumar, P.; Kumari, S.; Hu, Z.; Fraser, I.D.C.; Moir, S.; et al. Immune regulation by glucocorticoids can be linked to cell type-dependent transcriptional responses. J. Exp. Med. 2019, 216, 384–406. [Google Scholar] [CrossRef] [Green Version]
- Kan, M.; Koziol-White, C.; Shumyatcher, M.; Johnson, M.; Jester, W.; Panettieri, R.A., Jr.; Himes, B.E. Airway Smooth Muscle-Specific Transcriptomic Signatures of Glucocorticoid Exposure. Am. J. Respir. Cell Mol. Biol. 2019, 61, 110–120. [Google Scholar] [CrossRef]
- Reddy, A.T.; Lakshmi, S.P.; Banno, A.; Reddy, R.C. Glucocorticoid Receptor alpha Mediates Roflumilast’s Ability to Restore Dexamethasone Sensitivity in COPD. Int. J. Chronic Obstr. Pulm. Dis. 2020, 15, 125–134. [Google Scholar] [CrossRef] [Green Version]
- Goleva, E.; Li, L.B.; Eves, P.T.; Strand, M.J.; Martin, R.J.; Leung, D.Y. Increased glucocorticoid receptor beta alters steroid response in glucocorticoid-insensitive asthma. Am. J. Respir. Crit. Care Med. 2006, 173, 607–616. [Google Scholar] [CrossRef] [Green Version]
- Leung, D.Y.; Hamid, Q.; Vottero, A.; Szefler, S.J.; Surs, W.; Minshall, E.; Chrousos, G.P.; Klemm, D.J. Association of glucocorticoid insensitivity with increased expression of glucocorticoid receptor beta. J. Exp. Med. 1997, 186, 1567–1574. [Google Scholar] [CrossRef] [PubMed]
- Hodge, G.; Roscioli, E.; Jersmann, H.; Tran, H.B.; Holmes, M.; Reynolds, P.N.; Hodge, S. Steroid resistance in COPD is associated with impaired molecular chaperone Hsp90 expression by pro-inflammatory lymphocytes. Respir. Res. 2016, 17, 135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, P.J.; Michaeloudes, C.; Zhu, J.; Shaikh, N.; Baker, J.; Chung, K.F.; Bhavsar, P.K. Impaired nuclear translocation of the glucocorticoid receptor in corticosteroid-insensitive airway smooth muscle in severe asthma. Am. J. Respir. Crit. Care Med. 2015, 191, 54–62. [Google Scholar] [CrossRef]
- Matthews, J.G.; Ito, K.; Barnes, P.J.; Adcock, I.M. Defective glucocorticoid receptor nuclear translocation and altered histone acetylation patterns in glucocorticoid-resistant patients. J. Allergy Clin. Immunol. 2004, 113, 1100–1108. [Google Scholar] [CrossRef]
- Sun, X.J.; Li, Z.H.; Zhang, Y.; Zhou, G.; Zhang, J.Q.; Deng, J.M.; Bai, J.; Liu, G.N.; Li, M.H.; MacNee, W.; et al. Combination of erythromycin and dexamethasone improves corticosteroid sensitivity induced by CSE through inhibiting PI3K-delta/Akt pathway and increasing GR expression. Am. J. Physiol. Lung Cell Mol. Physiol. 2015, 309, L139–L146. [Google Scholar] [CrossRef]
- Randall, M.J.; Haenen, G.R.; Bouwman, F.G.; van der Vliet, A.; Bast, A. The tobacco smoke component acrolein induces glucocorticoid resistant gene expression via inhibition of histone deacetylase. Toxicol. Lett. 2016, 240, 43–49. [Google Scholar] [CrossRef] [Green Version]
- Ferraro, M.; Gjomarkaj, M.; Siena, L.; Di Vincenzo, S.; Pace, E. Formoterol and fluticasone propionate combination improves histone deacetylation and anti-inflammatory activities in bronchial epithelial cells exposed to cigarette smoke. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1718–1727. [Google Scholar] [CrossRef]
- Galliher-Beckley, A.J.; Cidlowski, J.A. Emerging roles of glucocorticoid receptor phosphorylation in modulating glucocorticoid hormone action in health and disease. IUBMB Life 2009, 61, 979–986. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Mercado, N.; Barnes, P.J.; Ito, K. Defects of protein phosphatase 2A causes corticosteroid insensitivity in severe asthma. PLoS ONE 2011, 6, e27627. [Google Scholar] [CrossRef] [PubMed]
- Bouazza, B.; Krytska, K.; Debba-Pavard, M.; Amrani, Y.; Honkanen, R.E.; Tran, J.; Tliba, O. Cytokines alter glucocorticoid receptor phosphorylation in airway cells: Role of phosphatases. Am. J. Respir. Cell Mol. Biol. 2012, 47, 464–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Dang, T.; Blind, R.D.; Wang, Z.; Cavasotto, C.N.; Hittelman, A.B.; Rogatsky, I.; Logan, S.K.; Garabedian, M.J. Glucocorticoid receptor phosphorylation differentially affects target gene expression. Mol. Endocrinol. 2008, 22, 1754–1766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Chen, W.; Kono, E.; Dang, T.; Garabedian, M.J. Modulation of glucocorticoid receptor phosphorylation and transcriptional activity by a C-terminal-associated protein phosphatase. Mol. Endocrinol. 2007, 21, 625–634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Frederick, J.; Garabedian, M.J. Deciphering the phosphorylation "code" of the glucocorticoid receptor in vivo. J. Biol. Chem. 2002, 277, 26573–26580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blind, R.D.; Garabedian, M.J. Differential recruitment of glucocorticoid receptor phospho-isoforms to glucocorticoid-induced genes. J. Steroid Biochem. Mol. Biol. 2008, 109, 150–157. [Google Scholar] [CrossRef] [Green Version]
- Itoh, M.; Adachi, M.; Yasui, H.; Takekawa, M.; Tanaka, H.; Imai, K. Nuclear export of glucocorticoid receptor is enhanced by c-Jun N-terminal kinase-mediated phosphorylation. Mol. Endocrinol. 2002, 16, 2382–2392. [Google Scholar] [CrossRef] [PubMed]
