1. Introduction
Tuberculosis (TB) remains a major cause of morbidity and mortality in underdeveloped and developing countries. While substantial progress has been made in understanding the biological basis of Mtb pathogenesis, precisely how Mtb physiology and metabolism contribute to persistence, pathogenesis, and drug resistance is poorly understood.
The gasotransmitters carbon monoxide (CO) and nitric oxide (NO) have important roles in
Mtb physiology [
1,
2,
3,
4]. A third gasotransmitter, hydrogen sulfide (H
2S), is involved in a wide variety of physiological processes in eukaryotes and prokaryotes [
5]. H
2S is a weak acid in solution and at physiological pH, it exists predominantly as a hydrosulfide anion (HS
−, 75–80%), with the rest as H
2S (20–25%) and only trace amounts of S
2− [
6]. Notably, H
2S diffuses easily through cell membranes unlike its deprotonated conjugate bases HS
− and S
2−. The complex nature of H
2S in solution and its volatility make this molecule very difficult to measure and challenging to work with [
6,
7]. Furthermore, H
2S can be generated non-enzymatically from media components [
8] and, therefore, methods for measuring H
2S require rigorous validation.
Cysteine (Cys) is frequently used as a substrate by bacterial Cys desulfhydrases for H
2S production. Intracellular Cys levels require careful control, since excessive levels can stimulate oxidative stress via the Fenton reaction, which produces hydroxyl radicals [
9]. On the other hand, H
2S is also a substrate for Cys biosynthesis, which is required for the formation of low molecular weight thiols such as mycothiol (MSH) and ergothioneine (EGT) and biogenesis of iron sulfur cluster proteins [
10,
11]. The ability of bacteria to produce H
2S has been widely used as a diagnostic test and for taxonomic purposes [
12,
13].
H
2S production in bacteria has been attributed mainly to the enzymatic activity of cystathionine β-synthase (CBS), cystathionine γ-lyase (CSE), and 3-mercaptopyruvate sulfurtransferase (3MST) [
14,
15]. Addition of Cys to bacterial cultures has been shown to stimulate H
2S production [
14,
15,
16,
17]. In both bacterial and mammalian cells, Cys is used as a substrate by CBS and CSE to produce H
2S [
15,
18,
19]. Although bacterial production of H
2S was initially considered to be a byproduct of sulfur metabolism with unclear physiological relevance, studies in several bacterial species have shown that disrupting H
2S-producing genes increases antibiotic susceptibility [
15]. Obviously, these findings have important implications for understanding drug resistance in bacterial pathogens. In
E. coli, 3MST produces the majority of cellular H
2S from Cys [
14] and genetic disruption of this gene leads to increased sensitivity to oxidative stress [
14], implicating 3MST-derived H
2S in maintaining redox homeostasis.
Several studies have shown that H
2S reversibly inhibits cytochrome c oxidase (Complex IV) at high concentrations and stimulates mitochondrial respiration at low concentrations [
20,
21]. We have recently shown that exogenous H
2S also targets the
Mtb electron transport chain to increase respiration and ATP levels leading to increased growth [
22], and that host-generated H
2S exacerbates TB in mice [
22,
23]. Further, CSE and 3MST protein levels are substantially increased in human tuberculous lung tissue, and it has been proposed that
Mtb triggers supraphysiological levels of host-generated H
2S at the site of infection to suppress host immunity, thereby exacerbating disease [
23]. Notably,
Mtb senses host-generated H
2S during infection and reprograms its metabolism accordingly [
22].
H
2S plays important roles in modulating mammalian immunity, although its effects have been shown to be both pro- and anti-inflammatory [
18,
24]. Thus, H
2S produced by bacterial pathogens could act as a signaling molecule in the host, potentially exacerbating disease. The presence of
Mtb homologues of H
2S-producing enzymes identified in KEGG [
25] and several biochemical studies [
26,
27,
28,
29,
30] suggest that
Mtb has the capacity to produce H
2S. However, formal genetic and biochemical demonstration of H
2S production by
Mtb cells and its functions in
Mtb physiology is lacking. Due to the diverse roles H
2S plays in prokaryotic biology and bacterial classification, clear evidence that
Mtb produces H
2S is expected to widely influence our understanding of
Mtb physiology, disease, and diagnostics.
Here, we tested the hypothesis that Mtb produces H2S by measuring H2S production in drug-susceptible (DS) and multidrug-resistant (MDR) clinical strains, laboratory Mtb strains, and non-pathogenic mycobacterial species. We then identified a gene and corresponding protein responsible for H2S production in Mtb. We generated an Mtb knockout strain and utilized flow cytometry, extracellular metabolic flux analysis, 13C stable isotope analyses, and drug studies to determine the effect of endogenously produced H2S on respiration, central metabolism, redox balance, and drug susceptibility. We expect our findings to broadly impact our understanding of Mtb physiology and drug resistance.
2. Materials and Methods
2.1. General
All mycobacteria strains (See
Supplementary Materials, Table S1) were cultured in Middlebrook 7H9 media (BD Difco, New York, NY, USA) supplemented with 0.01% tyloxapol (Sigma–Aldrich, USA), 0.2% glycerol (Sigma–Aldrich, New York, NY, USA), and 10% (oleic acid, bovine albumin fraction V, dextrose, and catalase (OADC, BD Difco, New York, NY, USA) unless stated otherwise. Cultures were placed in a shaking incubator (100 rpm) at 37 °C. Strains examined included
Mb H37Rv,
Mtb CDC1551,
Mycobacterium bovis (supplemented media with 100 µM sodium pyruvate),
Mycobacterium bovis BCG,
M. smegmatis, two drug susceptible
Mtb strains (i.e., TKK-01-0027 and TKK-01-0047), and two multi-drug resistant
Mtb strains (i.e., TKK-01-0035 and TKK-01-0001). The drugs’ MIC
50 values used during this study were as follows: clofazimine (CFZ), 211 nM; rifampicin (RIF), 486 nM; isoniazid (INH), 240 nM. Where required, the following antibiotics were used; hygromycin B (100 µg/mL for
E. coli, 50 µg/mL for mycobacteria) and kanamycin (50 µg/mL for
E. coli, 25 µg/mL for mycobacteria). Dihydroethidium (DHE) was purchased from Thermo Fisher Scientific (New York, NY, USA) (Cat# D11347). Restriction enzymes were obtained from Thermo Fisher Scientific (Germany). The KOD Xtreme Hotstart DNA polymerase kit was obtained from Merck (Darmstadt, Germany). T4 DNA ligase was obtained from New England Biolabs (NEB, New York, NY, USA).
E. coli DH5α, used for cloning and DNA manipulation, was routinely cultured in Luria-Bertani liquid media at 37 °C. Oligonucleotides were synthesized by Thermo Fisher Scientific (USA). All other reagents were purchased from Merck or Sigma–Aldrich.
2.2. Preparation of Mycobacterial Lysates
All cultures were grown to an OD600 of ~0.8. The cells were then harvested from 30 mL of culture and centrifuged at 4000× g for 5 min. The supernatant was discarded, and the pellet was resuspended in 1–2 mL of lysis buffer (50 mM Tris-HCl, pH 8.0; 150 mM NaCl; protease inhibitor (Roche, New York, NY, USA)), depending on the size of the cell pellet. Cells were lysed in a MagNA Lyser (Roche, USA) at 7000 rpm for 1 min and then placed on ice for 4 min. This was repeated 3–4 times. The lysates were then centrifuged at 15,000× g for 10 min. The supernatant was collected and passed through a 0.22 µm filter. Protein concentrations were determined using the Micro BCA Protein Assay Kit (Thermo Fisher Scientific, USA), and the absorbance at 562 nm was measured using a Biotek Synergy H4 Hybrid Reader (BioTek, New York, NY, USA. Lysates were stored at 80 °C until use.
