
Interventricular Differences of Signaling Pathways-Mediated Regulation of Cardiomyocyte Function in Response to High Oxidative Stress in the Post-Ischemic Failing Rat Heart

 ,
,  , ,
, ,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Animal Model
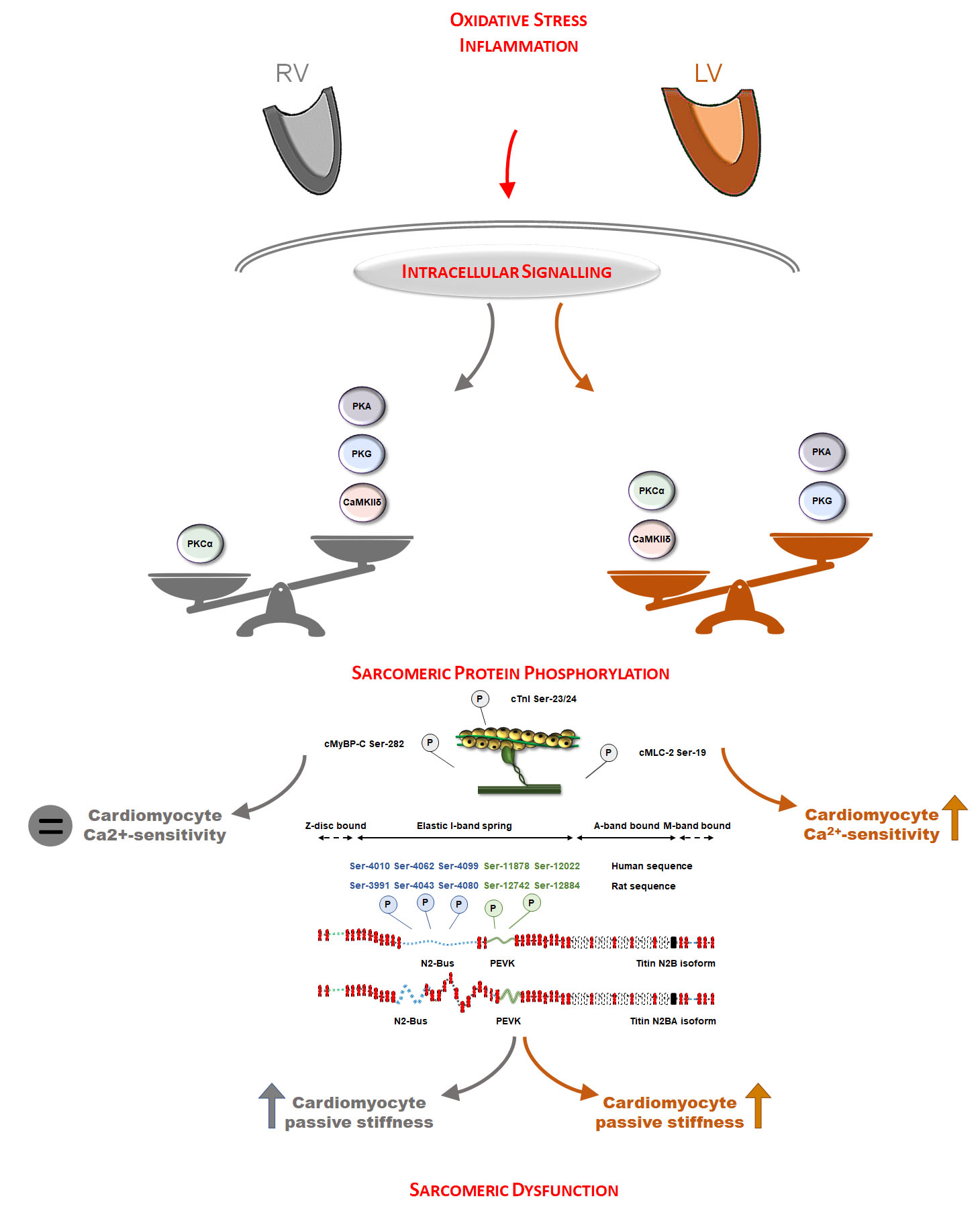
2.2. Quantification of Tissue Oxidative Stress and Inflammation
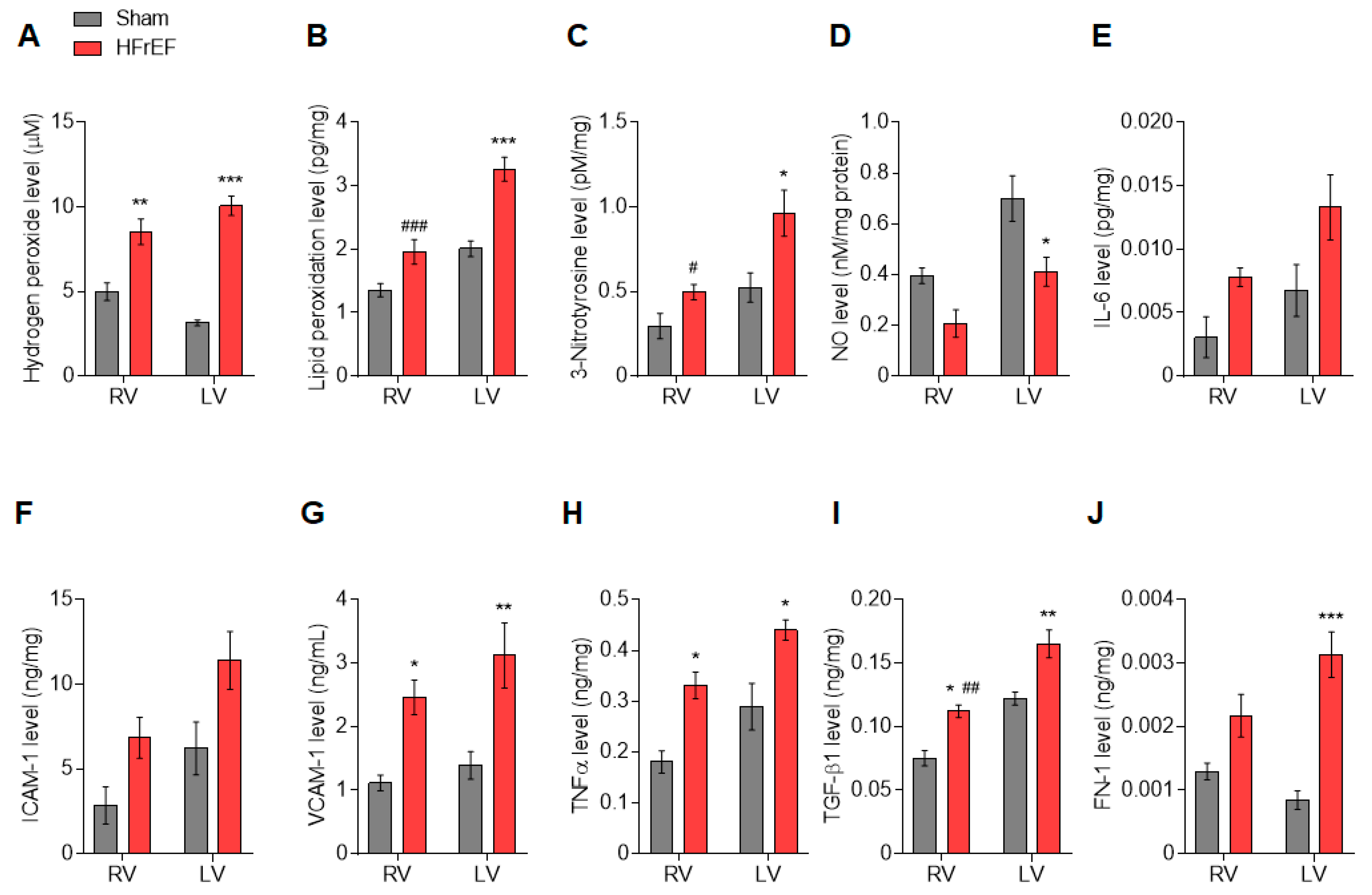
2.3. Force Measurements in Isolated Cardiomyocytes
2.4. Myocardial CaMKII Activity
2.5. Myocardial PKG Activity
2.6. Myocardial PKA and PKC Activities
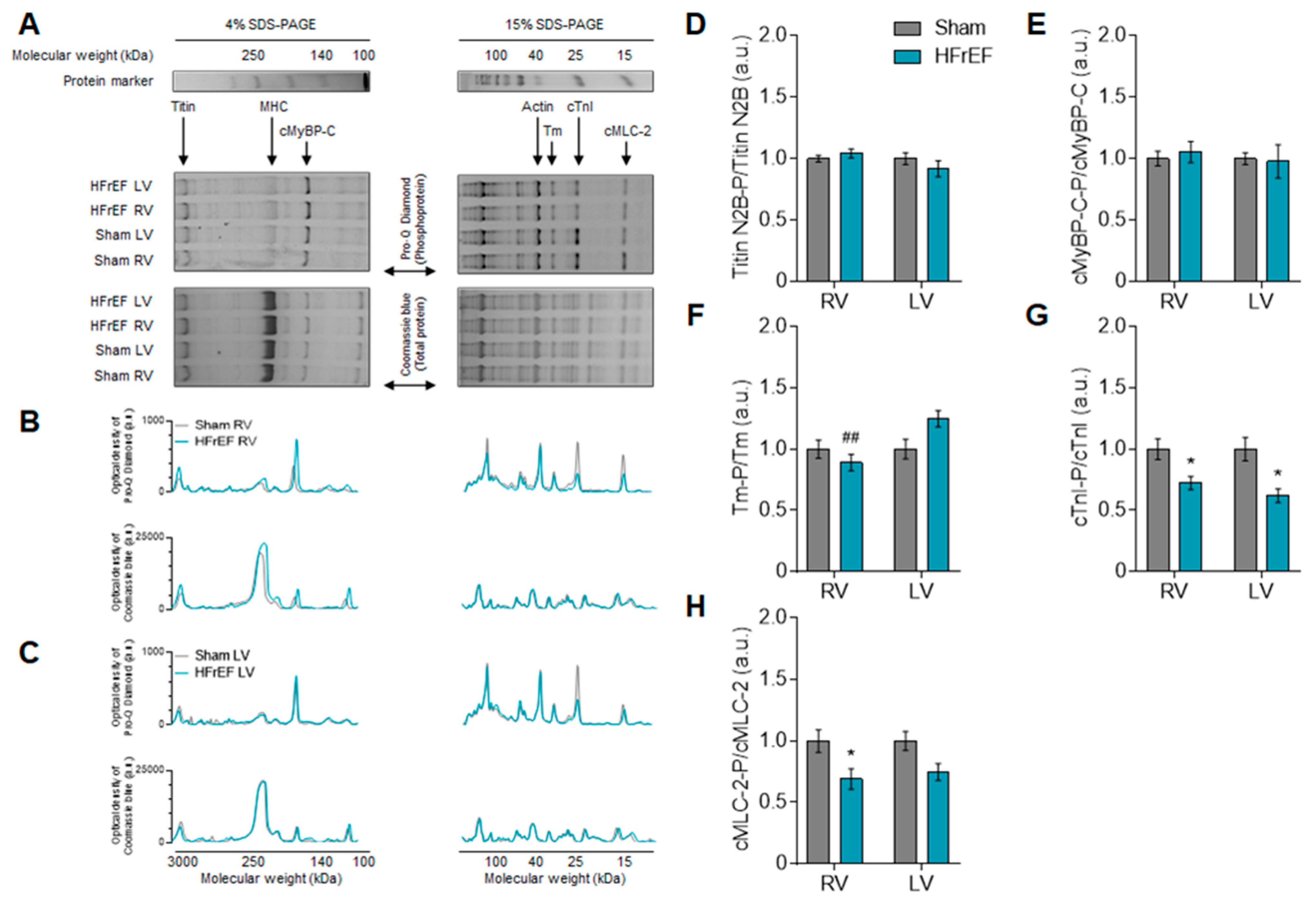
2.7. Determination of Myofilament Protein Phosphorylation
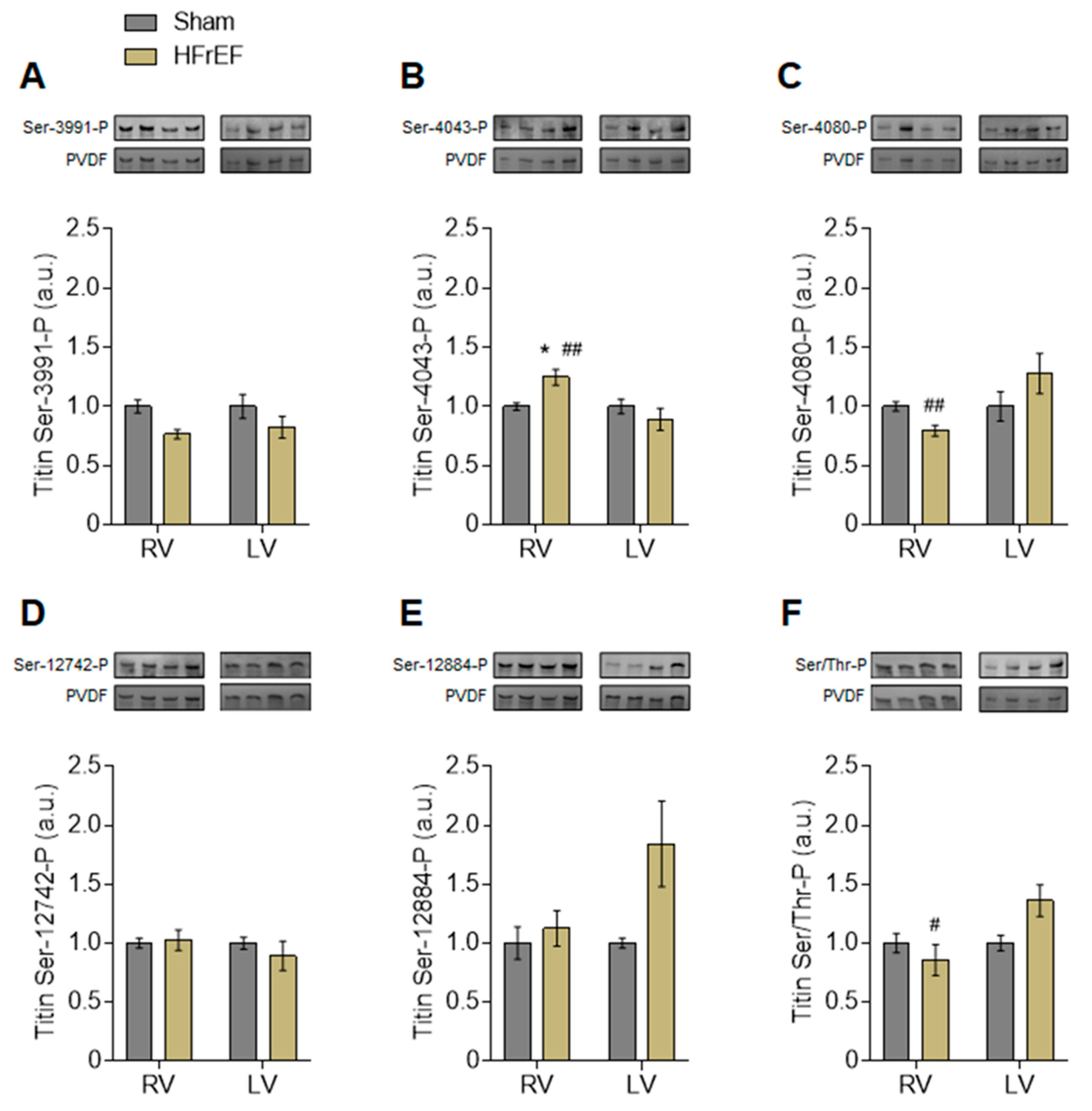
2.8. Phospho-Titin Analysis by Western Immunoblot
- Anti-phospho-N2Bus (Ser-3991) against EEGKS (PO3H2) LSFPLA (dilution 1:500);
- Anti-phospho-N2Bus (Ser-4043) against QELLS (PO3H2) KETLFP (dilution 1:100);
- Anti-phospho-N2Bus (Ser-4080) against LFS (PO3H2) EWLRNI (dilution 1:500);
- Anti-phospho-PEVK (Ser-12742) against EVVLKS (PO3H2) VLRK (dilution 1:100);
- Anti-phospho-PEVK (Ser-12884) against KLRPGS (PO3H2) GGEKPP (dilution 1:500).
2.9. Site-Specific Phosphorylation of Myofilament Proteins
2.10. Collagen Gene Expression Analysis
2.11. Data and Statistical Analysis
3. Results
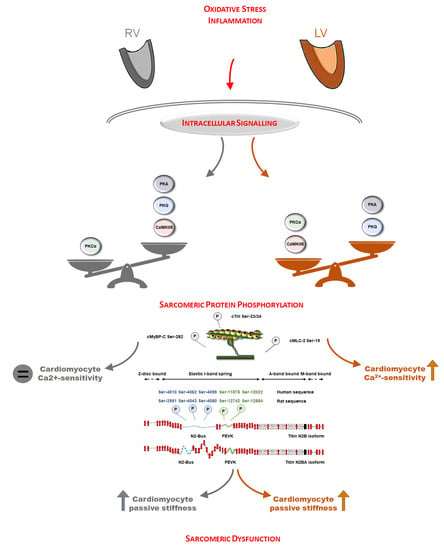
3.1. Oxidative Stress and Inflammation Show Interventricular Differences in the Post-Ischemic Rat Heart
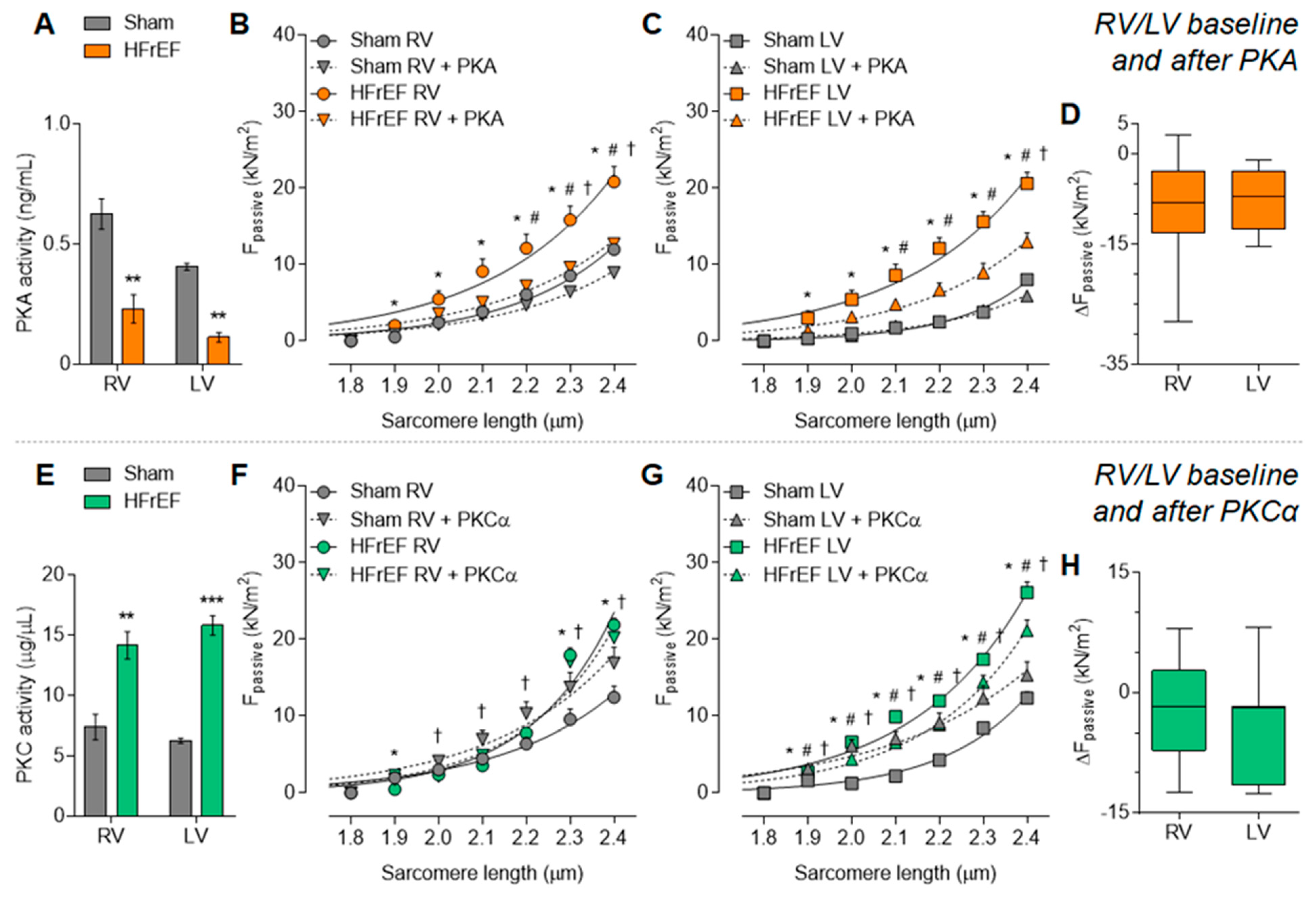
3.2. Protein Kinase-Mediated Cardiomyocyte Passive Stiffness Show Interventricular Differences in HFrEF Rats
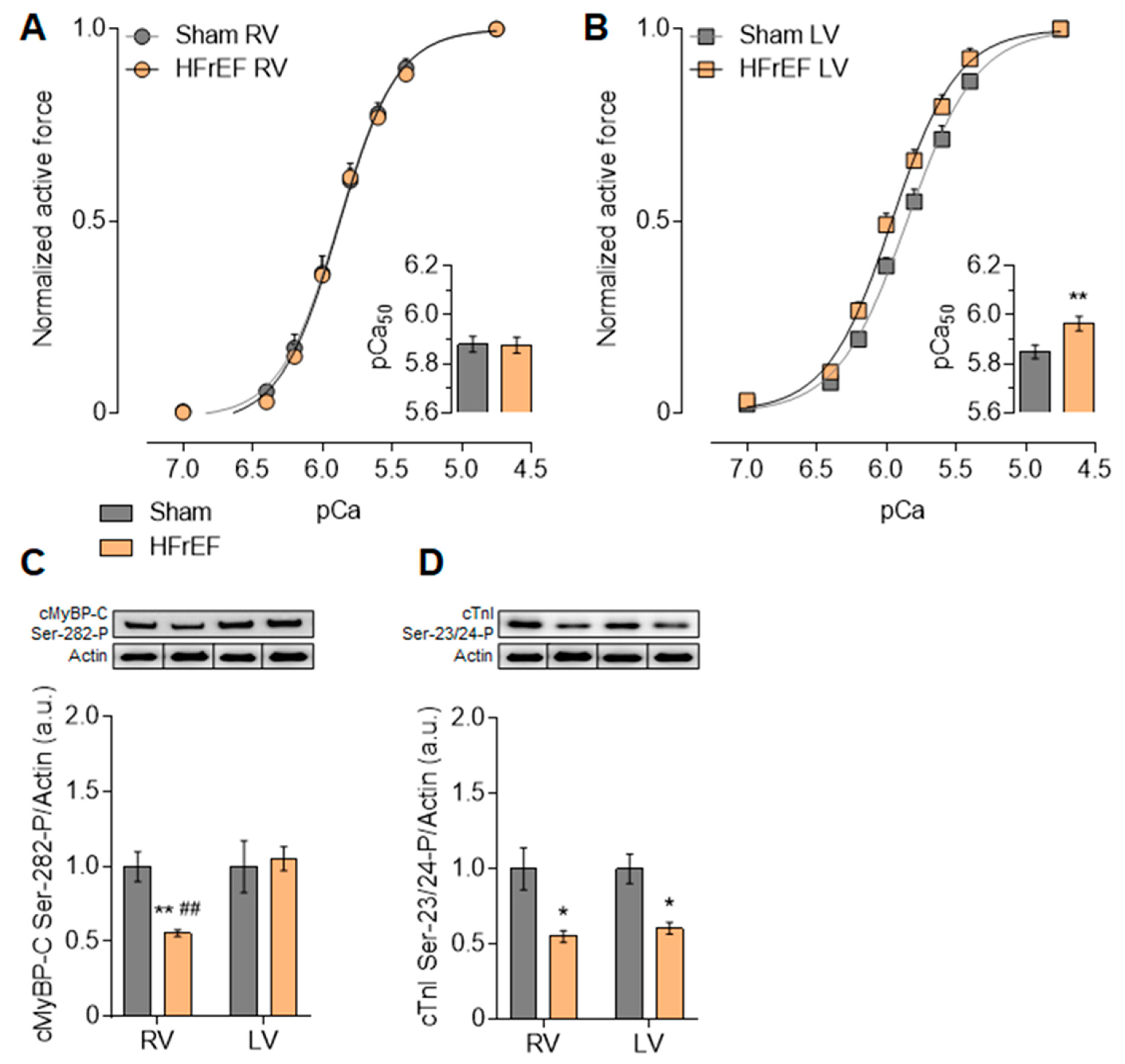
3.3. Myofilament Ca2+-Sensitivity and Protein Phosphorylation Show Interventricular Differences in the Failing Rat Heart
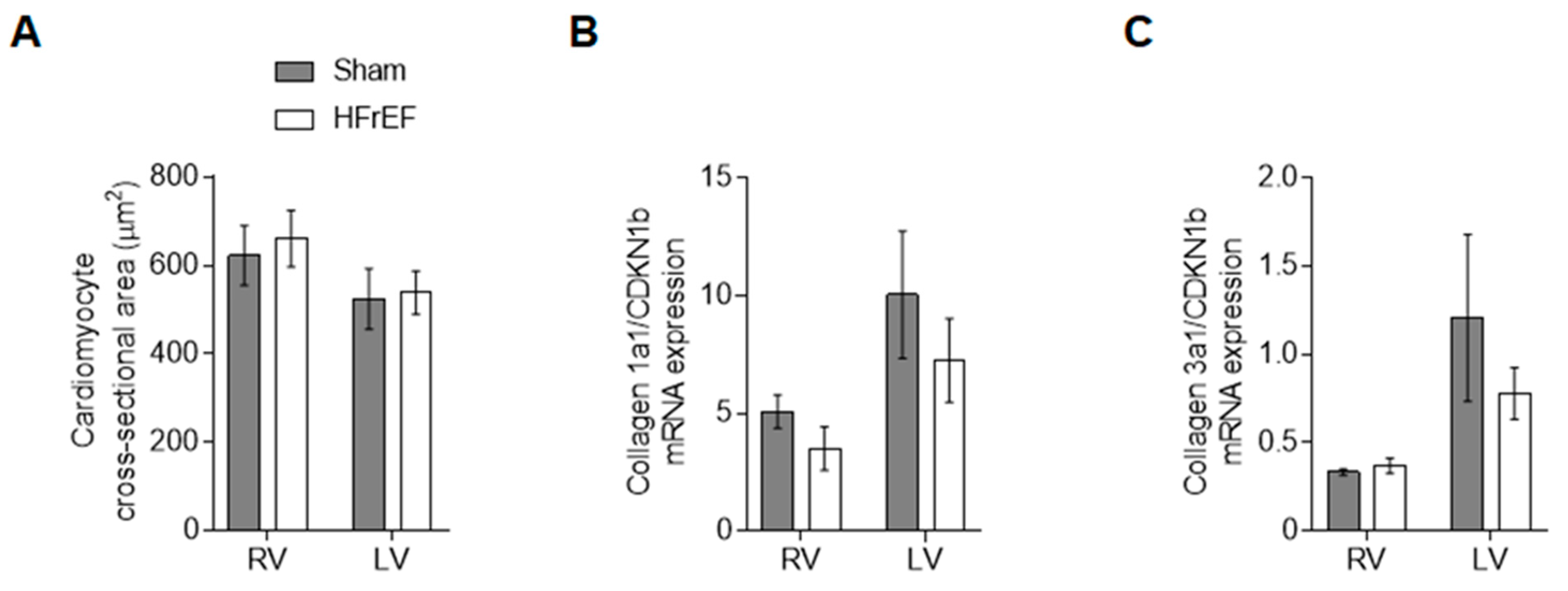
3.4. RV and LV Structure Show No Differences in Post-Ischemic HFrEF Rats
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ghio, S.; Gavazzi, A.; Campana, C.; Inserra, C.; Klersy, C.; Sebastiani, R.; Arbustini, E.; Recusani, F.; Tavazzi, L. Independent and additive prognostic value of right ventricular systolic function and pulmonary artery pressure in patients with chronic heart failure. J. Am. Coll. Cardiol. 2001, 37, 183–188. [Google Scholar] [CrossRef] [Green Version]
- Bosch, L.; Lam, C.S.P.; Gong, L.; Chan, S.P.; Sim, D.; Yeo, D.; Jaufeerally, F.; Leong, K.T.G.; Ong, H.Y.; Ng, T.P.; et al. Right ventricular dysfunction in left-sided heart failure with preserved versus reduced ejection fraction. Eur. J. Heart Fail 2017, 19, 1664–1671. [Google Scholar] [CrossRef] [Green Version]
- Konstam, M.A.; Kiernan, M.S.; Bernstein, D.; Bozkurt, B.; Jacob, M.; Kapur, N.K.; Kociol, R.D.; Lewis, E.F.; Mehra, M.R.; Pagani, F.D.; et al. Evaluation and management of right-sided heart failure: A scientific statement from the American Heart Association. Circulation 2018, 137, e578–e622. [Google Scholar] [CrossRef] [PubMed]