- Lea, S.; Li, J.; Plumb, J.; Gaffey, K.; Mason, S.; Gaskell, R.; Harbron, C.; Singh, D. P38 MAPK and glucocorticoid receptor crosstalk in bronchial epithelial cells. J. Mol. Med. 2020, 98, 361–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouazza, B.; Debba-Pavard, M.; Amrani, Y.; Isaacs, L.; O’Connell, D.; Ahamed, S.; Formella, D.; Tliba, O. Basal p38 mitogen-activated protein kinase regulates unliganded glucocorticoid receptor function in airway smooth muscle cells. Am. J. Respir. Cell Mol. Biol. 2014, 50, 301–315. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y.; Ito, K.; Kanda, A.; Tomoda, K.; Miller-Larsson, A.; Barnes, P.J.; Mercado, N. Protein tyrosine phosphatase PTP-RR regulates corticosteroid sensitivity. Respir. Res. 2016, 17, 30. [Google Scholar] [CrossRef] [Green Version]
- Qian, L.; Xu, D.; Xue, F.; Li, M.; Wang, X.; Liu, G. Interleukin-35 sensitizes monocytes from patients with asthma to glucocorticoid therapy by regulating p38 MAPK. Exp. Ther. Med. 2020, 19, 3247–3258. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Leung, D.Y.; Goleva, E. Anti-inflammatory and corticosteroid-enhancing actions of vitamin D in monocytes of patients with steroid-resistant and those with steroid-sensitive asthma. J. Allergy Clin. Immunol. 2014, 133, 1744–1752.e1741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mercado, N.; Hakim, A.; Kobayashi, Y.; Meah, S.; Usmani, O.S.; Chung, K.F.; Barnes, P.J.; Ito, K. Restoration of corticosteroid sensitivity by p38 mitogen activated protein kinase inhibition in peripheral blood mononuclear cells from severe asthma. PLoS ONE 2012, 7, e41582. [Google Scholar] [CrossRef] [Green Version]
- Chachi, L.; Abbasian, M.; Gavrila, A.; Alzahrani, A.; Tliba, O.; Bradding, P.; Wardlaw, A.J.; Brightling, C.; Amrani, Y. Protein phosphatase 5 mediates corticosteroid insensitivity in airway smooth muscle in patients with severe asthma. Allergy 2017, 72, 126–136. [Google Scholar] [CrossRef]
- Ito, K.; Yamamura, S.; Essilfie-Quaye, S.; Cosio, B.; Ito, M.; Barnes, P.J.; Adcock, I.M. Histone deacetylase 2-mediated deacetylation of the glucocorticoid receptor enables NF-kappaB suppression. J. Exp. Med. 2006, 203, 7–13. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, Y.; Bossley, C.; Gupta, A.; Akashi, K.; Tsartsali, L.; Mercado, N.; Barnes, P.J.; Bush, A.; Ito, K. Passive smoking impairs histone deacetylase-2 in children with severe asthma. Chest 2014, 145, 305–312. [Google Scholar] [CrossRef] [Green Version]
- Hew, M.; Bhavsar, P.; Torrego, A.; Meah, S.; Khorasani, N.; Barnes, P.J.; Adcock, I.; Chung, K.F. Relative corticosteroid insensitivity of peripheral blood mononuclear cells in severe asthma. Am. J. Respir. Crit. Care Med. 2006, 174, 134–141. [Google Scholar] [CrossRef] [Green Version]
- Ito, K.; Barnes, P.J.; Adcock, I.M. Glucocorticoid receptor recruitment of histone deacetylase 2 inhibits interleukin-1beta-induced histone H4 acetylation on lysines 8 and 12. Mol. Cell Biol. 2000, 20, 6891–6903. [Google Scholar] [CrossRef] [Green Version]
- Mercado, N.; To, Y.; Ito, K.; Barnes, P.J. Nortriptyline reverses corticosteroid insensitivity by inhibition of phosphoinositide-3-kinase-delta. J. Pharmacol. Exp. Ther. 2011, 337, 465–470. [Google Scholar] [CrossRef] [Green Version]
- Kim, R.Y.; Horvat, J.C.; Pinkerton, J.W.; Starkey, M.R.; Essilfie, A.T.; Mayall, J.R.; Nair, P.M.; Hansbro, N.G.; Jones, B.; Haw, T.J.; et al. MicroRNA-21 drives severe, steroid-insensitive experimental asthma by amplifying phosphoinositide 3-kinase-mediated suppression of histone deacetylase 2. J. Allergy Clin. Immunol. 2017, 139, 519–532. [Google Scholar] [CrossRef] [Green Version]
- Spears, M.; Donnelly, I.; Jolly, L.; Brannigan, M.; Ito, K.; McSharry, C.; Lafferty, J.; Chaudhuri, R.; Braganza, G.; Adcock, I.M.; et al. Effect of low-dose theophylline plus beclometasone on lung function in smokers with asthma: A pilot study. Eur. Respir. J. 2009, 33, 1010–1017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ford, P.A.; Durham, A.L.; Russell, R.E.; Gordon, F.; Adcock, I.M.; Barnes, P.J. Treatment effects of low-dose theophylline combined with an inhaled corticosteroid in COPD. Chest 2010, 137, 1338–1344. [Google Scholar] [CrossRef]
- Cosio, B.G.; Iglesias, A.; Rios, A.; Noguera, A.; Sala, E.; Ito, K.; Barnes, P.J.; Agusti, A. Low-dose theophylline enhances the anti-inflammatory effects of steroids during exacerbations of COPD. Thorax 2009, 64, 424–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devereux, G.; Cotton, S.; Fielding, S.; McMeekin, N.; Barnes, P.J.; Briggs, A.; Burns, G.; Chaudhuri, R.; Chrystyn, H.; Davies, L.; et al. Effect of Theophylline as Adjunct to Inhaled Corticosteroids on Exacerbations in Patients With COPD: A Randomized Clinical Trial. JAMA 2018, 320, 1548–1559. [Google Scholar] [CrossRef]
- Okros, Z.; Endreffy, E.; Novak, Z.; Maroti, Z.; Monostori, P.; Varga, I.S.; Kiraly, A.; Turi, S. Changes in NADPH oxidase mRNA level can be detected in blood at inhaled corticosteroid treated asthmatic children. Life Sci. 2012, 91, 907–911. [Google Scholar] [CrossRef]