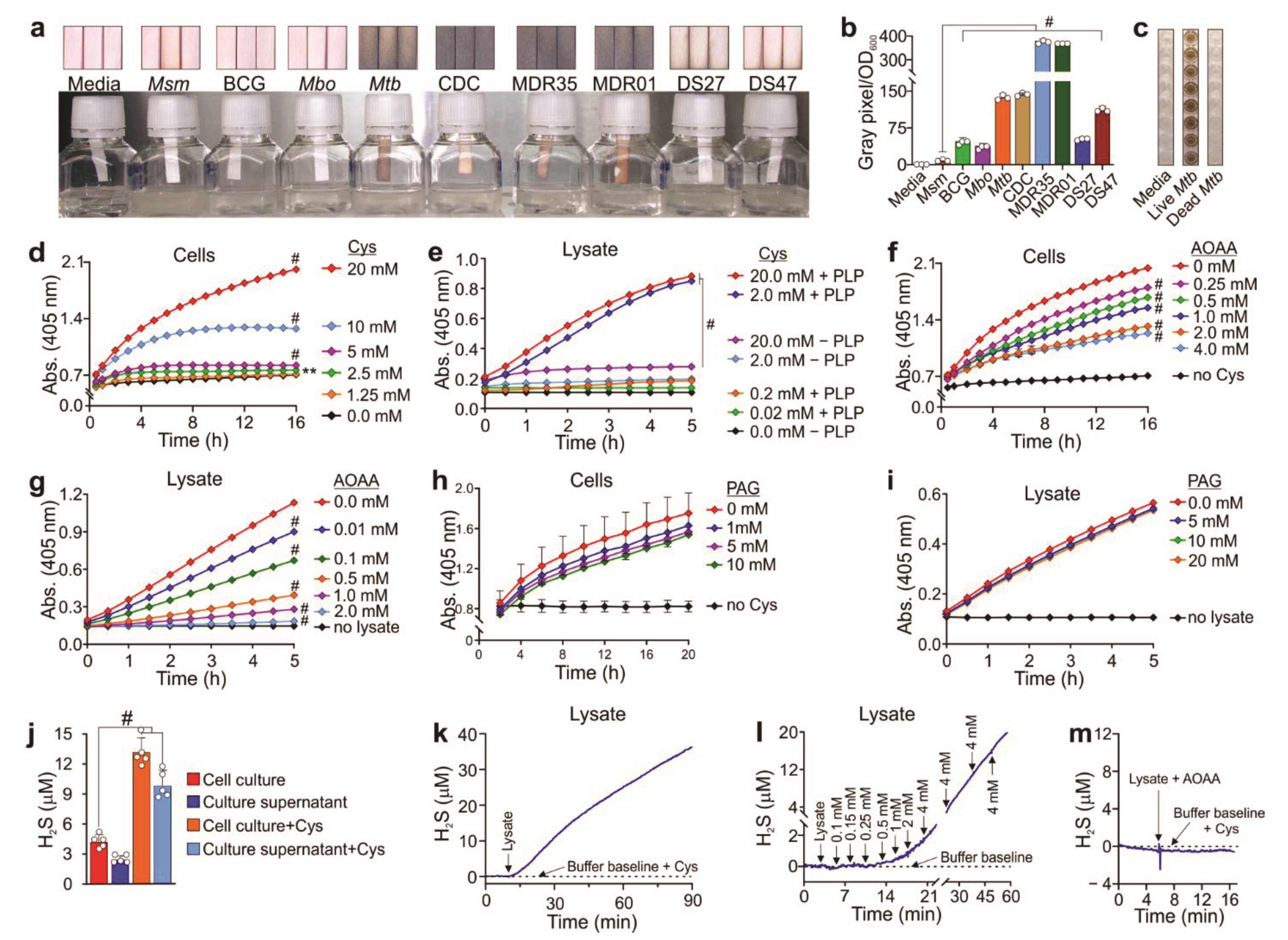
2.3. H2S Measurement Using the Lead Acetate Assay
Mycobacterial cultures were harvested at an OD
600 of 0.8–1 and centrifuged at 3500×
g for 5 min. The bacterial pellet was resuspended in an equal volume of 7H9 media containing 0.01% tyloxapol, 0.2% glycerol, and 10% OADC. 10 mL of diluted culture at an OD
600 of 0.1 was then transferred to a 30 mL culture bottle. Lead acetate strips (Thermo Fisher Scientific, USA) were affixed to the inner wall of the culture bottles. The strips were monitored for the formation of dark colored lead sulfide precipitate and scanned after 48 h. The intensity of the dark lead sulfide stain is proportional to the amount of H
2S present. The lead sulfide stain was then scanned and quantified by measuring the grayscale values for a specific area of each strip and normalized to OD
600 using ImageJ software version 1.53a (Java 1.8.8_12 (64 bit)) [
31].
2.4. H2S Measurement Using the Bismuth (III) Chloride (BiCl3) Assay
The BiCl
3 (BC) assay is used to measure H
2S based on the reaction of H
2S with a bismuth (III) salt to form bismuth (III) sulfide (Bi
2S
3), which appears as a brown-to-black precipitate [
32]. The microplate BC assay was performed in 96-well plates using intact H37Rv bacteria and lysates as described by Basic et al. (2015) [
33]. Once the OD
600 of cultures reached ~0.8–1, the cultures were centrifuged at 3500×
g for 5 min. The supernatant was discarded, and the culture pellet was resuspended in the original volume of media before centrifugation. The BC assay solution (2×) contains 0.4 M triethanolamine–HCl/Tris–HCl (Sigma–Aldrich, USA), pH 8.0; 1 mM BiCl
3 (Sigma–Aldrich, New York, NY, USA); 20 µM pyridoxal 5-phosphate monohydrate (PLP) (Sigma–Aldrich, USA), 20 mM EDTA (Sigma–Aldrich, USA), and 40 mM
l-cysteine (Cys) (Sigma–Aldrich, USA). One hundred microliters of the mycobacteria cell suspension (OD
600 = 1.0) or lysate (5 μg), with or without the inhibitors AOAA and PAG, was mixed with 100 µL of freshly prepared 2× bismuth solution in clear, flat-bottomed 96-well microtiter plates (Corning Inc., New York, NY, USA). For H
2S measurements when
Mtb was grown on different carbon sources, 100 µL of mycobacterial cell suspension in 7H9 medium containing 0.01% tyloxapol was supplemented with either: 0.4% glycerol, 1 mM sodium butyrate (Sigma–Aldrich, USA), 8 mM sodium acetate (Sigma), 2 mM sodium propionate (Sigma–Aldrich, USA) or 0.02% (
w/
v) cholesterol (Sigma–Aldrich, USA). These cell suspensions were then mixed with 100 µL of freshly prepared 2× bismuth solution in clear flat-bottomed 96-well microtiter plates. The working stock cholesterol was initially dissolved at 100 mg/mL in a solution of tyloxapol:ethanol (1:1) at 80 °C. Bi
2S
3 formation was determined by measuring the absorbance at 405 nm. The kinetics for mycobacterial cells was measured every 30 min for 15–20 h at 37 °C with shaking using a Hidex Sense Plate reader (Hidex, Finland). Enzymatic kinetics using lysates was measured every 5 min for 5–20 h with shaking at room temperature (~20–22 °C) using a Biotek Synergy H4 Hybrid Reader, (BioTek, New York, NY, USA).
2.5. Mtb Growth in Fatty Acids or Cholesterol as the Sole Carbon Source
One hundred microliters of mycobacterial cell suspension of OD600 ~0.1 in 7H9 containing 0.01% tyloxapol were mixed with 100 µL of 7H9 media containing 0.01% tyloxapol supplemented with either: 0.4% glycerol, 1 mM sodium butyrate, 8 mM sodium acetate, 2 mM sodium propionate, or 0.02% (w/v) cholesterol in clear, flat-bottomed 96-well microtiter plates. The plates were placed in an incubator at 37 °C. OD600 measurements were taken after 7 days.
2.6. H2S Measurement Using the Unisense Amperometric Microsensor
H2S released by cell cultures and lysates was measured at room temperature with a sensitive sulfide amperometric microsensor, H2S-500 (Unisense A/S, Denmark), connected to a microsensor multimeter (Unisense, A/S, Denmark) as an amplifier for data acquisition. The signal for H2S was collected in mV and converted to µM using a NaHS (Thermo Fisher Scientific) standard curve generated from a concentration range of 0–100 μM (freshly prepared in an anaerobic glovebox). The H2S microsensor was calibrated in accordance with the manufacturer’s instructions. Bacteria at an OD600 of 0.2 were cultured in media with or without 1 mM Cys. After 72 h, H2S concentrations were measured using the microsensor in the cell culture and the cell-free culture supernatants (referred to as “cleared supernatants”). Alternatively, Mtb strains were cultured to OD600 of ~0.8 without L-Cys and H2S measured after mixing 700 µL Mtb culture and 300 µL assay buffer (4 mL of 1.0 M Tris-HCl, pH 7.0, 1.0 mL of 400 mM L-Cys, and 1.0 mL of 200 mM EDTA) using the microsensor at different time points. H2S levels were normalized according to optical density.
For real-time H2S measurement in lysates, the microsensor was placed in a 2 mL tube containing 200 µL of 2× assay buffer (0.4 M triethanolamine-HCl, pH 8.0; 20 µM PLP, 20 mM EDTA) with or without 40 mM L-Cys and 160 µL lysis buffer (50 mM Tris-HCl, pH 8.0; 150 mM NaCl). The signal was allowed to generate a stable buffer baseline for ~5 min after which 40 µg (adjusted volume to 40 µL) of mycobacterial lysate was added to the reaction. When appropriate, L-Cys (0.1–4 mM) was subsequently added to the tube at different time intervals. To confirm AOAA inhibition, Mtb lysate preincubated with AOAA (4 mM) was added to the assay buffer containing 20 mM L-Cys. OASS activity was measured by placing the microsensor in 1 mL PBS solution. A 10 µL aliquot of 25 mM NaHS was added to the reaction tube twice, and after the signal stabilized, 30 ng of OASS was added to the reaction. After 2 min, OAS was added to the reaction to a final concentration of 10 mM, and the H2S signal was monitored in real time.