- Zaffran, S.; Kelly, R.G.; Meilhac, S.M.; Buckingham, M.E.; Brown, N.A. Right ventricular myocardium derives from the anterior heart field. Circ. Res. 2004, 95, 261–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellofiore, A.; Chesler, N.C. Methods for measuring right ventricular function and hemodynamic coupling with the pulmonary vasculature. Ann. Biomed. Eng. 2013, 41, 1384–1398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Littlejohns, B.; Heesom, K.; Angelini, G.D.; Suleiman, M.S. The effect of disease on human cardiac protein expression profiles in paired samples from right and left ventricles. Clin. Proteom. 2014, 11, 34. [Google Scholar] [CrossRef] [Green Version]
- Fields, J.Z.; Roeske, W.R.; Morkin, E.; Yamamura, H.I. Cardiac muscarinic cholinergic receptors. Biochemical identification and characterization. J. Biol. Chem. 1978, 253, 3251–3258. [Google Scholar] [CrossRef]
- Reddy, S.; Bernstein, D. Molecular mechanisms of right ventricular failure. Circulation 2015, 132, 1734–1742. [Google Scholar] [CrossRef]
- Haddad, F.; Doyle, R.; Murphy, D.J.; Hunt, S.A. Right ventricular function in cardiovascular disease, part II: Pathophysiology, clinical importance, and management of right ventricular failure. Circulation 2008, 117, 1717–1731. [Google Scholar] [CrossRef]
- Dini, F.L.; Carluccio, E.; Simioniuc, A.; Biagioli, P.; Reboldi, G.; Galeotti, G.G.; Raineri, C.; Gargani, L.; Scelsi, L.; Mandoli, G.E.; et al. Network labs ultrasound (NEBULA) in heart failure study group. Right ventricular recovery during follow-up is associated with improved survival in patients with chronic heart failure with reduced ejection fraction. Eur. J. Heart Fail. 2016, 18, 1462–1471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.C.; Sanderson, J.E. Different prognostic significance of right and left ventricular diastolic dysfunction in heart failure. Clin. Cardiol. 1999, 22, 504–512. [Google Scholar] [CrossRef]
- Schlüter, K.D.; Kutsche, H.S.; Hirschhäuser, C.; Schreckenberg, R.; Schulz, R. Review on chamber-specific differences in right and left heart reactive oxygen species handling. Front. Physiol. 2018, 9, 1799. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Arroyo, J.; Mizuno, S.; Szczepanek, K.; Van Tassell, B.; Natarajan, R.; Dos Remedios, C.G.; Drake, J.I.; Farkas, L.; Kraskauskas, D.; Wijesinghe, D.S.; et al. Metabolic gene remodeling and mitochondrial dysfunction in failing right ventricular hypertrophy secondary to pulmonary arterial hypertension. Circ. Heart Fail. 2013, 6, 136–144. [Google Scholar] [CrossRef] [Green Version]