- Lv, Y.; Li, Y.; Zhang, D.; Zhang, A.; Guo, W.; Zhu, S. HMGB1-induced asthmatic airway inflammation through GRP75-mediated enhancement of ER-mitochondrial Ca(2+) transfer and ROS increased. J. Cell. Biochem. 2018, 119, 4205–4215. [Google Scholar] [CrossRef]
- Marks-Konczalik, J.; Chu, S.C.; Moss, J. Cytokine-mediated transcriptional induction of the human inducible nitric oxide synthase gene requires both activator protein 1 and nuclear factor kappaB-binding sites. J. Biol. Chem. 1998, 273, 22201–22208. [Google Scholar] [CrossRef] [Green Version]
- Sen, C.K.; Packer, L. Antioxidant and redox regulation of gene transcription. FASEB J. 1996, 10, 709–720. [Google Scholar] [CrossRef]
- Pourazar, J.; Mudway, I.S.; Samet, J.M.; Helleday, R.; Blomberg, A.; Wilson, S.J.; Frew, A.J.; Kelly, F.J.; Sandstrom, T. Diesel exhaust activates redox-sensitive transcription factors and kinases in human airways. Am. J. Physiol. Lung Cell Mol. Physiol. 2005, 289, L724–L730. [Google Scholar] [CrossRef]
- Liu, X.; Lin, R.; Zhao, B.; Guan, R.; Li, T.; Jin, R. Correlation between oxidative stress and the NF-kappaB signaling pathway in the pulmonary tissues of obese asthmatic mice. Mol. Med. Rep. 2016, 13, 1127–1134. [Google Scholar] [CrossRef] [Green Version]
- Rahman, I.; Smith, C.A.; Lawson, M.F.; Harrison, D.J.; MacNee, W. Induction of gamma-glutamylcysteine synthetase by cigarette smoke is associated with AP-1 in human alveolar epithelial cells. FEBS Lett. 1996, 396, 21–25. [Google Scholar] [CrossRef] [Green Version]
- Fu, L.; Fei, J.; Tan, Z.X.; Chen, Y.H.; Hu, B.; Xiang, H.X.; Zhao, H.; Xu, D.X. Low Vitamin D Status Is Associated with Inflammation in Patients with Chronic Obstructive Pulmonary Disease. J. Immunol. 2021, 206, 515–523. [Google Scholar] [CrossRef]
- Gagliardo, R.; Chanez, P.; Profita, M.; Bonanno, A.; Albano, G.D.; Montalbano, A.M.; Pompeo, F.; Gagliardo, C.; Merendino, A.M.; Gjomarkaj, M. IkappaB kinase-driven nuclear factor-kappaB activation in patients with asthma and chronic obstructive pulmonary disease. J. Allergy Clin. Immunol. 2011, 128, 635–645.e2. [Google Scholar] [CrossRef] [PubMed]
- Edwards, M.R.; Bartlett, N.W.; Clarke, D.; Birrell, M.; Belvisi, M.; Johnston, S.L. Targeting the NF-kappaB pathway in asthma and chronic obstructive pulmonary disease. Pharmacol. Ther. 2009, 121, 1–13. [Google Scholar] [CrossRef]
- Gagliardo, R.; Chanez, P.; Mathieu, M.; Bruno, A.; Costanzo, G.; Gougat, C.; Vachier, I.; Bousquet, J.; Bonsignore, G.; Vignola, A.M. Persistent activation of nuclear factor-kappaB signaling pathway in severe uncontrolled asthma. Am. J. Respir. Crit. Care Med. 2003, 168, 1190–1198. [Google Scholar] [CrossRef] [Green Version]
- Villarino, A.V.; Kanno, Y.; O’Shea, J.J. Mechanisms and consequences of Jak-STAT signaling in the immune system. Nat. Immunol. 2017, 18, 374–384. [Google Scholar] [CrossRef]
- Simon, A.R.; Rai, U.; Fanburg, B.L.; Cochran, B.H. Activation of the JAK-STAT pathway by reactive oxygen species. Am. J. Physiol. 1998, 275, C1640–C1652. [Google Scholar] [CrossRef]
- Guo, F.H.; Uetani, K.; Haque, S.J.; Williams, B.R.; Dweik, R.A.; Thunnissen, F.B.; Calhoun, W.; Erzurum, S.C. Interferon gamma and interleukin 4 stimulate prolonged expression of inducible nitric oxide synthase in human airway epithelium through synthesis of soluble mediators. J. Clin. Investig. 1997, 100, 829–838. [Google Scholar] [CrossRef] [Green Version]
- Britt, R.D., Jr.; Thompson, M.A.; Sasse, S.; Pabelick, C.M.; Gerber, A.N.; Prakash, Y.S. Th1 cytokines TNF-alpha and IFN-gamma promote corticosteroid resistance in developing human airway smooth muscle. Am. J. Physiol. Lung Cell Mol. Physiol. 2019, 316, L71–L81. [Google Scholar] [CrossRef]
- Gauthier, M.; Chakraborty, K.; Oriss, T.B.; Raundhal, M.; Das, S.; Chen, J.; Huff, R.; Sinha, A.; Fajt, M.; Ray, P.; et al. Severe asthma in humans and mouse model suggests a CXCL10 signature underlies corticosteroid-resistant Th1 bias. JCI Insight 2017, 2, e94580. [Google Scholar] [CrossRef]
- Patel, S. Danger-Associated Molecular Patterns (DAMPs): The Derivatives and Triggers of Inflammation. Curr. Allergy Asthma Rep. 2018, 18, 63. [Google Scholar] [CrossRef]
- Roan, F.; Obata-Ninomiya, K.; Ziegler, S.F. Epithelial cell-derived cytokines: More than just signaling the alarm. J. Clin. Investig. 2019, 129, 1441–1451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmitz, J.; Owyang, A.; Oldham, E.; Song, Y.; Murphy, E.; McClanahan, T.K.; Zurawski, G.; Moshrefi, M.; Qin, J.; Li, X.; et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity 2005, 23, 479–490. [Google Scholar] [CrossRef] [Green Version]
- van der Ploeg, E.K.; Golebski, K.; van Nimwegen, M.; Fergusson, J.R.; Heesters, B.A.; Martinez-Gonzalez, I.; Kradolfer, C.M.A.; van Tol, S.; Scicluna, B.P.; de Bruijn, M.J.W.; et al. Steroid-resistant human inflammatory ILC2s are marked by CD45RO and elevated in type 2 respiratory diseases. Sci. Immunol. 2021, 6, eabd3489. [Google Scholar] [CrossRef] [PubMed]
- Saglani, S.; Lui, S.; Ullmann, N.; Campbell, G.A.; Sherburn, R.T.; Mathie, S.A.; Denney, L.; Bossley, C.J.; Oates, T.; Walker, S.A.; et al. IL-33 promotes airway remodeling in pediatric patients with severe steroid-resistant asthma. J. Allergy Clin. Immunol. 2013, 132, 676–685.e613. [Google Scholar] [CrossRef] [Green Version]