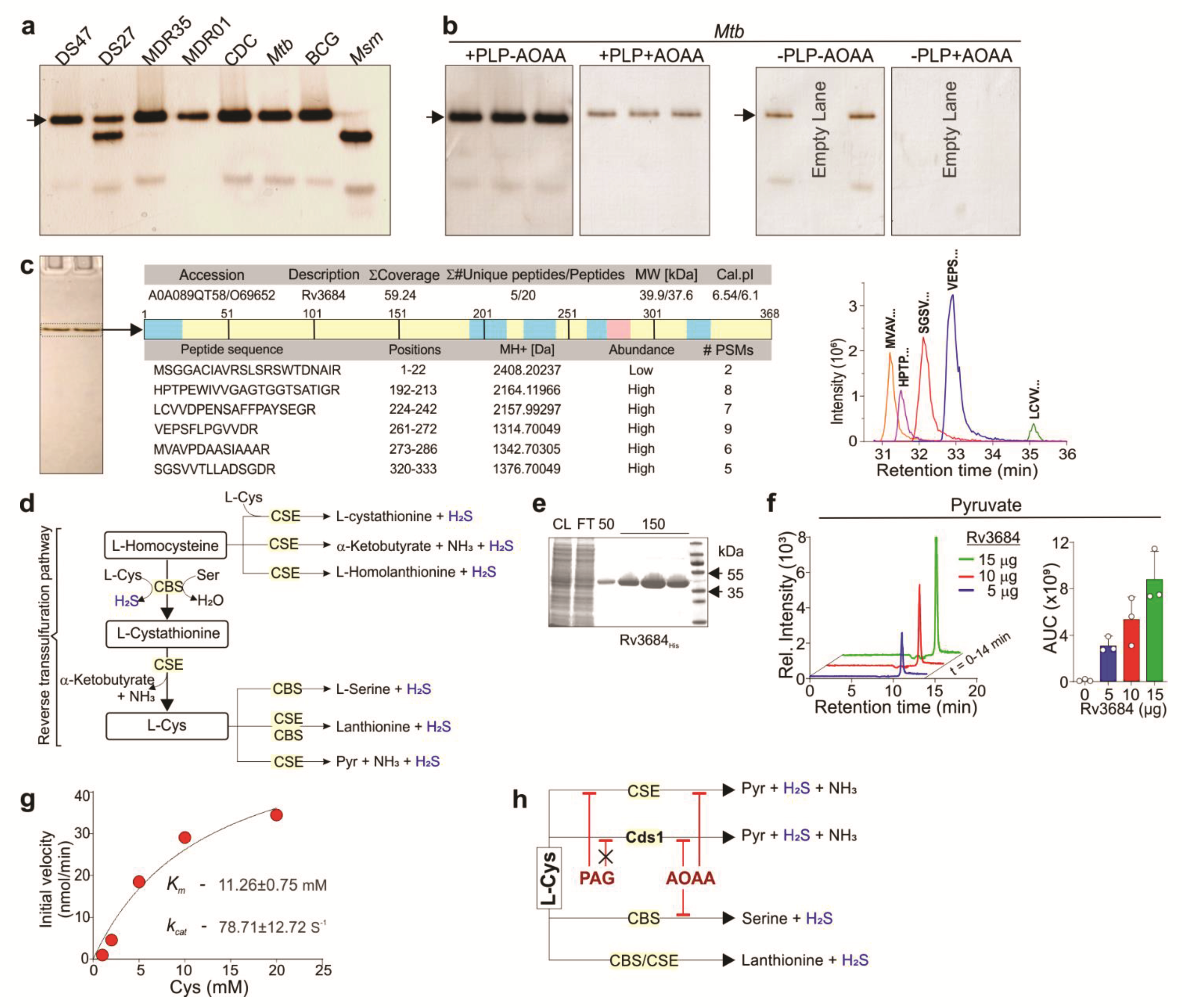
2.7. Native PAGE Analysis and In-Gel BC Assay
Equal amounts (15–25 µg per lane) of mycobacterial lysate were resolved on 10% PAGE gels (Bio-Rad, New York, NY, USA) under non-denaturing conditions using running buffer containing 25 mM Tris-base and 190 mM glycine. To detect the presence of H2S-producing proteins, gels were incubated in 20–50 mL of BC solution and incubated at room temperature with shaking. Gels were monitored every 20–60 min for the appearance of dark-colored Bi2S3. For gels exposed to AOAA, gels were incubated in 20 mL of 2 mM AOAA in 50 mM Tris-HCl pH 8.0 with shaking at room temperature for 5 min, followed by BC solution containing 2 mM AOAA overnight.
2.8. Extracellular Flux Analysis
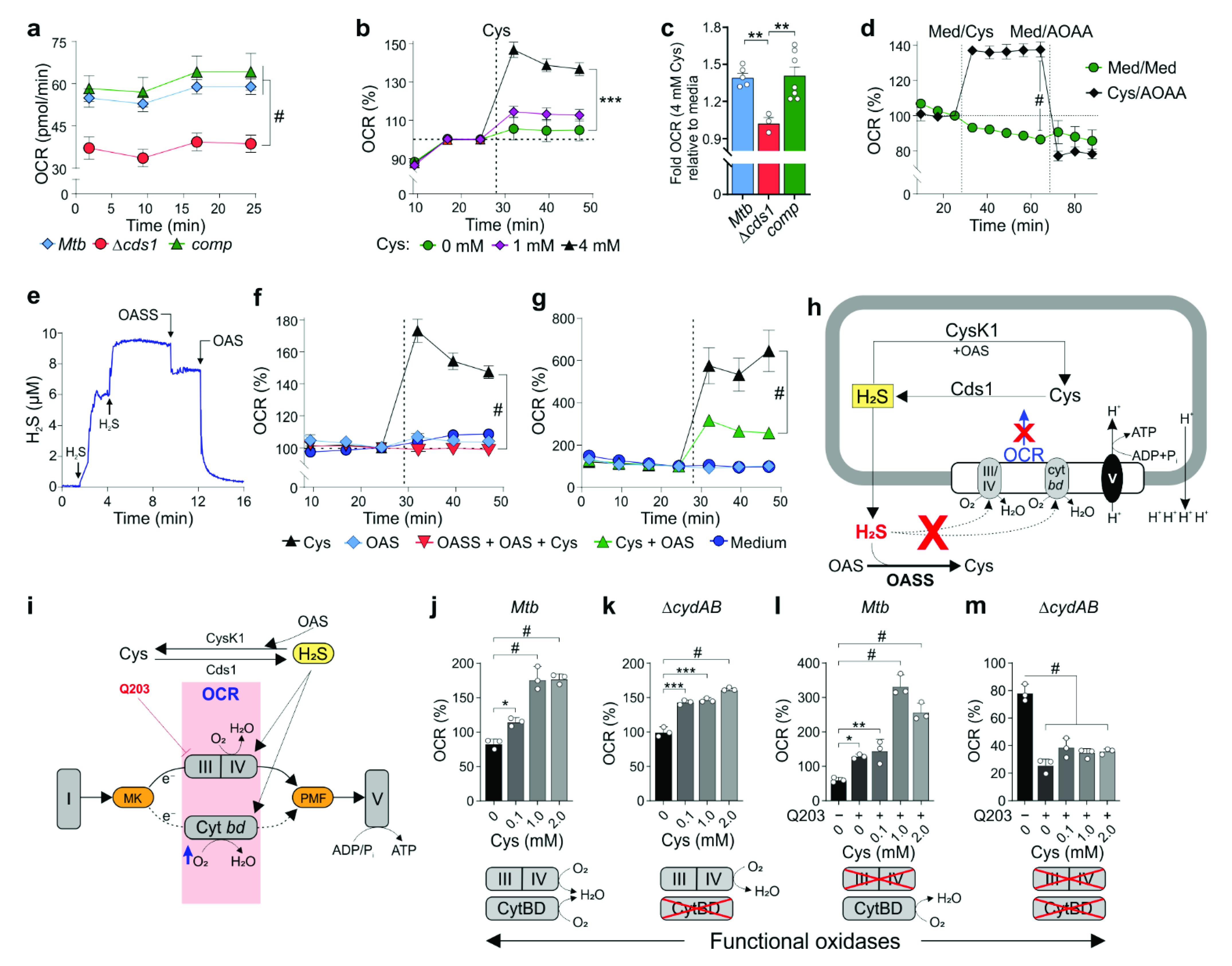
The oxygen consumption rates (OCR) of
Mtb strains were measured using a Seahorse XFe96 Extracellular Flux Analyzer (Agilent Technologies Inc., New York, NY, USA).
Mtb bacilli were adhered to the bottom of a Cell-Tak-coated XF cell culture microplate at 2 × 10
6 bacilli per well. Cell-Tak has no effect on
Mtb basal respiration [
34]. Assays were carried out in unbuffered 7H9 media (pH 7.35) with no carbon source.
Mtb bacilli were grown in this unbuffered 7H9 media, containing only 0.01% Tyloxapol, for 24 h before being seeded into the XF cell culture microplate at the start of the experiment. In general, basal OCR was measured for ~25 min before automatic sequential injection of various compounds through the drug ports of the sensor cartridge. The duration of OCR measurements after compound addition and the concentrations used varied by experiment. OASS modulation of
Mtb OCR in the presence of L-Cys was performed by the simultaneous addition of Cys, OASS, and substrate OAS (final concentration of 4 mM, 0.03 µg/mL, and 4 mM, respectively). Q203-based modulation of the OCR in
Mtb and Δ
cydAB cells was performed in the presence of the indicated Cys concentration followed by the addition of Q203 (300 × MIC
50; MIC
50 for Q203 is 3 nM) [
34]. To chemically complement Δ
cds1 cells with H
2S, different concentrations of NaHS were added to cells after measuring the baseline OCR. All OCR data figures indicate the time of each addition as dotted lines. OCR data points are representative of the average OCR during 4 min of continuous measurement in the transient microchamber, with the error being calculated from the OCR measurements taken from at least three replicate wells by the Seahorse Wave Desktop 2.2 software (Agilent Technologies Inc., New York, NY, USA). The transient microchamber was automatically re-equilibrated between measurements through the up and down mixing of the probes of the XF96 sensor cartridge in the wells of the XF cell culture microplate.
2.9. CFU-Based Assay
Mid-log phase mycobacterial cultures were diluted to an OD600 of 0.01 in 7H9 media. For survival studies in the presence of NaHS, bacterial cultures (7H9 with 10% OAD) were untreated or treated with anti-TB drugs and NaHS at indicated concentrations. For survival studies in the presence of antioxidants, bacterial cultures (7H9 with 10% OAD) were treated with or without 0.25 mM cumene hydroperoxide (CHP) for 16 h. For survival studies in the presence of CFZ, bacterial cultures (7H9 with 10% OADC) were treated with or without clofazimine at MIC 60×, 100× and 300× for 8 days. For all studies, samples were taken at indicated time points, serially diluted in PBS containing 0.01% tyloxapol and plated onto 7H11 OADC agar plates. Plates were incubated at 37 °C for 4 weeks to determine CFU counts.