- Tsutsui, H.; Ide, T.; Hayashidani, S.; Suematsu, N.; Utsumi, H.; Nakamura, R.; Egashira, K.; Takeshita, A. Greater susceptibility of failing cardiac myocytes to oxygen free radical-mediated injury. Cardiovasc. Res. 2001, 49, 103–109. [Google Scholar] [CrossRef]
- Abbate, A.; Bussani, R.; Sinagra, G.; Barresi, E.; Pivetta, A.; Perkan, A.; Hoke, N.H.; Salloum, F.N.; Kontos, M.C.; Biondi-Zoccai, G.G.; et al. Right ventricular cardiomyocyte apoptosis in patients with acute myocardial infarction of the left ventricular wall. Am. J. Cardiol. 2008, 102, 658–662. [Google Scholar] [CrossRef] [Green Version]
- Dewachter, L.; Dewachter, C. Inflammation in right ventricular failure: Does it matter? Front. Physiol. 2018, 9, 1056. [Google Scholar] [CrossRef] [PubMed]
- Sydykov, A.; Mamazhakypov, A.; Petrovic, A.; Kosanovic, D.; Sarybaev, A.S.; Weissmann, N.; Ghofrani, H.A.; Schermuly, R.T. Inflammatory mediators drive adverse right ventricular remodeling and dysfunction and serve as potential biomarkers. Front. Physiol. 2018, 9, 609. [Google Scholar] [CrossRef]
- Di Salvo, T.G.; Yang, K.C.; Brittain, E.; Absi, T.; Maltais, S.; Hemnes, A. Right ventricular myocardial biomarkers in human heart failure. J. Card. Fail. 2015, 21, 398–411. [Google Scholar] [CrossRef]
- Su, Y.R.; Chiusa, M.; Brittain, E.; Hemnes, A.R.; Absi, T.S.; Lim, C.C.; Di Salvo, T.G. Right ventricular protein expression profile in end-stage heart failure. Pulm. Circ. 2015, 5, 481–497. [Google Scholar] [CrossRef] [Green Version]
- Linke, W.A.; Hamdani, N. Gigantic business: Titin properties and function through thick and thin. Circ. Res. 2014, 114, 1052–1068. [Google Scholar] [CrossRef]
- Sanganalmath, S.K.; Barta, J.; Takeda, N.; Kumamoto, H.; Dhalla, N.S. Antiplatelet therapy mitigates cardiac remodeling and dysfunction in congestive heart failure due to myocardial infarction. Can. J. Physiol. Pharmacol. 2008, 86, 180–189. [Google Scholar] [CrossRef] [PubMed]
- Hamdani, N.; Bishu, K.G.; Von Frieling-Salewsky, M.; Redfield, M.M.; Linke, W.A. Deranged myofilament phosphorylation and function in experimental heart failure with preserved ejection fraction. Cardiovasc. Res. 2013, 97, 464–471. [Google Scholar] [CrossRef] [Green Version]
- Papp, Z.; Szabó, A.; Barends, J.P.; Stienen, G.J. The mechanism of the force enhancement by MgADP under simulated ischaemic conditions in rat cardiac myocytes. J. Physiol. 2002, 543, 177–189. [Google Scholar] [CrossRef]
- Van Heerebeek, L.; Hamdani, N.; Falcão-Pires, I.; Leite-Moreira, A.F.; Begieneman, M.P.; Bronzwaer, J.G.; Van der Velden, J.; Stienen, G.J.; Laarman, G.J.; Somsen, A.; et al. Low myocardial protein kinase G activity in heart failure with preserved ejection fraction. Circulation 2012, 126, 830–839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaremba, R.; Merkus, D.; Hamdani, N.; Lamers, J.M.; Paulus, W.J.; Dos Remedios, C.; Duncker, D.J.; Stienen, G.J.; Van der Velden, J. Quantitative analysis of myofilament protein phosphorylation in small cardiac biopsies. Proteom. Clin. Appl 2007, 1, 1285–1290. [Google Scholar] [CrossRef] [PubMed]