- Kabata, H.; Moro, K.; Fukunaga, K.; Suzuki, Y.; Miyata, J.; Masaki, K.; Betsuyaku, T.; Koyasu, S.; Asano, K. Thymic stromal lymphopoietin induces corticosteroid resistance in natural helper cells during airway inflammation. Nat. Commun. 2013, 4, 2675. [Google Scholar] [CrossRef] [Green Version]
- Brandelius, A.; Mahmutovic Persson, I.; Calven, J.; Bjermer, L.; Persson, C.G.; Andersson, M.; Uller, L. Selective inhibition by simvastatin of IRF3 phosphorylation and TSLP production in dsRNA-challenged bronchial epithelial cells from COPD donors. Br. J. Pharmacol. 2013, 168, 363–374. [Google Scholar] [CrossRef] [Green Version]
- Smelter, D.F.; Sathish, V.; Thompson, M.A.; Pabelick, C.M.; Vassallo, R.; Prakash, Y.S. Thymic stromal lymphopoietin in cigarette smoke-exposed human airway smooth muscle. J. Immunol. 2010, 185, 3035–3040. [Google Scholar] [CrossRef]
- Menzies-Gow, A.; Wechsler, M.E.; Brightling, C.E. Unmet need in severe, uncontrolled asthma: Can anti-TSLP therapy with tezepelumab provide a valuable new treatment option? Respir. Res. 2020, 21, 268. [Google Scholar] [CrossRef]
- Martinon, F. Signaling by ROS drives inflammasome activation. Eur. J. Immunol. 2010, 40, 616–619. [Google Scholar] [CrossRef] [PubMed]
- Birrell, M.A.; Eltom, S. The role of the NLRP3 inflammasome in the pathogenesis of airway disease. Pharmacol. Ther. 2011, 130, 364–370. [Google Scholar] [CrossRef] [PubMed]
- dos Santos, G.; Kutuzov, M.A.; Ridge, K.M. The inflammasome in lung diseases. Am. J. Physiol. Lung Cell Mol. Physiol. 2012, 303, L627–L633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, F.; Wen, Z.; Shi, X.; Fan, J. Inflammasome in the Pathogenesis of Pulmonary Diseases. Exp. Suppl. 2018, 108, 111–151. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.S.; Chhabra, S.K. A study on the serum levels of interleukin-1beta in bronchial asthma. J. Indian Med. Assoc. 2003, 101, 282–284. [Google Scholar]
- Wang, C.C.; Fu, C.L.; Yang, Y.H.; Lo, Y.C.; Wang, L.C.; Chuang, Y.H.; Chang, D.M.; Chiang, B.L. Adenovirus expressing interleukin-1 receptor antagonist alleviates allergic airway inflammation in a murine model of asthma. Gene Ther. 2006, 13, 1414–1421. [Google Scholar] [CrossRef] [PubMed]
- Konno, S.; Gonokami, Y.; Kurokawa, M.; Kawazu, K.; Asano, K.; Okamoto, K.; Adachi, M. Cytokine concentrations in sputum of asthmatic patients. Int. Arch. Allergy Immunol. 1996, 109, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.Y.; Pinkerton, J.W.; Essilfie, A.T.; Robertson, A.A.B.; Baines, K.J.; Brown, A.C.; Mayall, J.R.; Ali, M.K.; Starkey, M.R.; Hansbro, N.G.; et al. Role for NLRP3 Inflammasome-mediated, IL-1beta-Dependent Responses in Severe, Steroid-Resistant Asthma. Am. J. Respir. Crit. Care Med. 2017, 196, 283–297. [Google Scholar] [CrossRef] [PubMed]
- Holguin, F. Oxidative stress in airway diseases. Ann. Am. Thorac Soc. 2013, 10, S150–S157. [Google Scholar] [CrossRef]
- Jarjour, N.N.; Calhoun, W.J. Enhanced production of oxygen radicals in asthma. J. Lab. Clin. Med. 1994, 123, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Calhoun, W.J.; Reed, H.E.; Moest, D.R.; Stevens, C.A. Enhanced superoxide production by alveolar macrophages and air-space cells, airway inflammation, and alveolar macrophage density changes after segmental antigen bronchoprovocation in allergic subjects. Am. Rev. Respir. Dis. 1992, 145, 317–325. [Google Scholar] [CrossRef]
- Zeng, M.; Li, Y.; Jiang, Y.; Lu, G.; Huang, X.; Guan, K. Local and systemic oxidative stress status in chronic obstructive pulmonary disease patients. Can. Respir. J. 2013, 20, 35–41. [Google Scholar] [CrossRef]
- Erzurum, S.C. New Insights in Oxidant Biology in Asthma. Ann. Am. Thorac Soc. 2016, 13 (Suppl. 1), S35–S39. [Google Scholar] [CrossRef]
- Stephenson, S.T.; Brown, L.A.; Helms, M.N.; Qu, H.; Brown, S.D.; Brown, M.R.; Fitzpatrick, A.M. Cysteine oxidation impairs systemic glucocorticoid responsiveness in children with difficult-to-treat asthma. J. Allergy Clin. Immunol. 2015, 136, 454–461.e459. [Google Scholar] [CrossRef] [Green Version]
- Reynaert, N.L. Glutathione biochemistry in asthma. Biochim. Biophys. Acta 2011, 1810, 1045–1051. [Google Scholar] [CrossRef]
- Roum, J.H.; Buhl, R.; McElvaney, N.G.; Borok, Z.; Crystal, R.G. Systemic deficiency of glutathione in cystic fibrosis. J. Appl Physiol. 1993, 75, 2419–2424. [Google Scholar] [CrossRef]
- Pizzorno, J. Glutathione! Integr. Med. (Encinitas) 2014, 13, 8–12. [Google Scholar]
- Deneke, S.M.; Fanburg, B.L. Regulation of cellular glutathione. Am. J. Physiol. 1989, 257, L163–L173. [Google Scholar] [CrossRef]
- Meister, A.; Anderson, M.E. Glutathione. Annu. Rev. Biochem. 1983, 52, 711–760. [Google Scholar] [CrossRef]
- Fitzpatrick, A.M.; Teague, W.G.; Holguin, F.; Yeh, M.; Brown, L.A.; Severe Asthma Research, P. Airway glutathione homeostasis is altered in children with severe asthma: Evidence for oxidant stress. J. Allergy Clin. Immunol. 2009, 123, 146–152.e148. [Google Scholar] [CrossRef] [Green Version]
- Corradi, M.; Folesani, G.; Andreoli, R.; Manini, P.; Bodini, A.; Piacentini, G.; Carraro, S.; Zanconato, S.; Baraldi, E. Aldehydes and glutathione in exhaled breath condensate of children with asthma exacerbation. Am. J. Respir. Crit. Care Med. 2003, 167, 395–399. [Google Scholar] [CrossRef]