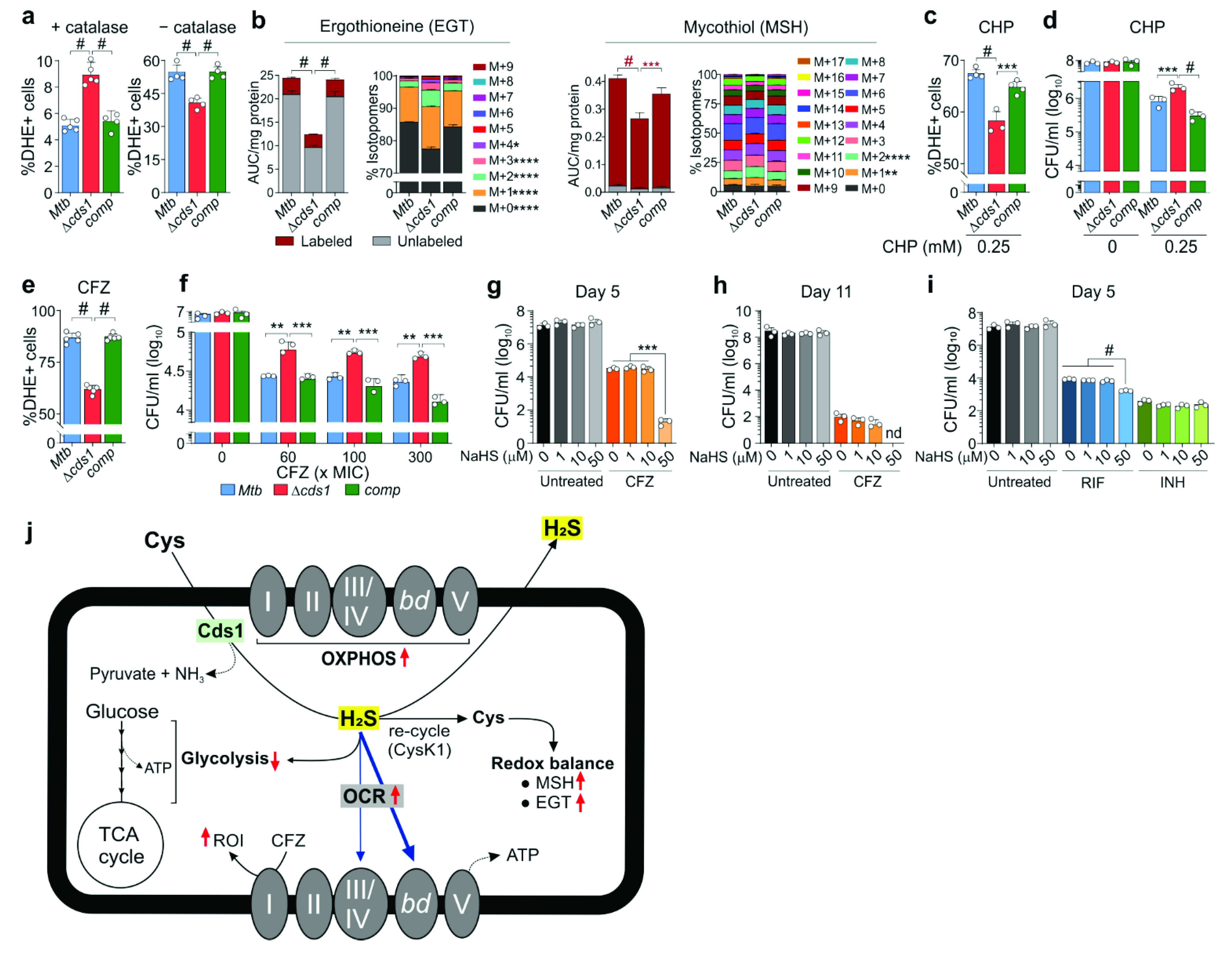
2.10. ROI Assay
ROI production in Mtb strains (OD600 ~1.0) was measured using the dihydroethidium ROI sensing dye (DHE, excitation/emission at 500/605 nm). Mtb strains were cultured in Middlebrook 7H9 media supplemented with 0.2% glycerol and 0.01% tyloxapol at 37 °C with either 10% OAD (oleic acid, albumin, dextrose) with/without 0.25 mM cumene hydroperoxide (CHP) or 10% OADC (oleic acid, albumin, dextrose, and catalase) with or without 60 × MIC of CFZ for 16 h in 4 replicates. After treatment, Mtb cultures were washed by centrifugation (3000× g) and resuspended in 1× PBS (pH 7.4) containing 10 µM DHE, incubated further for 20 min at 37 °C followed by two washes with PBS to remove residual extracellular dye. The fluorescence of DHE-stained cells was acquired with a FACS Aria III cell sorter using the 500 nm laser excitation, and BP 610/20 nm for emission acquisition (PerCP-CyTM5.5). The cells were acquired at a constant flow rate of setting 4, a threshold rate of approximately 1000–2000 events per second, and 100,000 total events were recorded per sample. For analysis, the bacterial population was identified according to the forward and side scattering property of the population (FSC versus SSC). To obtain single cell populations, bacterial aggregation was removed from the data analysis using doublet discrimination from the FCS-height versus FSC-area plots. The percentage of DHE+ cells and mean fluorescent intensity were calculated with FlowJoTM v10.4.2 (Tree Star, Ashland, OR, USA).
2.11. Identification of Proteins by LC-MS/MS
The entire Bi2S3-stained protein band was excised from the gel, rinsed with water, and cut into approximately 1 mm × 4mm pieces using a sterile scalpel. The gel slices were then rinsed with 100 mM ammonium bicarbonate solution and collectively transferred into a sterile Eppendorf® LoBind (Eppendorf, Germany) 1.5 mL microcentrifuge tube. Five hundred microliters of acetonitrile (ACN) was added, and the sample was incubated on ice for 10 min. The sample was then briefly centrifuged, the acetonitrile removed, and 100 µL of 10 mM dithiothreitol (DTT) solution was added to rehydrate the gel pieces and reduce the proteins. The sample was incubated in 10 mM DTT solution at 56 °C for 30 min, removed, cooled to room temperature, and then 500 µL of ACN was added and the sample was incubated on ice for 10 min. The sample was then centrifuged, and the supernatant removed, then 100 µL of 55 mM iodoacetamide solution was added and the sample was incubated at room temperature for 30 min in the dark to facilitate protein alkylation. Following alkylation, 500 µL of ACN was added and the sample was incubated on ice for 10 min. All solution was then removed, and 200 µL of trypsin (Promega, sequence grade) solution at a concentration of 13 ng/mL was added to the gel slices; the sample was mixed gently and incubated at 4 °C for 2 h to allow the gel slices to re-hydrate and for the slow diffusion of trypsin into the polyacrylamide gel matrix. The samples were incubated in the trypsin solution at 37 °C overnight (18–24 h) for optimum in-gel protein digestion. The resulting peptides were extracted by adding 400 µL of 5% formic acid/acetonitrile (1:2, v/v) solution to the sample followed by 15 min incubation at 37 °C on a shaking heating block set at 450 rpm. The sample was briefly centrifuged, the supernatant transferred to a sterile microcentrifuge tube and dried using a SpeedVac concentrator (Labconco, New York, NY, USA) set at 40 °C. The extracted, dried peptides were then reconstituted in 50 µL of 5% formic acid solution, transferred to a glass vial, and 1 µL of sample was injected for nano-LC-MS/MS analysis.
The peptide digests were analyzed using a Thermo Q Exactive Orbitrap mass spectrometer coupled to a DionexTM UltiMateTM 3000 UPLC system. The tryptic peptides were maintained at 6 °C in the autosampler and were separated on a 15 cm nano-capillary column (ID 75 μM) packed in the laboratory with Supelco® (Supelco Inc., New York, NY, USA) 3.5 μM C18 stationary phase. A 45 min gradient from 1% acetonitrile, 99% water/0.1% formic acid, to 50% acetonitrile/water, 0.1% formic acid, flow rate 300 nL/min, was used for the analysis. Peptide fragment mass spectra were acquired using a full MS, data dependent MS2 Top 10 method. The MS RAW files were processed using Thermo Scientific™ Proteome Discoverer™ (Thermo Fisher Scientific, USA) 2.2 software and SEQUEST™ (Thermo Fisher Scientific, USA) peak-finding search engine application to compare the mass spectra to the Mtb FASTA database to identify relevant proteins and peptides. The method was set to consider carbamidomethyl modifications and methionine oxidation. The protein candidates were then screened for pyridoxal phosphate binding domains and potential roles in sulfur metabolism. A targeted method was constructed using the 5 strongest peptide fragment ions for the most likely candidate, Rv3684/Cds1, and the samples re-analyzed using this method to confirm the presence of this protein.
2.12. Preparation of Mycobacterial Genomic DNA
Genomic DNA was isolated from Mtb H37Rv as follows: Mtb H37Rv was grown to late log phase (OD600 = 1.0) in 50 mL 7H9 liquid media. Cells were harvested (2000× g, 20 min), the supernatant was discarded, and 6 mL of a freshly prepared solution of 3 parts chloroform to 1 part methanol added. Tubes were then vortexed for 1 min. Tris-buffered phenol (6 mL) was then added and the tube vortexed for a further 30 s. Finally, 9 mL of 4 mM guanidine thiocyanate solution was added, and the tubes inverted several times. After centrifuging at 2000× g for 15 min, the upper phase was removed, and an equal volume of isopropanol was added to precipitate genomic DNA. The DNA was collected by centrifugation and washed with 70% ethanol before being air-dried and suspended in 100 µL Tris-EDTA, pH 7.5.