- Krüger, M.; Kötter, S.; Grützner, A.; Lang, P.; Andresen, C.; Redfield, M.M.; Butt, E.; Dos Remedios, C.G.; Linke, W.A. Protein kinase G modulates human myocardial passive stiffness by phosphorylation of the titin springs. Circ. Res. 2009, 104, 87–94. [Google Scholar] [CrossRef] [Green Version]
- Van Linthout, S.; Spillmann, F.; Riad, A.; Trimpert, C.; Lievens, J.; Meloni, M.; Escher, F.; Filenberg, E.; Demir, O.; Li, J.; et al. Human apolipoprotein A-I gene transfer reduces the development of experimental diabetic cardiomyopathy. Circulation 2008, 117, 1563–1573. [Google Scholar] [CrossRef] [Green Version]
- Naeije, R.; Badagliacca, R. The overloaded right heart and ventricular interdependence. Cardiovasc. Res. 2017, 113, 1474–1485. [Google Scholar] [CrossRef]
- Capasso, J.M.; Li, P.; Zhang, X.; Anversa, P. Coronary artery narrowing in rats: Mechanical alterations of left and right myocardium. Am. J. Physiol. 1991, 261, 1802–1810. [Google Scholar] [CrossRef]
- Toldo, S.; Bogaard, H.J.; Van Tassell, B.W.; Mezzaroma, E.; Seropian, I.M.; Robati, R.; Salloum, F.N.; Voelkel, N.F.; Abbate, A. Right ventricular dysfunction following acute myocardial infarction in the absence of pulmonary hypertension in the mouse. PLoS ONE 2011, 6, e18102. [Google Scholar] [CrossRef] [Green Version]
- Chung, J.H.; Martin, B.L.; Canan, B.D.; Elnakish, M.T.; Milani-Nejad, N.; Saad, N.S.; Repas, S.J.; Schultz, J.E.J.; Murray, J.D.; Slabaugh, J.L.; et al. Etiology-dependent impairment of relaxation kinetics in right ventricular end-stage failing human myocardium. J. Mol. Cell Cardiol. 2018, 121, 81–93. [Google Scholar] [CrossRef] [Green Version]
- Paulus, W.J.; Tschöpe, C. A novel paradigm for heart failure with preserved ejection fraction: Comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J. Am. Coll. Cardiol. 2013, 62, 263–271. [Google Scholar] [CrossRef] [Green Version]
- Hamdani, N.; Kooij, V.; Van Dijk, S.; Merkus, D.; Paulus, W.J.; Remedios, C.D.; Duncker, D.J.; Stienen, G.J.; Van der Velden, J. Sarcomeric dysfunction in heart failure. Cardiovasc. Res. 2008, 77, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Borbély, A.; Van der Velden, J.; Papp, Z.; Bronzwaer, J.G.; Edes, I.; Stienen, G.J.; Paulus, W.J. Cardiomyocyte stiffness in diastolic heart failure. Circulation 2005, 111, 774–781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kötter, S.; Gout, L.; Von Frieling-Salewsky, M.; Müller, A.E.; Helling, S.; Marcus, K.; Dos Remedios, C.; Linke, W.A.; Krüger, M. Differential changes in titin domain phosphorylation increase myofilament stiffness in failing human hearts. Cardiovasc. Res. 2013, 99, 648–656. [Google Scholar] [CrossRef] [PubMed]