- Corradi, M.; Rubinstein, I.; Andreoli, R.; Manini, P.; Caglieri, A.; Poli, D.; Alinovi, R.; Mutti, A. Aldehydes in exhaled breath condensate of patients with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2003, 167, 1380–1386. [Google Scholar] [CrossRef]
- Nadeem, A.; Siddiqui, N.; Alharbi, N.O.; Alharbi, M.M.; Imam, F. Acute glutathione depletion leads to enhancement of airway reactivity and inflammation via p38MAPK-iNOS pathway in allergic mice. Int. Immunopharmacol. 2014, 22, 222–229. [Google Scholar] [CrossRef]
- Bowler, R.P.; Crapo, J.D. Oxidative stress in airways: Is there a role for extracellular superoxide dismutase? Am. J. Respir. Crit. Care Med. 2002, 166, S38–S43. [Google Scholar] [CrossRef] [PubMed]
- Crapo, J.D.; Oury, T.; Rabouille, C.; Slot, J.W.; Chang, L.Y. Copper, Zinc superoxide dismutase is primarily a cytosolic protein in human cells. Proc. Natl. Acad. Sci. USA 1992, 89, 10405–10409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, F.; Koenitzer, J.R.; Tobolewski, J.M.; Jiang, D.; Liang, J.; Noble, P.W.; Oury, T.D. Extracellular superoxide dismutase inhibits inflammation by preventing oxidative fragmentation of hyaluronan. J. Biol. Chem. 2008, 283, 6058–6066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Comhair, S.A.; Xu, W.; Ghosh, S.; Thunnissen, F.B.; Almasan, A.; Calhoun, W.J.; Janocha, A.J.; Zheng, L.; Hazen, S.L.; Erzurum, S.C. Superoxide dismutase inactivation in pathophysiology of asthmatic airway remodeling and reactivity. Am. J. Pathol. 2005, 166, 663–674. [Google Scholar] [CrossRef] [Green Version]
- De Raeve, H.R.; Thunnissen, F.B.; Kaneko, F.T.; Guo, F.H.; Lewis, M.; Kavuru, M.S.; Secic, M.; Thomassen, M.J.; Erzurum, S.C. Decreased Cu, Zn-SOD activity in asthmatic airway epithelium: Correction by inhaled corticosteroid in vivo. Am. J. Physiol. 1997, 272, L148–L154. [Google Scholar] [CrossRef]
- Comhair, S.A.; Bhathena, P.R.; Dweik, R.A.; Kavuru, M.; Erzurum, S.C. Rapid loss of superoxide dismutase activity during antigen-induced asthmatic response. Lancet 2000, 355, 624. [Google Scholar] [CrossRef]
- Alvarez, B.; Demicheli, V.; Duran, R.; Trujillo, M.; Cervenansky, C.; Freeman, B.A.; Radi, R. Inactivation of human Cu, Zn superoxide dismutase by peroxynitrite and formation of histidinyl radical. Free Radic. Biol. Med. 2004, 37, 813–822. [Google Scholar] [CrossRef]
- MacMillan-Crow, L.A.; Crow, J.P.; Thompson, J.A. Peroxynitrite-mediated inactivation of manganese superoxide dismutase involves nitration and oxidation of critical tyrosine residues. Biochemistry 1998, 37, 1613–1622. [Google Scholar] [CrossRef]
- MacPherson, J.C.; Comhair, S.A.; Erzurum, S.C.; Klein, D.F.; Lipscomb, M.F.; Kavuru, M.S.; Samoszuk, M.K.; Hazen, S.L. Eosinophils are a major source of nitric oxide-derived oxidants in severe asthma: Characterization of pathways available to eosinophils for generating reactive nitrogen species. J. Immunol. 2001, 166, 5763–5772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, W.; Samoszuk, M.K.; Comhair, S.A.; Thomassen, M.J.; Farver, C.F.; Dweik, R.A.; Kavuru, M.S.; Erzurum, S.C.; Hazen, S.L. Eosinophils generate brominating oxidants in allergen-induced asthma. J. Clin. Investig. 2000, 105, 1455–1463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cantin, A.; Crystal, R.G. Oxidants, antioxidants and the pathogenesis of emphysema. Eur. J. Respir. Dis. Suppl. 1985, 139, 7–17. [Google Scholar] [PubMed]
- Kirkman, H.N.; Rolfo, M.; Ferraris, A.M.; Gaetani, G.F. Mechanisms of protection of catalase by NADPH. Kinetics and stoichiometry. J. Biol. Chem. 1999, 274, 13908–13914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, S.; Janocha, A.J.; Aronica, M.A.; Swaidani, S.; Comhair, S.A.; Xu, W.; Zheng, L.; Kaveti, S.; Kinter, M.; Hazen, S.L.; et al. Nitrotyrosine proteome survey in asthma identifies oxidative mechanism of catalase inactivation. J. Immunol. 2006, 176, 5587–5597. [Google Scholar] [CrossRef] [Green Version]
- Hoshino, T.; Okamoto, M.; Takei, S.; Sakazaki, Y.; Iwanaga, T.; Aizawa, H. Redox-regulated mechanisms in asthma. Antioxid. Redox Signal. 2008, 10, 769–783. [Google Scholar] [CrossRef]
- Xu, J.; Li, T.; Wu, H.; Xu, T. Role of thioredoxin in lung disease. Pulm. Pharmacol. Ther. 2012, 25, 154–162. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Wang, C.; Wu, J.; Fukunaga, A.; Cheng, Z.; Wang, J.; Yamauchi, A.; Yodoi, J.; Tian, H. Anti-Allergic and Anti-Inflammatory Effects and Molecular Mechanisms of Thioredoxin on Respiratory System Diseases. Antioxid. Redox Signal. 2020, 32, 785–801. [Google Scholar] [CrossRef]
- Imaoka, H.; Hoshino, T.; Okamoto, M.; Sakazaki, Y.; Sawada, M.; Takei, S.; Kinoshita, T.; Kawayama, T.; Kato, S.; Aizawa, H. Endogenous and exogenous thioredoxin 1 prevents goblet cell hyperplasia in a chronic antigen exposure asthma model. Allergol. Int. 2009, 58, 403–410. [Google Scholar] [CrossRef] [Green Version]