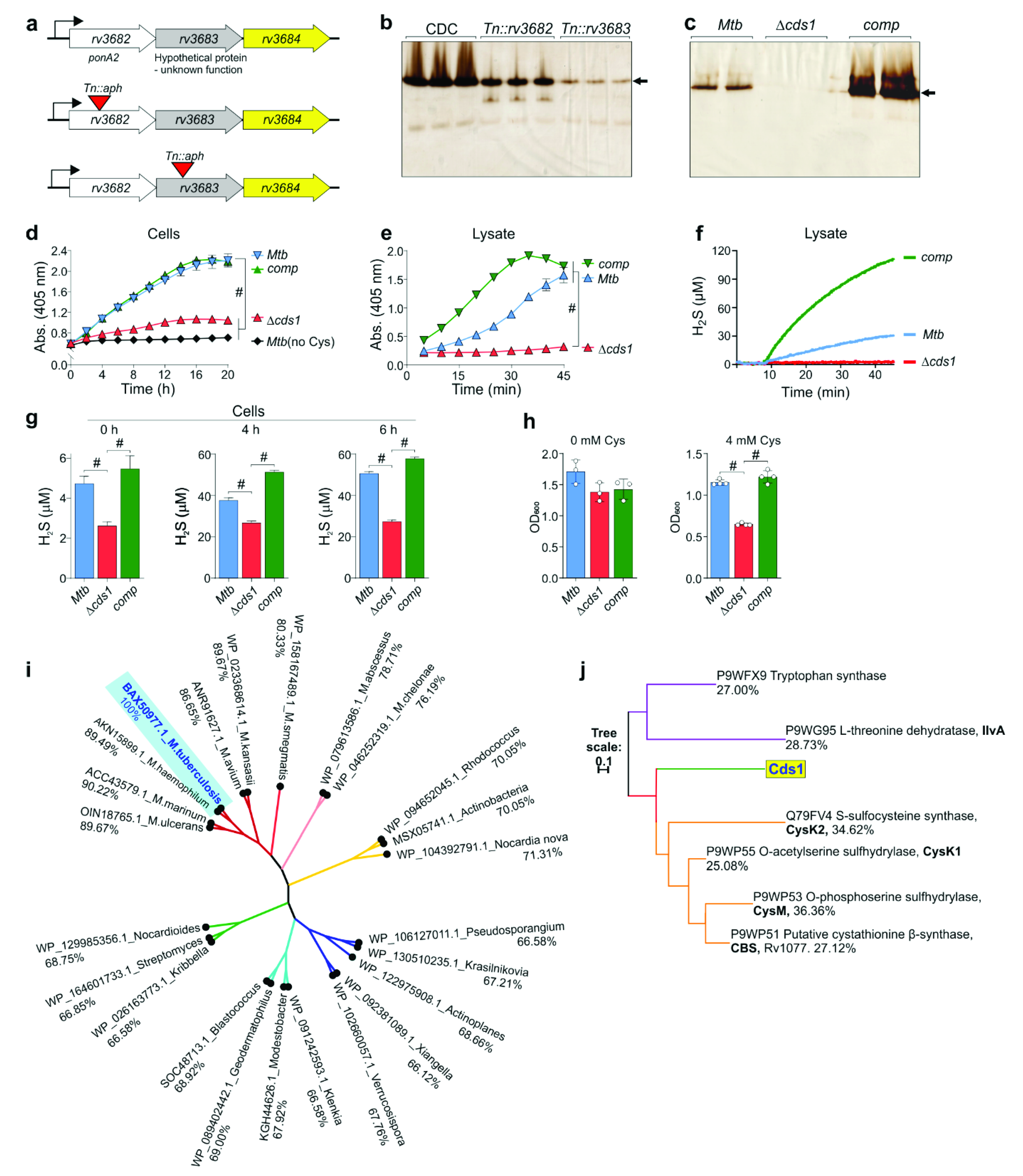
2.13. Construction of Δcds1 and Δrv1077 Mycobacterial Strains
The
rv1077 (
cbs) and
rv3684 (cds1) knockout
Mtb strains were generated by homologous recombination using specialized phage transduction according to Badarov, et al. (2002) [
35] (see
Supplementary Materials, Table S2). The
cds1 allelic exchange substrate (AES) phasmid was a kind gift from Michelle Larsson (Albert Einstein College of Medicine). The AES contained
cds1 disrupted by the hygromycin resistance gene. Briefly, the AES phasmid was amplified in
E. coli DH5α and purified using a DNA Plasmid Miniprep Kit (Thermo).
M. smegmatis was then transduced with the AES, and a high titer phage lysate was prepared.
Mtb H37Rv was grown to an OD
600 of ~0.8 and washed twice with buffer (50 mM Tris-HCl, pH 7.6, 150 mM NaCl, 10 mM MgCl
2, 2 mM CaCl
2), then mixed with the high titer phage lysate in a 1:1 (CFU:plaque forming units) ratio, and incubated at 37 °C overnight. After centrifugation (16,000×
g, 10 min, 4 °C) the pellet was resuspended in 0.2 mL 7H9 media and plated on hygromycin-containing 7H10 agar. After 3 weeks at 37 °C, 5 individual colonies were inoculated in 7H9 media supplemented with 50 µg/mL hygromycin. The genomic DNA of each colony was extracted and gene deletion confirmed using PCR with primers Rv3684CF, Rv3684CR (
rv3684) or Rv1077CF, Rv1077CR (
rv1077), and UUT (See
Supplementary Materials, Table S3).
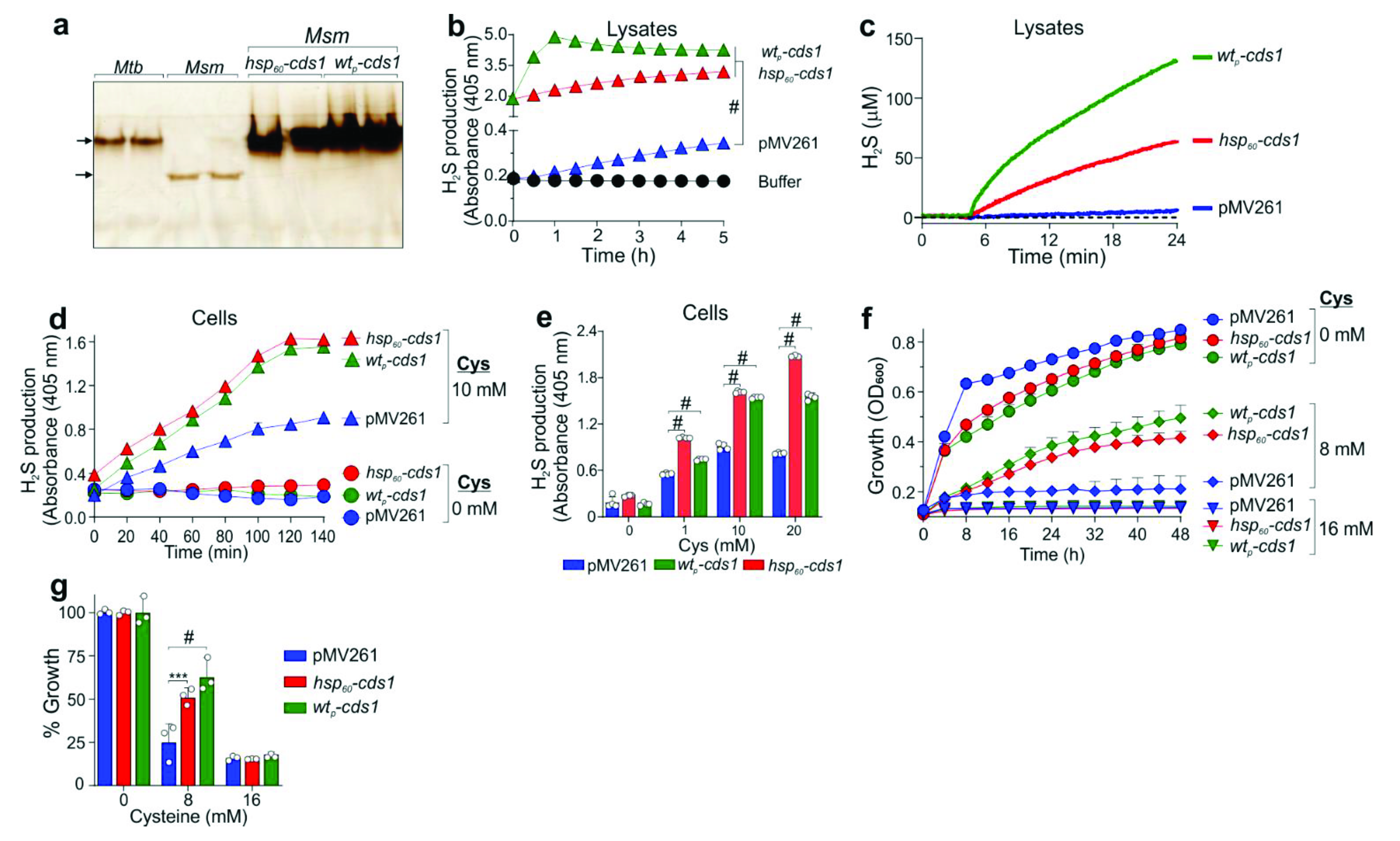
2.14. Mycobacterial Complementation
The
cds1 ORF was PCR amplified from genomic
Mtb DNA using KOD Xtreme HotStart DNA polymerase (Roche, New York, NY, USA) according to the manufacturer’s protocol and primers (Rv3684F and Rv3684R); see
Supplementary Materials Table S3. The PCR product and pMV261 vector were digested with
Bam HI and
Cla I (Thermo Fisher Scientific, USA), isolated using agarose purification, and ligated using T4 DNA ligase (NEB, USA) to produce pMV261::
hsp60-cds1 (see
Supplementary Materials, Table S2). A second complementation vector was constructed. The ORF of
rv3682,
rv3683 and
cds1, with an additional 500 bp upstream region, was PCR amplified using primers ponABCF and ponABCR (see
Supplementary Materials, Table S3). The amplicon was digested using
BamHI and
ClaI and ligated into the pMV261 vector. Complementation vectors expressing
cds1 under the control of either the
hsp60 or native promoter were electroporated (Gene Pulser Xcell, Bio-Rad, USA) into the
Mtb Δ
cds1 strain and transformants selected on 7H10 agar plates containing hygromycin (50 µg/mL) and kanamycin (25 µg/mL). These vectors were similarly electroporated into
M. smegmatis. Complemented strains were grown in 7H9 media containing 25 µg/mL kanamycin.
2.15. Purification of Recombinant Proteins
The
cds1 1041 bp open reading frame was PCR amplified using
Mtb genomic DNA and the primers Rv3684CEF and Rv3684CER (see
Supplementary Materials, Table S3). The PCR product was digested with
Nde I and
Bam HI and then ligated into the pET15b expression vector previously digested with
Nde I and
Bam HI. These restriction sites are in the pET15b MCS downstream of a 6-His coding region, resulting in the addition of a 6-His tag to the N-terminus of the encoded protein. The ligated construct (i.e., pET15b-
cds1) was then verified by sequencing.