- Hamdani, N.; Krysiak, J.; Kreusser, M.M.; Neef, S.; Dos Remedios, C.G.; Maier, L.S.; Krüger, M.; Backs, J.; Linke, W.A. Crucial role for Ca2(+)/calmodulin-dependent protein kinase-II in regulating diastolic stress of normal and failing hearts via titin phosphorylation. Circ. Res. 2013, 112, 664–674. [Google Scholar] [CrossRef] [Green Version]
- Warren, M.; Sciuto, K.J.; Taylor, T.G.; Garg, V.; Torres, N.S.; Shibayama, J.; Spitzer, K.W.; Zaitsev, A.V. Blockade of CaMKII depresses conduction preferentially in the right ventricular outflow tract and promotes ischemic ventricular fibrillation in the rabbit heart. Am. J. Physiol. Heart Circ. Physiol. 2017, 312, H752–H767. [Google Scholar] [CrossRef] [Green Version]
- Andersen, A.; Povlsen, J.A.; Johnsen, J.; Jespersen, N.R.; Bøtker, H.E.; Nielsen-Kudsk, J.E. sGC-cGMP-PKG pathway stimulation protects the healthy but not the failing right ventricle of rats against ischemia and reperfusion injury. Int. J. Cardiol. 2016, 223, 674–680. [Google Scholar] [CrossRef]
- Bonderman, D.; Ghio, S.; Felix, S.B.; Ghofrani, H.A.; Michelakis, E.; Mitrovic, V.; Oudiz, R.J.; Boateng, F.; Scalise, A.V.; Roessig, L.; et al. Left ventricular systolic dysfunction associated with pulmonary hypertension riociguat trial (LEPHT) study group. Riociguat for patients with pulmonary hypertension caused by systolic left ventricular dysfunction: A phase IIb double-blind, randomized, placebo-controlled, dose-ranging hemodynamic study. Circulation 2013, 128, 502–511. [Google Scholar]
- Guazzi, M.; Vicenzi, M.; Arena, R.; Guazzi, M.D. Pulmonary hypertension in heart failure with preserved ejection fraction: A target of phosphodiesterase-5 inhibition in a 1-year study. Circulation 2011, 124, 164–174. [Google Scholar] [CrossRef]
- Hoendermis, E.S.; Liu, L.C.; Hummel, Y.M.; Van der Meer, P.; De Boer, R.A.; Berger, R.M.; Van Veldhuisen, D.J.; Voors, A.A. Effects of sildenafil on invasive haemodynamics and exercise capacity in heart failure patients with preserved ejection fraction and pulmonary hypertension: A randomized controlled trial. Eur. Heart J. 2015, 36, 2565–2573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamasaki, R.; Wu, Y.; McNabb, M.; Greaser, M.; Labeit, S.; Granzier, H. Protein kinase A phosphorylates titin’s cardiac-specific N2B domain and reduces passive tension in rat cardiac myocytes. Circ. Res. 2002, 90, 1181–1188. [Google Scholar] [CrossRef] [Green Version]