- Torii, M.; Wang, L.; Ma, N.; Saito, K.; Hori, T.; Sato-Ueshima, M.; Koyama, Y.; Nishikawa, H.; Katayama, N.; Mizoguchi, A.; et al. Thioredoxin suppresses airway inflammation independently of systemic Th1/Th2 immune modulation. Eur. J. Immunol. 2010, 40, 787–796. [Google Scholar] [CrossRef]
- Imaoka, H.; Hoshino, T.; Takei, S.; Sakazaki, Y.; Kinoshita, T.; Okamoto, M.; Kawayama, T.; Yodoi, J.; Kato, S.; Iwanaga, T.; et al. Effects of thioredoxin on established airway remodeling in a chronic antigen exposure asthma model. Biochem. Biophys. Res. Commun. 2007, 360, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Tanabe, N.; Hoshino, Y.; Marumo, S.; Kiyokawa, H.; Sato, S.; Kinose, D.; Uno, K.; Muro, S.; Hirai, T.; Yodoi, J.; et al. Thioredoxin-1 protects against neutrophilic inflammation and emphysema progression in a mouse model of chronic obstructive pulmonary disease exacerbation. PLoS ONE 2013, 8, e79016. [Google Scholar] [CrossRef] [Green Version]
- Sato, A.; Hoshino, Y.; Hara, T.; Muro, S.; Nakamura, H.; Mishima, M.; Yodoi, J. Thioredoxin-1 ameliorates cigarette smoke-induced lung inflammation and emphysema in mice. J. Pharmacol. Exp. Ther. 2008, 325, 380–388. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, T.; Hoshino, T.; Imaoka, H.; Ichiki, H.; Okamoto, M.; Kawayama, T.; Yodoi, J.; Kato, S.; Aizawa, H. Thioredoxin prevents the development and progression of elastase-induced emphysema. Biochem. Biophys. Res. Commun. 2007, 354, 712–719. [Google Scholar] [CrossRef]
- Yamada, Y.; Nakamura, H.; Adachi, T.; Sannohe, S.; Oyamada, H.; Kayaba, H.; Yodoi, J.; Chihara, J. Elevated serum levels of thioredoxin in patients with acute exacerbation of asthma. Immunol. Lett. 2003, 86, 199–205. [Google Scholar] [CrossRef]
- Liu, Q.; Gao, Y.; Ci, X. Role of Nrf2 and Its Activators in Respiratory Diseases. Oxid. Med. Cell Longev. 2019, 2019, 7090534. [Google Scholar] [CrossRef] [Green Version]
- Mizumura, K.; Maruoka, S.; Shimizu, T.; Gon, Y. Role of Nrf2 in the pathogenesis of respiratory diseases. Respir. Investig. 2020, 58, 28–35. [Google Scholar] [CrossRef]
- Iizuka, T.; Ishii, Y.; Itoh, K.; Kiwamoto, T.; Kimura, T.; Matsuno, Y.; Morishima, Y.; Hegab, A.E.; Homma, S.; Nomura, A.; et al. Nrf2-deficient mice are highly susceptible to cigarette smoke-induced emphysema. Genes Cells 2005, 10, 1113–1125. [Google Scholar] [CrossRef] [PubMed]
- Ishii, Y.; Itoh, K.; Morishima, Y.; Kimura, T.; Kiwamoto, T.; Iizuka, T.; Hegab, A.E.; Hosoya, T.; Nomura, A.; Sakamoto, T.; et al. Transcription factor Nrf2 plays a pivotal role in protection against elastase-induced pulmonary inflammation and emphysema. J. Immunol. 2005, 175, 6968–6975. [Google Scholar] [CrossRef]
- Rangasamy, T.; Guo, J.; Mitzner, W.A.; Roman, J.; Singh, A.; Fryer, A.D.; Yamamoto, M.; Kensler, T.W.; Tuder, R.M.; Georas, S.N.; et al. Disruption of Nrf2 enhances susceptibility to severe airway inflammation and asthma in mice. J. Exp. Med. 2005, 202, 47–59. [Google Scholar] [CrossRef]
- Sussan, T.E.; Gajghate, S.; Chatterjee, S.; Mandke, P.; McCormick, S.; Sudini, K.; Kumar, S.; Breysse, P.N.; Diette, G.B.; Sidhaye, V.K.; et al. Nrf2 reduces allergic asthma in mice through enhanced airway epithelial cytoprotective function. Am. J. Physiol. Lung Cell Mol. Physiol. 2015, 309, L27–L36. [Google Scholar] [CrossRef] [Green Version]
- Blake, D.J.; Singh, A.; Kombairaju, P.; Malhotra, D.; Mariani, T.J.; Tuder, R.M.; Gabrielson, E.; Biswal, S. Deletion of Keap1 in the lung attenuates acute cigarette smoke-induced oxidative stress and inflammation. Am. J. Respir. Cell Mol. Biol. 2010, 42, 524–536. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Sun, D.; Zhu, Y. Expression of nuclear factor erythroid-2-related factor 2, broad complex-tramtrack-bric a brac and Cap‘n’collar homology 1 and gamma-glutamic acid cysteine synthase in peripheral blood of patients with chronic obstructive pulmonary disease and its clinical significance. Exp. Ther. Med. 2021, 21, 516. [Google Scholar] [CrossRef] [PubMed]
- Fratta Pasini, A.M.; Stranieri, C.; Ferrari, M.; Garbin, U.; Cazzoletti, L.; Mozzini, C.; Spelta, F.; Peserico, D.; Cominacini, L. Oxidative stress and Nrf2 expression in peripheral blood mononuclear cells derived from COPD patients: An observational longitudinal study. Respir. Res. 2020, 21, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, M.; Betsuyaku, T.; Ito, Y.; Nagai, K.; Nasuhara, Y.; Kaga, K.; Kondo, S.; Nishimura, M. Down-regulated NF-E2-related factor 2 in pulmonary macrophages of aged smokers and patients with chronic obstructive pulmonary disease. Am. J. Respir. Cell Mol. Biol. 2008, 39, 673–682. [Google Scholar] [CrossRef] [Green Version]
- Michaeloudes, C.; Chang, P.J.; Petrou, M.; Chung, K.F. Transforming growth factor-beta and nuclear factor E2-related factor 2 regulate antioxidant responses in airway smooth muscle cells: Role in asthma. Am. J. Respir. Crit. Care Med. 2011, 184, 894–903. [Google Scholar] [CrossRef] [Green Version]
- Janciauskiene, S. The Beneficial Effects of Antioxidants in Health And Diseases. Chronic Obstr. Pulm. Dis. 2020, 7, 182–202. [Google Scholar] [CrossRef]