E. coli BL21 (DE3) cells were transformed with pET15b-
cds1 and grown until the OD
600 reached 0.5–0.6. Protein expression was induced by the addition of 0.4 mM of IPTG followed by growth overnight at 18 °C. The cells were pelleted by centrifugation at 5000 rpm for 10 min, sonicated, and the lysate used for protein purification using nickel-affinity resin (Bio-Rad, USA) by gravity chromatography. pET28b-EhOASS expressing recombinant OASS containing an N-terminal 6×His tag was overexpressed and purified from
E. coli as described previously [
36].
2.16. Cds1 Enzyme Kinetics
The rate of H
2S production by purified recombinant Cds1 was monitored via formation of Bi
2S
3, which has an absorbance maximum at 405 nm [
32]. Briefly, purified Cds1 enzyme (1 µg of enzyme in 20 µL buffer) and 180 µL of BC solution were mixed in a well of a flat bottom, clear 96-well plate (Corning Inc., USA). Bi
2S
3 formation was monitored at room temperature over 30 min (readings taken once per minute) using a Biotek Synergy H1 hybrid plate reader. The absorbance at 405 nm was converted into product concentrations using the Beer–Lambert equation. The molar absorption coefficient for Bi
2S
3 was determined to be 3156.9 M
−1 cm
−1 using Na
2S.9H
2O (Sigma–Aldrich, USA) as a standard. Initial velocities were calculated and plotted against Cys concentrations. The K
m and V
max values were determined in GraphPad Prism (version 8.4.3) using the Michaelis–Menten equation. The k
cat values were calculated by dividing V
max by the nanomoles of enzyme used in the reaction.
2.17. Identification of Cds1 Enzymatic Products by LC-MS/MS
Purified Cds1 was added to three separate reaction tubes containing 50 µL of 20 mM L-Cys in PBS (20 mM Na2HPO4, 100 mM NaCl, pH 7.5, and 20 µM PLP), and the reaction was allowed to proceed for 15 min at room temperature. The reactions were stopped by heating to 80 °C for 5 min and the tubes centrifuged at 15,000 rpm for 5 min. Ten microliter aliquots of each reaction solution were taken from the supernatant and analyzed by LC-MS/MS. We then identified the enzymatic products of Cds1 using ultra-high liquid chromatography coupled to high-resolution/high-accuracy mass spectrometry based on the analyte’s exact mass and HPLC retention time compared to authentic metabolite reference standards. A reference standard mixture of important negatively charged metabolites, including pyruvate and the study samples, were analyzed on the Q-Exactive LCMS system, as described elsewhere, using high-resolution molecular ion scans. The RAW files were subjected to post-run analysis using Skyline software along with a template constructed to monitor the relevant analytes. The exact theoretical mass for 12C pyruvate standard is 87.008768 (87.0088) Da. The standard was present in the molecular ion scans with the same mass in the standard mixture when subjected to analysis. The retention time for the pyruvate in the standard mix on the Aminex column (Bio-Rad, USA) used was 11.0 min. In the samples, a peak was observed with the same retention time and exact mass as the pyruvate standard, and this was considered sufficient evidence for verification of identity. Each sample was assayed in triplicate.
2.18. In Vitro CFU Assay
Thioglycollate-elicited peritoneal macrophages were isolated from C57BL/6 mice [
23] and were plated at 1.0 × 10
6 cells per well in 6-well plates. Cells were infected with
Mtb strains at an MOI of 0.2 and incubated at 37 °C for 2 h, followed by washing cells twice to remove non-internalized bacilli (day 0 post infection). At 0, 1, 2, and 4 days, cells were lysed with DPBS containing 0.05% SDS, and CFU were determined by plating serial dilutions of lysates on 7H11 agar plates supplemented with 10% OADC. Plates were incubated at 37 °C with 5% CO
2 for 4 weeks to determine CFU counts.
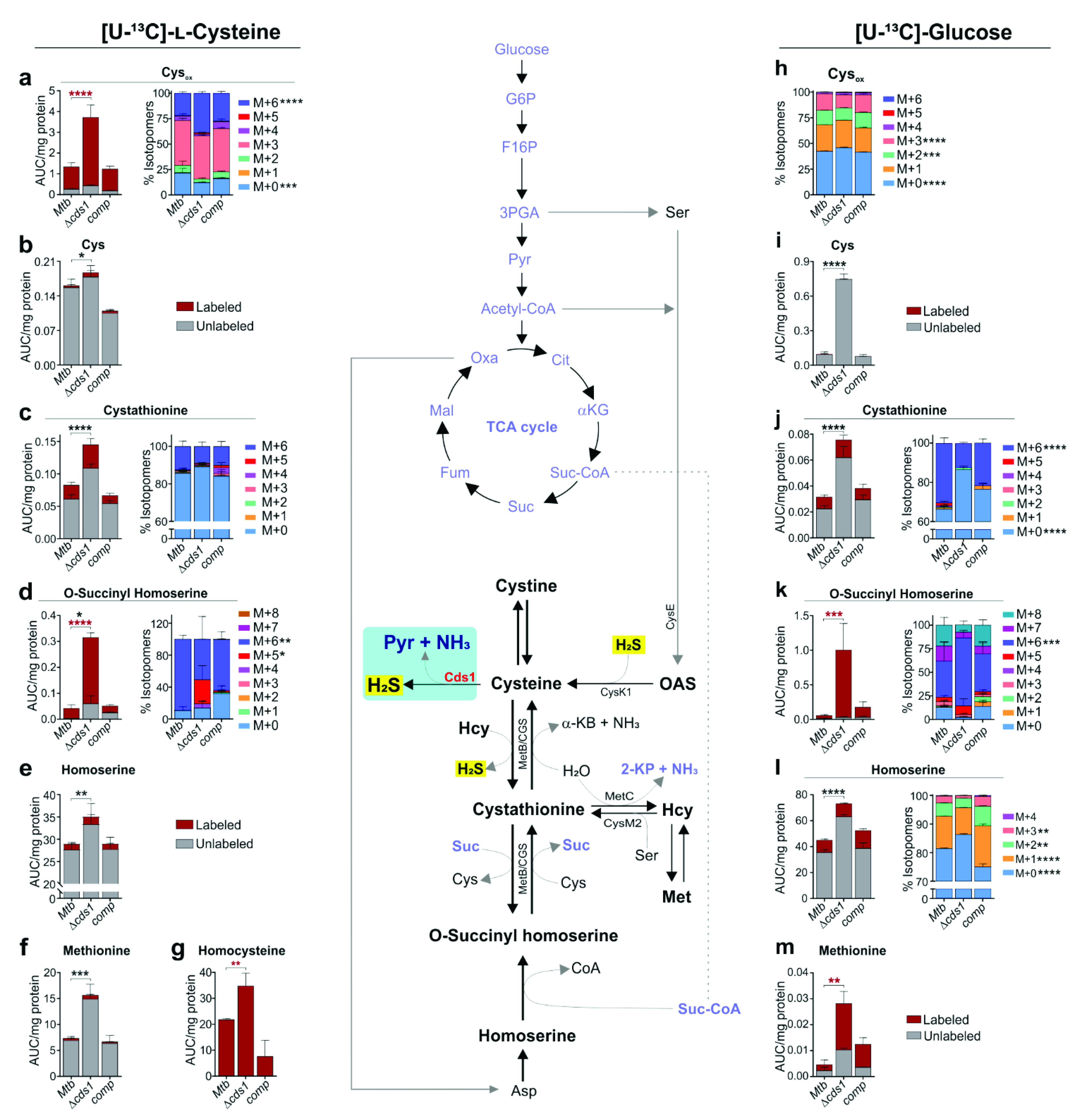
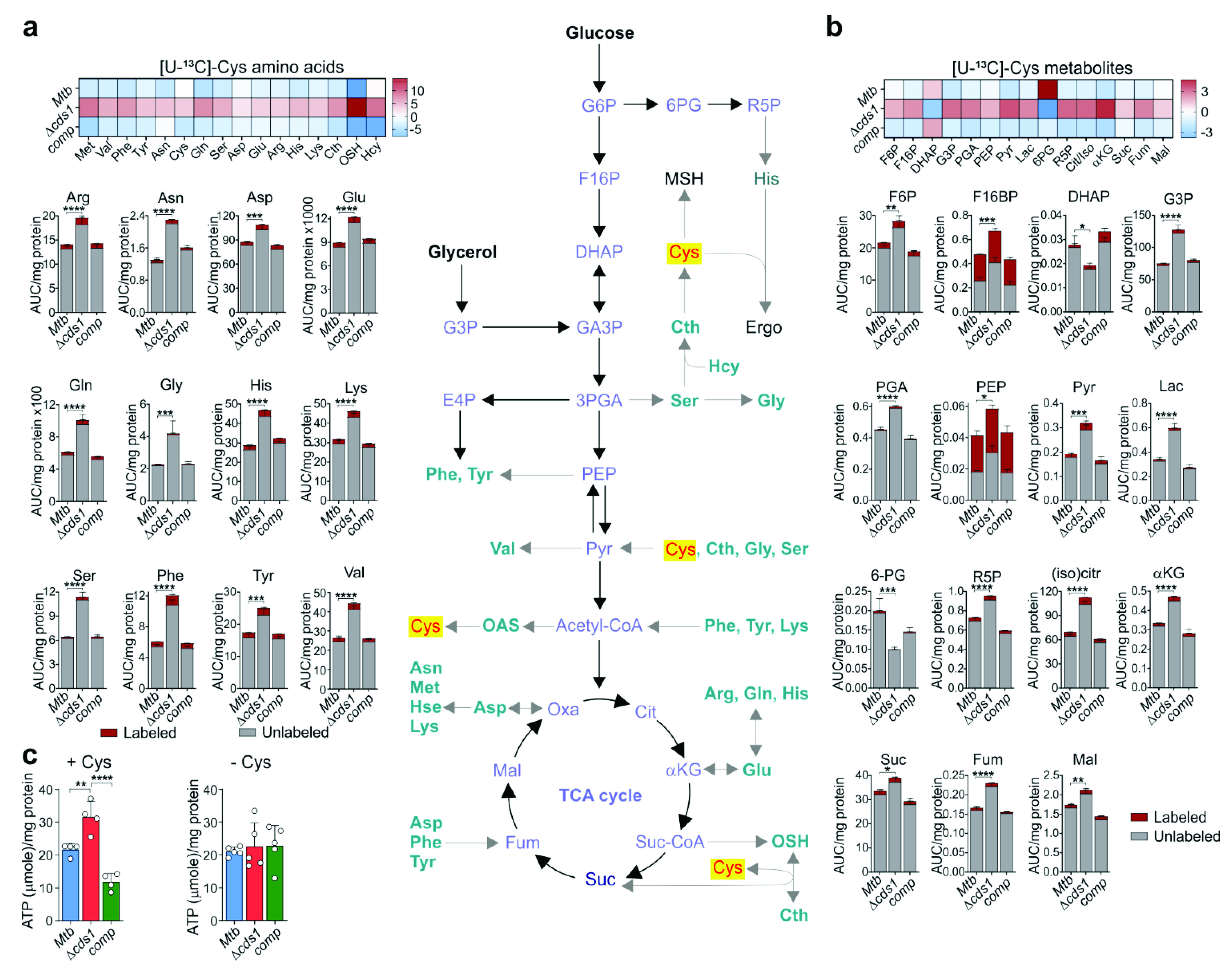
2.19. LC-MS/MS Targeted Metabolomics Analyses
Mtb strains in replicate were inoculated from freshly grown culture in 7H9 media (containing 0.01% tyloxapol, 0.2% glycerol, and 10% OADC) with 4 mM cysteine at an OD600 of approximately 0.05. These strains were cultured (~30 mL each) in inkwell bottles to an OD600 of ~0.8 at 37 °C with shaking (150 rpm). Each culture was pelleted by centrifuging at 3500× g for 10 min. The culture supernatant was discarded, and cells were washed three times with 7H9 containing 0.01% tyloxapol. After the last wash, the pelleted cells were resuspended in 5 mL of 7H9 media containing 0.01% tyloxapol, 0.2% glycerol, 10% OAS (oleic acid, bovine albumin, and NaCl), 0.2% [U-13C]-Glucose (Sigma–Aldrich, USA) or 0.01% tyloxapol, 0.2% glycerol, 10% OADS (oleic acid, bovine albumin, dextrose, and NaCl), 100 µM [U-13C]-cysteine (Cambridge Isotope Laboratories Inc., USA), or 7H9 media containing 0.01% tyloxapol, 0.2% glycerol, 10% OAD, and 0.25 mM CHP. After resuspension of the pellet, the cultures were then incubated overnight at 37 °C with shaking (150 rpm) and harvested by centrifugation at 3500× g for 3 min. Cells pellets were immediately snap-frozen on dry-ice for approximately 5 min and then thawed and prepared for lysis using a MagNA Lyser (Roche, USA) at 7000 rpm for 1 min, followed by cooling on ice for 4 min, repeated 3–4 times. The lysis was performed using a 1.8 mL solution of methanol, acetonitrile, and water in the ratio of 2:2:1). The lysate was then centrifuged at 15,000–17,000× g for 10 min. The supernatant was collected and filtered through a 0.22 µm filter. The recovered lysate was then vacuum concentrated to dryness (Eppendorf Concentrator Plus, Eppendorf, Germany) at 30 °C for 12 h. The dried lysate pellets were then resuspended in 200 µL of purified water. 100 µL of this suspension for each replicate was submitted for LC-MS/MS targeted organic acid (metabolites) analysis and 100 µL (50 µL resuspension plus 50 µL acetonitrile) was submitted for LC-MS/MS targeted amino acid analysis.