- Krüger, M.; Linke, W.A. Protein kinase-A phosphorylates titin in human heart muscle and reduces myofibrillar passive tension. J. Muscle Res. Cell Motil. 2006, 27, 435–444. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo, C.; Hudson, B.; Bogomolovas, J.; Zhu, Y.; Anderson, B.; Greaser, M.; Labeit, S.; Granzier, H. PKC phosphorylation of titin’s PEVK element: A novel and conserved pathway for modulating myocardial stiffness. Circ. Res. 2009, 105, 631–638. [Google Scholar] [CrossRef] [PubMed]
- Kovács, Á.; Fülöp, G.Á.; Kovács, A.; Csípő, T.; Bódi, B.; Priksz, D.; Juhász, B.; Beke, L.; Hendrik, Z.; Méhes, G.; et al. A. Renin overexpression leads to increased titin-based stiffness contributing to diastolic dysfunction in hypertensive mRen2 rats. Am. J. Physiol. Heart Circ. Physiol. 2016, 310, H1671–H1682. [Google Scholar] [CrossRef] [Green Version]
- Belin, R.J.; Sumandea, M.P.; Allen, E.J.; Schoenfelt, K.; Wang, H.; Solaro, R.J.; De Tombe, P.P. Augmented protein kinase C-alpha-induced myofilament protein phosphorylation contributes to myofilament dysfunction in experimental congestive heart failure. Circ. Res. 2007, 101, 195–204. [Google Scholar] [CrossRef] [Green Version]
- Belin, R.J.; Sumandea, M.P.; Allen, E.J.; Schoenfelt, K.; Wang, H.; Solaro, R.J.; De Tombe, P.P. Interventricular differences in myofilament function in experimental congestive heart failure. Pflugers Arch. 2011, 462, 795–809. [Google Scholar] [CrossRef] [Green Version]
- Solaro, R.J.; Henze, M.; Kobayashi, T. Integration of troponin I phosphorylation with cardiac regulatory networks. Circ. Res. 2013, 112, 355–366. [Google Scholar] [CrossRef] [Green Version]
- Avner, B.S.; Shioura, K.M.; Scruggs, S.B.; Grachoff, M.; Geenen, D.L.; Helseth, D.L., Jr.; Farjah, M.; Goldspink, P.H.; Solaro, R.J. Myocardial infarction in mice alters sarcomeric function via post-translational protein modification. Mol. Cell Biochem. 2012, 363, 203–215. [Google Scholar] [CrossRef] [Green Version]
- Balogh, A.; Santer, D.; Pásztor, E.T.; Tóth, A.; Czuriga, D.; Podesser, B.K.; Trescher, K.; Jaquet, K.; Erdodi, F.; Edes, I.; et al. Myofilament protein carbonylation contributes to the contractile dysfunction in the infarcted LV region of mouse hearts. Cardiovasc. Res. 2014, 101, 108–119. [Google Scholar] [CrossRef] [Green Version]
- Czuriga, D.; Tóth, A.; Pásztor, E.T.; Balogh, A.; Bodnár, A.; Nizsalóczki, E.; Lionetti, V.; Recchia, F.A.; Czuriga, I.; Edes, I.; et al. Cell-to-cell variability in troponin I phosphorylation in a porcine model of pacing-induced heart failure. Basic Res. Cardiol 2012, 107, 244. [Google Scholar] [CrossRef] [Green Version]
- Sheikh, F.; Ouyang, K.; Campbell, S.G.; Lyon, R.C.; Chuang, J.; Fitzsimons, D.; Tangney, J.; Hidalgo, C.G.; Chung, C.S.; Cheng, H.; et al. Mouse and computational models link Mlc2v dephosphorylation to altered myosin kinetics in early cardiac disease. J. Clin. Invest. 2012, 122, 1209–1221. [Google Scholar] [CrossRef] [Green Version]