- Kianian, F.; Karimian, S.M.; Kadkhodaee, M.; Takzaree, N.; Seifi, B.; Adeli, S.; Harati, E.; Sadeghipour, H.R. Combination of ascorbic acid and calcitriol attenuates chronic asthma disease by reductions in oxidative stress and inflammation. Respir. Physiol. Neurobiol. 2019, 270, 103265. [Google Scholar] [CrossRef] [PubMed]
- Kianian, F.; Karimian, S.M.; Kadkhodaee, M.; Takzaree, N.; Seifi, B.; Sadeghipour, H.R. Protective effects of ascorbic acid and calcitriol combination on airway remodelling in ovalbumin-induced chronic asthma. Pharm. Biol. 2020, 58, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Peh, H.Y.; Ho, W.E.; Cheng, C.; Chan, T.K.; Seow, A.C.; Lim, A.Y.; Fong, C.W.; Seng, K.Y.; Ong, C.N.; Wong, W.S. Vitamin E Isoform gamma-Tocotrienol Downregulates House Dust Mite-Induced Asthma. J. Immunol. 2015, 195, 437–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burbank, A.J.; Duran, C.G.; Pan, Y.; Burns, P.; Jones, S.; Jiang, Q.; Yang, C.; Jenkins, S.; Wells, H.; Alexis, N.; et al. Gamma tocopherol-enriched supplement reduces sputum eosinophilia and endotoxin-induced sputum neutrophilia in volunteers with asthma. J. Allergy Clin. Immunol. 2018, 141, 1231–1238.e1231. [Google Scholar] [CrossRef] [Green Version]
- Peh, H.Y.; Tan, W.S.D.; Chan, T.K.; Pow, C.W.; Foster, P.S.; Wong, W.S.F. Vitamin E isoform gamma-tocotrienol protects against emphysema in cigarette smoke-induced COPD. Free Radic. Biol. Med. 2017, 110, 332–344. [Google Scholar] [CrossRef]
- Jafarinia, M.; Sadat Hosseini, M.; Kasiri, N.; Fazel, N.; Fathi, F.; Ganjalikhani Hakemi, M.; Eskandari, N. Quercetin with the potential effect on allergic diseases. Allergy Asthma Clin. Immunol. 2020, 16, 36. [Google Scholar] [CrossRef]
- Joskova, M.; Sadlonova, V.; Nosalova, G.; Novakova, E.; Franova, S. Polyphenols and their components in experimental allergic asthma. Adv. Exp. Med. Biol. 2013, 756, 91–98. [Google Scholar] [CrossRef]
- Chen, J.; Zhou, H.; Wang, J.; Zhang, B.; Liu, F.; Huang, J.; Li, J.; Lin, J.; Bai, J.; Liu, R. Therapeutic effects of resveratrol in a mouse model of HDM-induced allergic asthma. Int. Immunopharmacol. 2015, 25, 43–48. [Google Scholar] [CrossRef]
- Park, H.J.; Lee, C.M.; Jung, I.D.; Lee, J.S.; Jeong, Y.I.; Chang, J.H.; Chun, S.H.; Kim, M.J.; Choi, I.W.; Ahn, S.C.; et al. Quercetin regulates Th1/Th2 balance in a murine model of asthma. Int. Immunopharmacol. 2009, 9, 261–267. [Google Scholar] [CrossRef]
- Wood, L.G.; Wark, P.A.; Garg, M.L. Antioxidant and anti-inflammatory effects of resveratrol in airway disease. Antioxid. Redox Signal. 2010, 13, 1535–1548. [Google Scholar] [CrossRef]
- Meja, K.K.; Rajendrasozhan, S.; Adenuga, D.; Biswas, S.K.; Sundar, I.K.; Spooner, G.; Marwick, J.A.; Chakravarty, P.; Fletcher, D.; Whittaker, P.; et al. Curcumin restores corticosteroid function in monocytes exposed to oxidants by maintaining HDAC2. Am. J. Respir. Cell Mol. Biol. 2008, 39, 312–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eftekhari, P.; Hajizadeh, S.; Raoufy, M.R.; Masjedi, M.R.; Yang, M.; Hansbro, N.; Li, J.J.; Foster, P.S. Preventive effect of N-acetylcysteine in a mouse model of steroid resistant acute exacerbation of asthma. EXCLI J. 2013, 12, 184–192. [Google Scholar]
- De Backer, J.; Vos, W.; Van Holsbeke, C.; Vinchurkar, S.; Claes, R.; Parizel, P.M.; De Backer, W. Effect of high-dose N-acetylcysteine on airway geometry, inflammation, and oxidative stress in COPD patients. Int. J. Chronic Obstr. Pulm. Dis. 2013, 8, 569–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Overveld, F.J.; Demkow, U.; Gorecka, D.; de Backer, W.A.; Zielinski, J. New developments in the treatment of COPD: Comparing the effects of inhaled corticosteroids and N-acetylcysteine. J. Physiol. Pharmacol. 2005, 56 (Suppl. 4), 135–142. [Google Scholar] [PubMed]
- Song, Y.; Lu, H.Z.; Xu, J.R.; Wang, X.L.; Zhou, W.; Hou, L.N.; Zhu, L.; Yu, Z.H.; Chen, H.Z.; Cui, Y.Y. Carbocysteine restores steroid sensitivity by targeting histone deacetylase 2 in a thiol/GSH-dependent manner. Pharmacol. Res. 2015, 91, 88–98. [Google Scholar] [CrossRef]
- Pace, E.; Ferraro, M.; Di Vincenzo, S.; Cipollina, C.; Gerbino, S.; Cigna, D.; Caputo, V.; Balsamo, R.; Lanata, L.; Gjomarkaj, M. Comparative cytoprotective effects of carbocysteine and fluticasone propionate in cigarette smoke extract-stimulated bronchial epithelial cells. Cell Stress Chaperones 2013, 18, 733–743. [Google Scholar] [CrossRef] [Green Version]
- Harvey, C.J.; Thimmulappa, R.K.; Sethi, S.; Kong, X.; Yarmus, L.; Brown, R.H.; Feller-Kopman, D.; Wise, R.; Biswal, S. Targeting Nrf2 signaling improves bacterial clearance by alveolar macrophages in patients with COPD and in a mouse model. Sci. Transl. Med. 2011, 3, 78ra32. [Google Scholar] [CrossRef] [Green Version]
- Starrett, W.; Blake, D.J. Sulforaphane inhibits de novo synthesis of IL-8 and MCP-1 in human epithelial cells generated by cigarette smoke extract. J. Immunotoxicol. 2011, 8, 150–158. [Google Scholar] [CrossRef] [Green Version]
- Jiao, Z.; Chang, J.; Li, J.; Nie, D.; Cui, H.; Guo, D. Sulforaphane increases Nrf2 expression and protects alveolar epithelial cells against injury caused by cigarette smoke extract. Mol. Med. Rep. 2017, 16, 1241–1247. [Google Scholar] [CrossRef] [Green Version]