LC-MS/MS was used for relative quantification of each organic acid and amino acid. The LC-MS/MS sample analysis was performed using a Thermo Scientific DionexTM UltiMateTM 3000 UHPLC system coupled to a Thermo Scientific Q-Exactive Mass Spectrometer with a HESI source. A sample volume of 1 µL was injected onto a Waters Xbridge® BEH HILIC HPLC column (2.5 µm particles, 2.1 × 100 mm), column oven set at 40 °C and chromatographic separation was performed using gradient elution at a flow rate of 200 µL/min and total run time of 26 min. Mobile phase A contained water with 0.1% formic acid and mobile phase B contained acetonitrile with 0.1% formic acid. Data were acquired using full-scan MS (without HCD fragmentation) in positive mode over the m/z range 50–750 Da at a 70,000 resolution. A QC sample was prepared using 21 different amino acids at a concentration of 500 ng/mL to monitor amino acid retention time consistency and MS sensitivity. Each sample was spiked with deuterated alanine (D4-Alanine, Sigma–Aldrich, USA) as an internal standard to monitor processing efficiency and data normalization. The data were processed, and peak areas were calculated using Skyline 3.7 (MacCoss Lab, University of Washington, Seattle, WA, USA).
2.20. Total ATP Quantitation
ATP quantitation was performed on cell lysates prepared for use in our metabolomics analysis (see
Section 2.19). ATP levels of cell lysates were measured using a Roche ATP Bioluminescence kit CLS II according to the manufacturer’s instructions. ATP levels were normalized to the protein concentration of each cell lysate.
2.21. Statistics
Unless specified otherwise in
Section 2, all experiments were performed on 3–8 biological replicates, and the data were expressed as the mean ± SD or the mean ± SEM. Statistical significance of the data was determined using GraphPad Prism 8.4.3 (GraphPad Software, Inc., USA). Specific statistical tests are noted in the figure legends and include the Student’s unpaired
t-test (two-tailed) and one-way or two-way ANOVA.
4. Discussion
This study elucidated several previously unrecognized physiological features of
Mtb. Using three different methods, we showed that laboratory and MDR and DS clinical
Mtb strains produced H
2S as do non-pathogenic slow- and fast-growing mycobacterial strains. We then identified the genetic locus
cds1 (
rv3684) and confirmed that the gene product, Cds1, is a PLP-dependent H
2S-producing enzyme. Importantly, we demonstrated that endogenous H
2S can influence
Mtb bioenergetics by enhancing respiration, primarily via CytBD, and by modulating the balance between respiration (OXPHOS) and glycolysis. Further, we described a plausible mechanism by which
Mtb mitigates oxidative stress by converting excess Cys into H
2S, which is released and then recycled. Our findings point to a paradigm whereby
Mtb-generated H
2S, together with host-generated H
2S, exacerbates TB disease by dysregulating host immunity [
23]. Lastly, since H
2S production by
Mtb has been an overlooked confounder in routine culturing of
Mtb, we anticipate our findings to have a broad practical impact in the TB field.
Since the discovery of
Mtb by Robert Koch in 1882, formal proof that
Mtb produces H
2S has been lacking. One likely reason is that since
Mtb is highly contagious and spread through aerosols, smelling of cultures is a significant health risk and extreme measures are taken to avoid inhalation. Nonetheless, several biochemical studies [
26,
27,
28,
29,
30] as well as homologues of H
2S-producing enzymes in the
Mtb genome have provided strong circumstantial evidence that
Mtb is a likely producer of H
2S. Furthermore, complicating factors are that H
2S is a difficult molecule to study because measuring it is complex [
6] and H
2S can be spontaneously generated from media components [
8]. Hence, we employed multiple approaches to provide compelling evidence that
Mtb produces H
2S.
Our findings that two clinical MDR
Mtb strains produced the highest levels of H
2S, followed by laboratory strains and clinical DS strains, have considerable clinical importance for several reasons. Firstly, many clinical MDR and XDR
Mtb strains are notoriously difficult to culture due to extremely slow growth. Since high concentrations of H
2S inhibit respiration [
20,
58], it is not unreasonable to propose that excessive H
2S production contributes to the slow growth of many of these strains, even when cultured in the absence of Cys. Secondly, our findings that endogenous levels of H
2S suppress central metabolism suggest that excessively high levels of H
2S produced by MDR
Mtb strains could reprogram metabolism by shifting the balance between OXPHOS and glycolysis, ultimately leading to an energetically impaired state that inhibits growth. Thirdly, the large variation in H
2S production among clinical strains likely reflects functional differences that are due to the fact of SNPs or genomic rearrangements that contribute to strain-specific transcriptional regulation. Lastly, since H
2S is widely used as a diagnostic test for bacteria, there is potential for the development of H
2S-based diagnostics for
Mtb, e.g., detection of H
2S in the exhaled breath or sputum of TB patients.