- Sadayappan, S.; Gulick, J.; Osinska, H.; Barefield, D.; Cuello, F.; Avkiran, M.; Lasko, V.M.; Lorenz, J.N.; Maillet, M.; Martin, J.L.; et al. A critical function for Ser-282 in cardiac Myosin binding protein-C phosphorylation and cardiac function. Circ. Res. 2011, 109, 141–150. [Google Scholar] [CrossRef] [Green Version]
- Sadayappan, S.; Osinska, H.; Klevitsky, R.; Lorenz, J.N.; Sargent, M.; Molkentin, J.D.; Seidman, C.E.; Seidman, J.G.; Robbins, J. Cardiac myosin binding protein C phosphorylation is cardioprotective. Proc. Natl. Acad. Sci. USA 2006, 103, 16918–16923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, C.W.; Nair, N.A.; Doersch, K.M.; Liu, Y.; Rosas, P.C. Cardiac myosin-binding protein-C is a critical mediator of diastolic function. Pflugers Arch. 2014, 466, 451–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reil, J.C.; Reil, G.H.; Kovács, Á.; Sequeira, V.; Waddingham, M.T.; Lodi, M.; Herwig, M.; Ghaderi, S.; Kreusser, M.M.; Papp, Z.; et al. CaMKII activity contributes to homeometric autoregulation of the heart: A novel mechanism for the Anrep effect. J. Physiol. 2020, 598, 3129–3153. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.; Thai, K.; Park, K.W.; Wang, P.; Makwana, O.; Lovett, D.H.; Simpson, P.C.; Baker, A.J. Intraventricular and interventricular cellular heterogeneity of inotropic responses to α(1)-adrenergic stimulation. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H946–H953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sethi, R.; Dhalla, K.S.; Beamish, R.E.; Dhalla, N.S. Differential changes in left and right ventricular adenylyl cyclase activities in congestive heart failure. Am. J. Physiol. 1997, 272, H884–H893. [Google Scholar] [CrossRef] [PubMed]
- Stefanon, I.; Valero-Muñoz, M.; Fernandes, A.A.; Ribeiro, R.F., Jr.; Rodríguez, C.; Miana, M.; Martínez-González, J.; Spalenza, J.S.; Lahera, V.; Vassallo, P.F.; et al. Left and right ventricle late remodeling following myocardial infarction in rats. PLoS ONE 2013, 8, e64986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rüdebusch, J.; Benkner, A.; Poesch, A.; Dörr, M.; Völker, U.; Grube, K.; Hammer, E.; Felix, S.B. Dynamic adaptation of myocardial proteome during heart failure development. PLoS ONE 2017, 12, e0185915. [Google Scholar] [CrossRef] [Green Version]