- Al-Harbi, N.O.; Nadeem, A.; Ahmad, S.F.; AlThagfan, S.S.; Alqinyah, M.; Alqahtani, F.; Ibrahim, K.E.; Al-Harbi, M.M. Sulforaphane treatment reverses corticosteroid resistance in a mixed granulocytic mouse model of asthma by upregulation of antioxidants and attenuation of Th17 immune responses in the airways. Eur. J. Pharmacol. 2019, 855, 276–284. [Google Scholar] [CrossRef]
- Sakurai, H.; Morishima, Y.; Ishii, Y.; Yoshida, K.; Nakajima, M.; Tsunoda, Y.; Hayashi, S.Y.; Kiwamoto, T.; Matsuno, Y.; Kawaguchi, M.; et al. Sulforaphane ameliorates steroid insensitivity through an Nrf2-dependent pathway in cigarette smoke-exposed asthmatic mice. Free Radic. Biol. Med. 2018, 129, 473–485. [Google Scholar] [CrossRef]
- Zhang, J.H.; Yang, X.; Chen, Y.P.; Zhang, J.F.; Li, C.Q. Nrf2 Activator RTA-408 Protects Against Ozone-Induced Acute Asthma Exacerbation by Suppressing ROS and gammadeltaT17 Cells. Inflammation 2019, 42, 1843–1856. [Google Scholar] [CrossRef]
- Wise, R.A.; Holbrook, J.T.; Criner, G.; Sethi, S.; Rayapudi, S.; Sudini, K.R.; Sugar, E.A.; Burke, A.; Thimmulappa, R.; Singh, A.; et al. Lack of Effect of Oral Sulforaphane Administration on Nrf2 Expression in COPD: A Randomized, Double-Blind, Placebo Controlled Trial. PLoS ONE 2016, 11, e0163716. [Google Scholar] [CrossRef] [Green Version]
- Duran, C.G.; Burbank, A.J.; Mills, K.H.; Duckworth, H.R.; Aleman, M.M.; Kesic, M.J.; Peden, D.B.; Pan, Y.; Zhou, H.; Hernandez, M.L. A proof-of-concept clinical study examining the NRF2 activator sulforaphane against neutrophilic airway inflammation. Respir. Res. 2016, 17, 89. [Google Scholar] [CrossRef] [Green Version]
- Sudini, K.; Diette, G.B.; Breysse, P.N.; McCormack, M.C.; Bull, D.; Biswal, S.; Zhai, S.; Brereton, N.; Peng, R.D.; Matsui, E.C. A Randomized Controlled Trial of the Effect of Broccoli Sprouts on Antioxidant Gene Expression and Airway Inflammation in Asthmatics. J. Allergy Clin. Immunol. Pract. 2016, 4, 932–940. [Google Scholar] [CrossRef] [Green Version]
- Khattak, S.; Zhang, Q.Q.; Sarfraz, M.; Muhammad, P.; Ngowi, E.E.; Khan, N.H.; Rauf, S.; Wang, Y.Z.; Qi, H.W.; Wang, D.; et al. The Role of Hydrogen Sulfide in Respiratory Diseases. Biomolecules 2021, 11, 682. [Google Scholar] [CrossRef]
- Schiliro, M.; Bartman, C.M.; Pabelick, C. Understanding hydrogen sulfide signaling in neonatal airway disease. Expert Rev. Respir. Med. 2021, 15, 351–372. [Google Scholar] [CrossRef]
- Kimura, Y.; Goto, Y.; Kimura, H. Hydrogen sulfide increases glutathione production and suppresses oxidative stress in mitochondria. Antioxid. Redox Signal. 2010, 12, 1–13. [Google Scholar] [CrossRef]
- Jia, G.; Yu, S.; Sun, W.; Yang, J.; Wang, Y.; Qi, Y.; Chen, Y. Hydrogen Sulfide Attenuates Particulate Matter-Induced Emphysema and Airway Inflammation Through Nrf2-Dependent Manner. Front. Pharmacol. 2020, 11, 29. [Google Scholar] [CrossRef]
- Guan, R.; Wang, J.; Cai, Z.; Li, Z.; Wang, L.; Li, Y.; Xu, J.; Li, D.; Yao, H.; Liu, W.; et al. Hydrogen sulfide attenuates cigarette smoke-induced airway remodeling by upregulating SIRT1 signaling pathway. Redox Biol. 2020, 28, 101356. [Google Scholar] [CrossRef]
- Bazhanov, N.; Ansar, M.; Ivanciuc, T.; Garofalo, R.P.; Casola, A. Hydrogen Sulfide: A Novel Player in Airway Development, Pathophysiology of Respiratory Diseases, and Antiviral Defenses. Am. J. Respir. Cell Mol. Biol. 2017, 57, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Wang, K.; Li, M.X.; He, W.; Chang, J.R.; Liao, C.C.; Lin, F.; Qi, Y.F.; Wang, R.; Chen, Y.H. Metabolic changes of H2S in smokers and patients of COPD which might involve in inflammation, oxidative stress and steroid sensitivity. Sci. Rep. 2015, 5, 14971. [Google Scholar] [CrossRef] [PubMed]
- Liao, W.; Lim, A.Y.H.; Tan, W.S.D.; Abisheganaden, J.; Wong, W.S.F. Restoration of HDAC2 and Nrf2 by andrographolide overcomes corticosteroid resistance in chronic obstructive pulmonary disease. Br. J. Pharmacol. 2020, 177, 3662–3673. [Google Scholar] [CrossRef] [PubMed]
- Li, J.J.; Tay, H.L.; Maltby, S.; Xiang, Y.; Eyers, F.; Hatchwell, L.; Zhou, H.; Toop, H.D.; Morris, J.C.; Nair, P.; et al. MicroRNA-9 regulates steroid-resistant airway hyperresponsiveness by reducing protein phosphatase 2A activity. J. Allergy Clin. Immunol. 2015, 136, 462–473. [Google Scholar] [CrossRef] [PubMed]
- Khorasani, N.; Baker, J.; Johnson, M.; Chung, K.F.; Bhavsar, P.K. Reversal of corticosteroid insensitivity by p38 MAPK inhibition in peripheral blood mononuclear cells from COPD. Int. J. Chronic Obstr. Pulm. Dis. 2015, 10, 283–291. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lewis, B.W.; Ford, M.L.; Rogers, L.K.; Britt, R.D., Jr. Oxidative Stress Promotes Corticosteroid Insensitivity in Asthma and COPD. Antioxidants 2021, 10, 1335. https://doi.org/10.3390/antiox10091335
Lewis BW, Ford ML, Rogers LK, Britt RD Jr. Oxidative Stress Promotes Corticosteroid Insensitivity in Asthma and COPD. Antioxidants. 2021; 10(9):1335. https://doi.org/10.3390/antiox10091335
Chicago/Turabian StyleLewis, Brandon W., Maria L. Ford, Lynette K. Rogers, and Rodney D. Britt, Jr. 2021. "Oxidative Stress Promotes Corticosteroid Insensitivity in Asthma and COPD" Antioxidants 10, no. 9: 1335. https://doi.org/10.3390/antiox10091335