
Oxidative Stress Signaling in Blast TBI-Induced Tau Phosphorylation

 , ,
, ,
Abstract
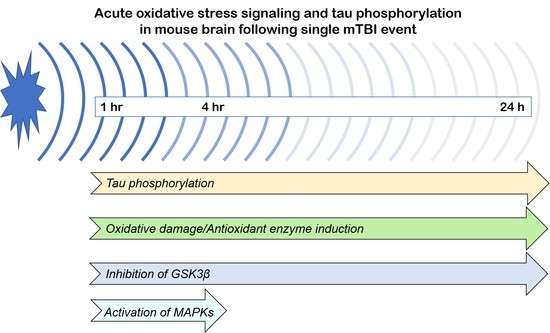
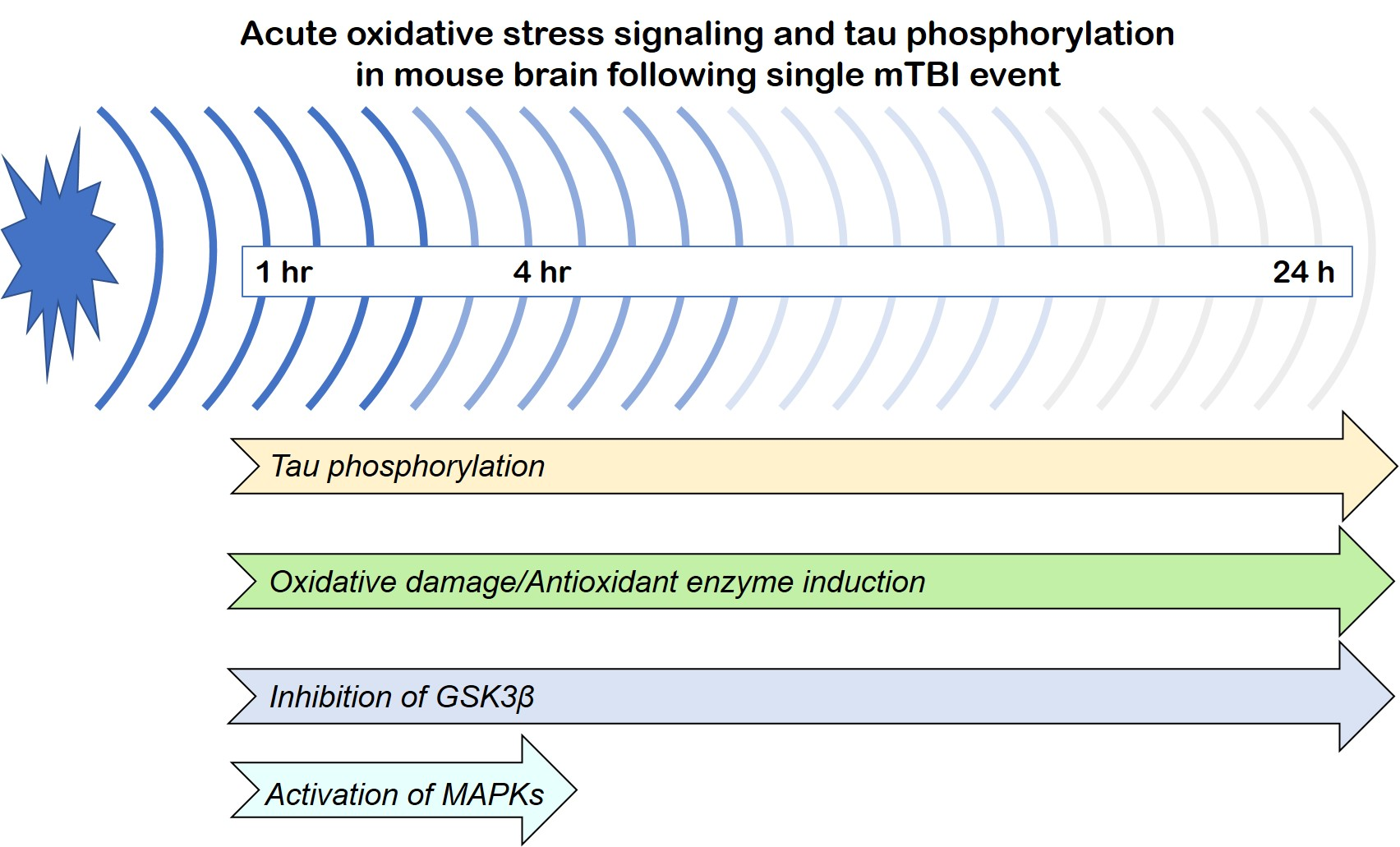