How do these findings contribute to a more accurate understanding of
Mtb physiology and pathogenesis? In two recent studies, it was shown that host-derived H
2S exacerbates
Mtb disease in the mouse model of TB [
22,
23] and that CSE and 3MST protein levels are markedly increased in human TB lesions that surround necrotic granulomas and cavities [
23]. Hence, it was proposed that because of the strong immunomodulatory activity of H
2S, excessive host H
2S production triggered by
Mtb infection dysregulates immunity to promote disease [
23]. Considering the findings in this study, we posit that
Mtb-generated H
2S may act as a signaling molecule in the host, further contributing to excessive H
2S levels in vivo to exacerbate disease. This is possible since Cys concentrations range from ~128–250 μM in human cells and plasma [
59,
60]. Although a role for
Mtb cds1 in TB disease has not yet been reported, our genetic knockout (
Figure 3d) and other data (
Supplementary Materials, Table S4) indicate that more than one enzyme contributes to H
2S production in
Mtb. Therefore, multiple gene knockouts may be necessary to convincingly demonstrate a role for endogenously-produced H
2S in TB disease.
Discovery of a gene responsible for H2S production in Mtb cells establishes a paradigm for how H2S modulates Mtb physiology. While homologues of CBS and CSE are encoded in the Mtb genome, these proteins were not identified in our non-denaturing in-gel BC assays. However, this could be explained by the fact that the conditions of our in-gel BC assay were suboptimal for certain enzymes, and that alterations in environmental factors (substrate, pH, temperature, oxygen, etc.) or redox-dependent cofactors (e.g., NADH, NADPH, and heme) would allow for the detection of additional enzymes.
Based on our cell-based assays, Cds1 contributes significantly to H
2S production and showed increased activity when cells were exposed to Cys. Since the total sulfur atom concentration in bacteria (e.g.,
E. coli) is ~130 mM and intracellular Cys serves as the primary supplier of sulfur atoms [
47], it is possible that Cds1 shuttles sulfur atoms among metabolites to meet cellular demand and to maintain redox balance. Indeed, Cds1 protects
Mtb and
Msm against toxic concentrations of Cys, providing insight into the mechanisms, whereby Cds1 mitigates intracellular oxidative stress triggered by Cys, which feeds the Fenton reaction to generate free radicals [
9]. Therefore, our data (
Figure 3f,
Figure 6 and
Figure 7) support the concept that the Cds1-mediated conversion of Cys into H
2S functions as a sink for excess Cys. This concept is further supported by studies showing that high levels of Cys inhibit growth [
61], induce ROI production, and lead to DNA damage in
E. coli [
9] and
Mtb [
49].
Like other small nonelectrolytes, such as •NO, CO, and O
2, at a gas/liquid interface H
2S in solution (as a dissolved solute) readily “escapes” solvation and volatilizes. Likewise, as a gas at this interface, it will readily dissolve and, so, with a confined headspace, a dynamic equilibrium of volatilization/dissolution will exist, defined quantitatively by Henry’s Law. As pointed out elsewhere [
18], the ability of H
2S to exist as a gas has functional biological relevance only in the presence of such a gas/liquid interface, which is also true for •NO, CO, and O
2 [
62]. As demonstrated previously [
63], the contribution of volatilization in vitro will be determined by the specific experimental configuration. In the lung, where such an interface is central to organ function, the “headspace” is not confined. Therefore, according to Le Chatelier’s principle, the process of ventilation will serve to pull this equilibrium in the direction of volatilization and, therefore, may well serve to amplify the importance of desulfhydration as a sink for H
2S (and its conjugate bases HS
− and S
2−) in vivo. The relative importance of volatilization as a sink will depend on its magnitude compared to other competing mechanisms of consumption in the organism and tissue [
64].
Our finding that Cds1 generates H
2S and pyruvate using Cys as a substrate suggests that Cds1 is catalytically similar to mammalian and bacterial CSE, which converts Cys into H
2S, pyruvate, and ammonia [
19]. Elegant biochemical studies have shown that
Mtb Rv1079 possesses both CSE and cystathionine γ-synthase (CGS) activity [
27]. However, Rv1079 lacks Cys desulfhydrase activity, and these authors speculated that an as yet uncharacterized enzyme performed this important role [
27]. We provide genetic and biochemical evidence that Cds1 fulfils this function through its Cys desulfhydrase activity, which adds to our understanding of how
Mtb detoxifies Cys.
An unusual feature of H
2S is its ability to stimulate bioenergetics at low concentrations and inhibit respiration at higher concentrations. In a recent study, we demonstrated that low concentrations of exogenous H
2S stimulate
Mtb respiration and growth [
22]. In mammals, H
2S has been implicated in reversibly inhibiting cytochrome c oxidase (Complex IV) at high concentrations, and, conversely, stimulating mitochondrial respiration at low concentrations [
20,
21,
65,
66]. Our finding that Cds1-generated H
2S is an effector molecule that modulates basal respiration reveals a previously unknown facet of
Mtb physiology. This is supported by genetic and bioenergetic data showing a ~40% reduction in the basal respiration of Δ
cds1 cells compared to WT
Mtb.
By exploiting a
Mtb cytBD mutant and pharmacological inhibition of cytochrome
bc1/aa3, our findings provide insight into how H
2S can stimulate respiration. Contrary to studies in
E. coli that show cytochrome
bo3 oxidase is susceptible to H
2S and its two
bd oxidases are resistant to H
2S [
36,
47], our current data suggest that Cys-generated H
2S stimulates respiration via both
Mtb cytochrome
bc1/aa3 and CytBD, consistent with our previous findings [
22]. For example, in the presence of Cys, respiration is dramatically increased when cytochrome
bc1/aa3 oxidase is inhibited with Q203 compared to respiration in the
Mtb cytBD mutant. Hence, CytBD plays a more prominent role than cytochrome
bc1/aa3 in H
2S-mediated stimulation. Determining whether H
2S directly binds to one or both oxidases in
Mtb to stimulate respiration will require further investigation.
Mtb and
E. coli respond differently to H
2S, likely because they occupy vastly different niches.
Mtb is an obligate aerobe that colonizes the human lung, whereas
E. coli is a facultative anaerobe and ubiquitous member of the human gut microbiota that is exposed to millimolar concentrations of H
2S. More importantly, unlike other bacteria,
Mtb has the unusual ability to rapidly reroute ETC flux to either cytochrome
bc1/aa3 or CytBD [
34] depending on which oxidase is inhibited. This remarkable plasticity allows
Mtb to effectively respond to host gases, such as NO and CO, which inhibit respiration, and H
2S, which can stimulate respiration.
Respiration is directly linked to central metabolism, and not surprisingly, Cds1-generated H
2S suppresses glycolysis in WT
Mtb as was evident by increased levels of numerous glycolytic metabolites in Δ
cds1 cells. We propose two plausible mechanisms whereby H
2S could modulate the balance between OXPHOS and glycolysis (
Figure 8j): Firstly, reduced respiration (OXPHOS) in Δ
cds1 cells triggers a compensatory glycolytic response, i.e., substrate level of phosphorylation to meet the bioenergetic demands for ATP. This is not an unusual metabolic response, as it was previously posited that bedaquiline-mediated inhibition of OXPHOS leads to a compensatory induction of glycolysis to meet the demand for ATP through substrate-level phosphorylation [
34,
67]. In addition, recent studies on
Bacillus and
Staphylococcus spp. showed that glycolysis can reverse polymyxin B-mediated ATP depletion that resulted from dysregulation of OXPHOS [
68,
69]. Secondly, H
2S can directly target enzymes in the glycolytic pathway through persulfidation to modulate their activity [
53,
54]. This is supported by recent studies showing that H
2S suppresses glycolysis in macrophages upon
Mtb infection [
23].
Respiration and central metabolism are also linked to redox balance (via MSH and EGT) and ROI production in bacteria [
70], and we demonstrated that Cds1-generated H
2S plays a role in maintaining
Mtb redox homeostasis. Our data show that catalase in growth media influences ROI production and that H
2S can function as an oxidant or reductant depending on the environmental conditions. In this regard, careful consideration should be given to experimental design and subsequent conclusions, since exogenous catalase in 7H9 medium is widely known to influence INH drug susceptibility and is routinely excluded from medium in INH susceptibility studies. It has been suggested that 3MST-derived H
2S protects
E. coli against oxidative stress via H
2S-mediated sequestration of Fe
2+ [
14]. In
Mtb, exogenous H
2S upregulates key members of the copper regulon, suggesting that copper, which has a high affinity for H
2S, may contribute to increased ROI when exposed to excess H
2S [
22].
The unusual effect of H
2S on
Mtb respiration and oxidative stress, as demonstrated in this study as well as previous studies on how H
2S impacts antibiotic resistance [
15], guided us toward considering a role for H
2S in the cellular response to anti-TB drugs, such as CFZ, which kills
Mtb via ROI production [
71]. Indeed, our data demonstrate that H
2S exacerbates oxidative stress in the presence of CFZ. Not surprisingly, Δ
cds1 cells are more resistant to CFZ, consistent with our data showing that addition of exogenous H
2S to CFZ-treated cells increases
Mtb killing. On this basis, we propose that endogenous H
2S stimulates respiration leading to increased ROI production that synergizes with ROI produced by CFZ, ultimately increasing killing. Whereas previous studies have shown that addition of Cys to
Mtb cells increases respiration and killing of
Mtb persisters treated with INH and RIF [
49], our study suggests that H
2S could be the effector molecule in that model. It should be recognized that exogenous Cys also generates H
2O
2 to trigger the Fenton reaction, which leads to continuous
•OH formation that damages DNA which may exacerbate the effect of INH, particularly in closed vessels where H
2O
2 accumulates [
50]. Exogenous H
2S in combination with RIF also increased
Mtb killing, likely because RIF also generates oxidative stress [
72], but this effect was less pronounced. These findings may have important implications for TB therapy, as they suggest that sulfur sources in vivo could influence anti-TB drug efficacy. Similarly, recent studies have shown that exogenous H
2S with RIF (and other antibiotics) confer hypersensitivity to
Acinetobactor baumannii [
73].


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}