- Schulz, E.M.; Correll, R.N.; Sheikh, H.N.; Lofrano-Alves, M.S.; Engel, P.L.; Newman, G.; Schultz, J.J.; Molkentin, J.D.; Wolska, B.M.; Solaro, R.J.; et al. Tropomyosin dephosphorylation results in compensated cardiac hypertrophy. J. Biol. Chem. 2012, 287, 44478–44489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Platt, M.J.; Huber, J.S.; Romanova, N.; Brunt, K.R.; Simpson, J.A. Pathophysiological mapping of experimental heart failure: Left and right ventricular remodeling in transverse aortic constriction is temporally, kinetically and structurally distinct. Front. Physiol. 2018, 9, 472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rain, S.; Handoko, M.L.; Trip, P.; Gan, C.T.; Westerhof, N.; Stienen, G.J.; Paulus, W.J.; Ottenheijm, C.A.; Marcus, J.T.; Dorfmüller, P.; et al. Right ventricular diastolic impairment in patients with pulmonary arterial hypertension. Circulation 2013, 128, 2016–2025. [Google Scholar] [CrossRef] [Green Version]
- Rain, S.; Bos, D.S.; Handoko, M.L.; Westerhof, N.; Stienen, G.; Ottenheijm, C.; Goebel, M.; Dorfmüller, P.; Guignabert, C.; Humbert, M.; et al. Protein changes contributing to right ventricular cardiomyocyte diastolic dysfunction in pulmonary arterial hypertension. J. Am. Heart Assoc. 2014, 3, e000716. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| RV | LV | |
|---|---|---|
| Molecular basic structure | different from LV | different from RV |
| Post-MI ischemic stress | no | yes |
| Transmyocardial scar formation | no | yes |
| Volume overload | yes | yes |
| Consequence of systemic humoral and inflammatory activation | yes | yes |
| Regulation of signaling pathways | ||
| CaMKII | down | up |
| PKG | down | down |
| PKA | down | down |
| PKC | up | up |
| Cardiomyocyte passive stiffness | increased | increased |
| Cardiomyocyte Ca2+-sensitivity | unaltered | increased |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kovács, Á.; Herwig, M.; Budde, H.; Delalat, S.; Kolijn, D.; Bódi, B.; Hassoun, R.; Tangos, M.; Zhazykbayeva, S.; Balogh, Á.; et al. Interventricular Differences of Signaling Pathways-Mediated Regulation of Cardiomyocyte Function in Response to High Oxidative Stress in the Post-Ischemic Failing Rat Heart. Antioxidants 2021, 10, 964. https://doi.org/10.3390/antiox10060964
Kovács Á, Herwig M, Budde H, Delalat S, Kolijn D, Bódi B, Hassoun R, Tangos M, Zhazykbayeva S, Balogh Á, et al. Interventricular Differences of Signaling Pathways-Mediated Regulation of Cardiomyocyte Function in Response to High Oxidative Stress in the Post-Ischemic Failing Rat Heart. Antioxidants. 2021; 10(6):964. https://doi.org/10.3390/antiox10060964
Chicago/Turabian StyleKovács, Árpád, Melissa Herwig, Heidi Budde, Simin Delalat, Detmar Kolijn, Beáta Bódi, Roua Hassoun, Melina Tangos, Saltanat Zhazykbayeva, Ágnes Balogh, and et al. 2021. "Interventricular Differences of Signaling Pathways-Mediated Regulation of Cardiomyocyte Function in Response to High Oxidative Stress in the Post-Ischemic Failing Rat Heart" Antioxidants 10, no. 6: 964. https://doi.org/10.3390/antiox10060964