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Animals and Treatment
2.2. Western Blot
2.3. Antibodies
2.4. Image Acquisition and Statistical Analysis
3. Results
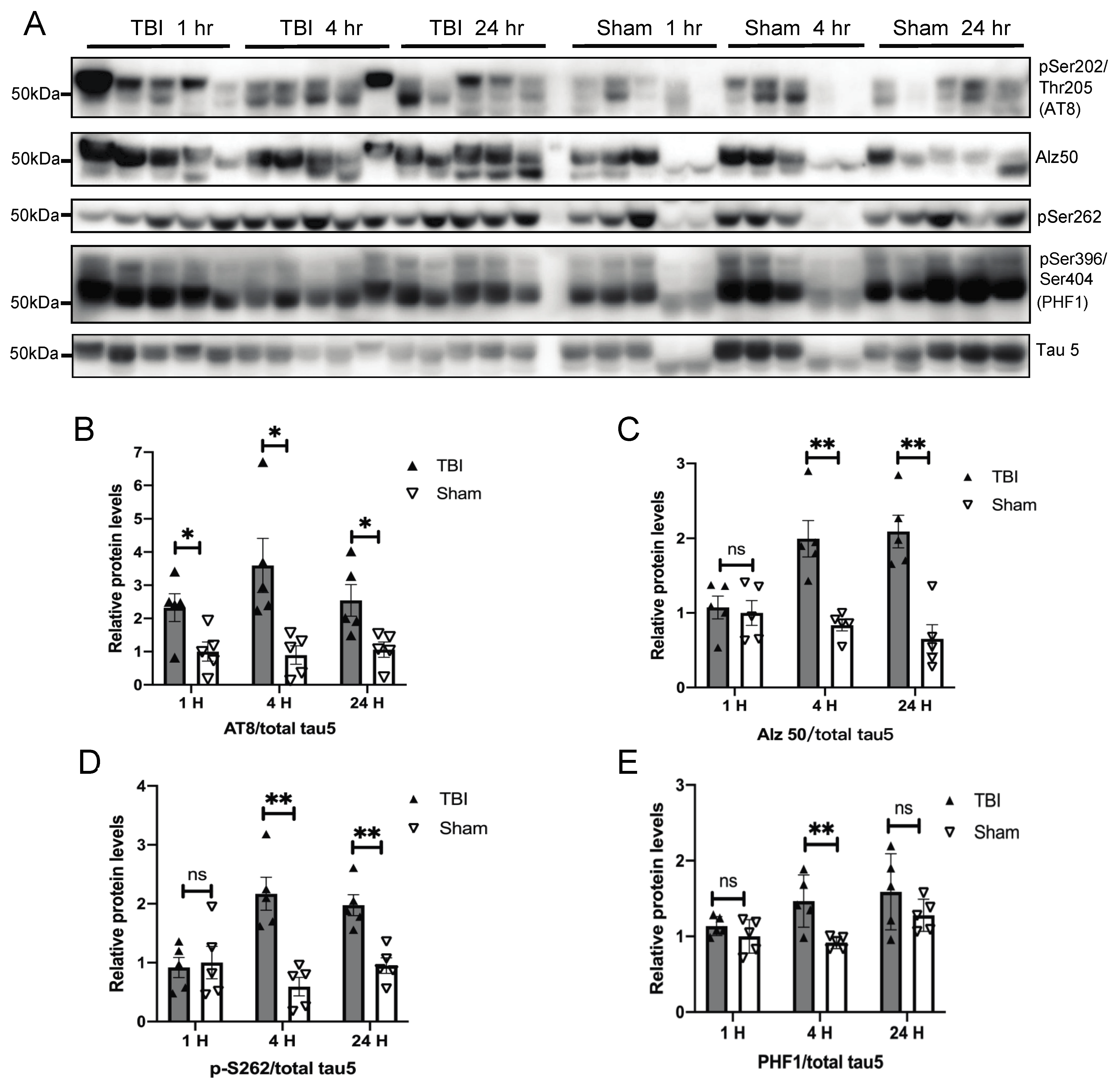
3.1. Effects of a Single Mild BOP Exposure on Tau Phosphorylation
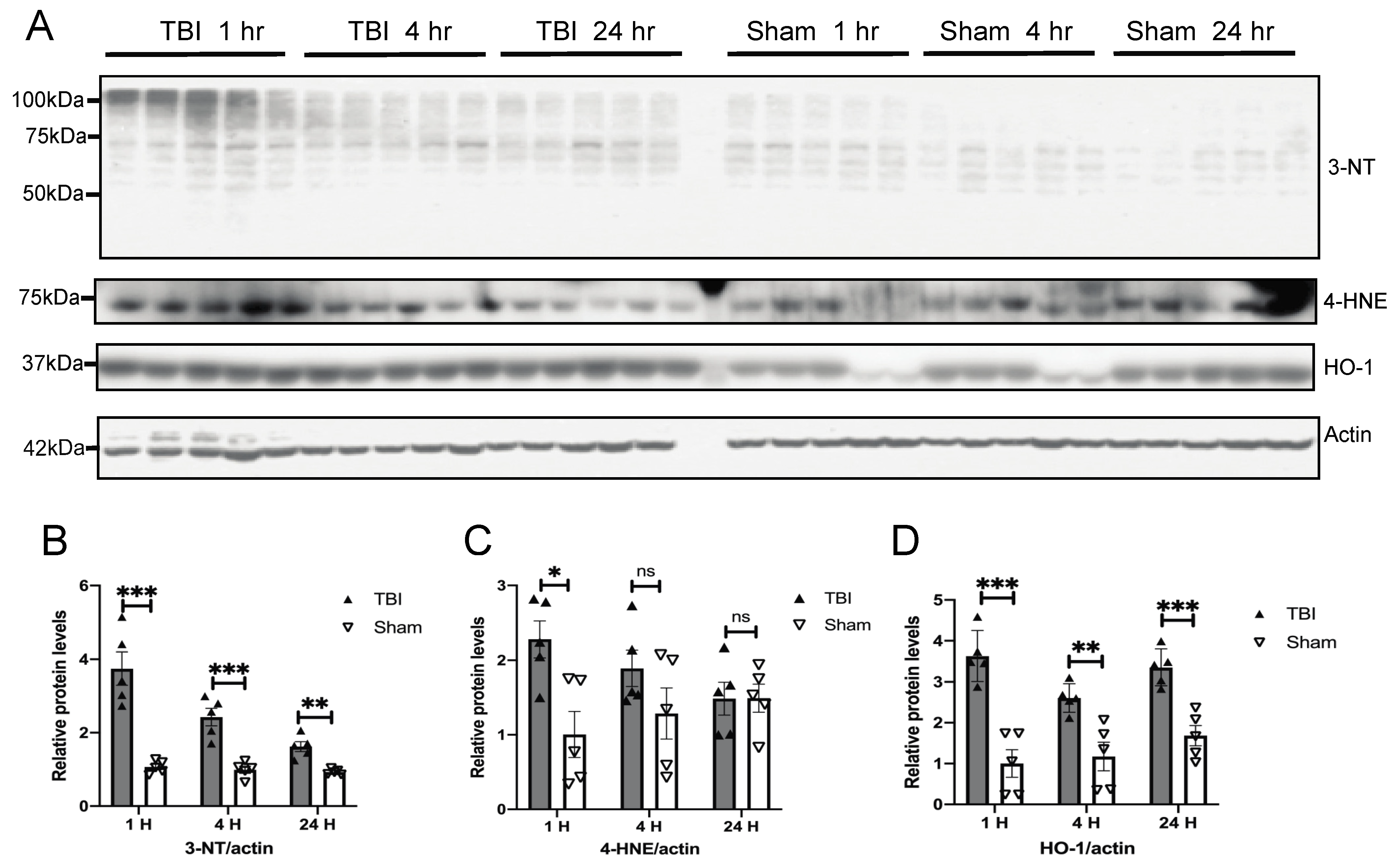
3.2. Effects of a Single Mild BOP Exposure on Oxidative Stress
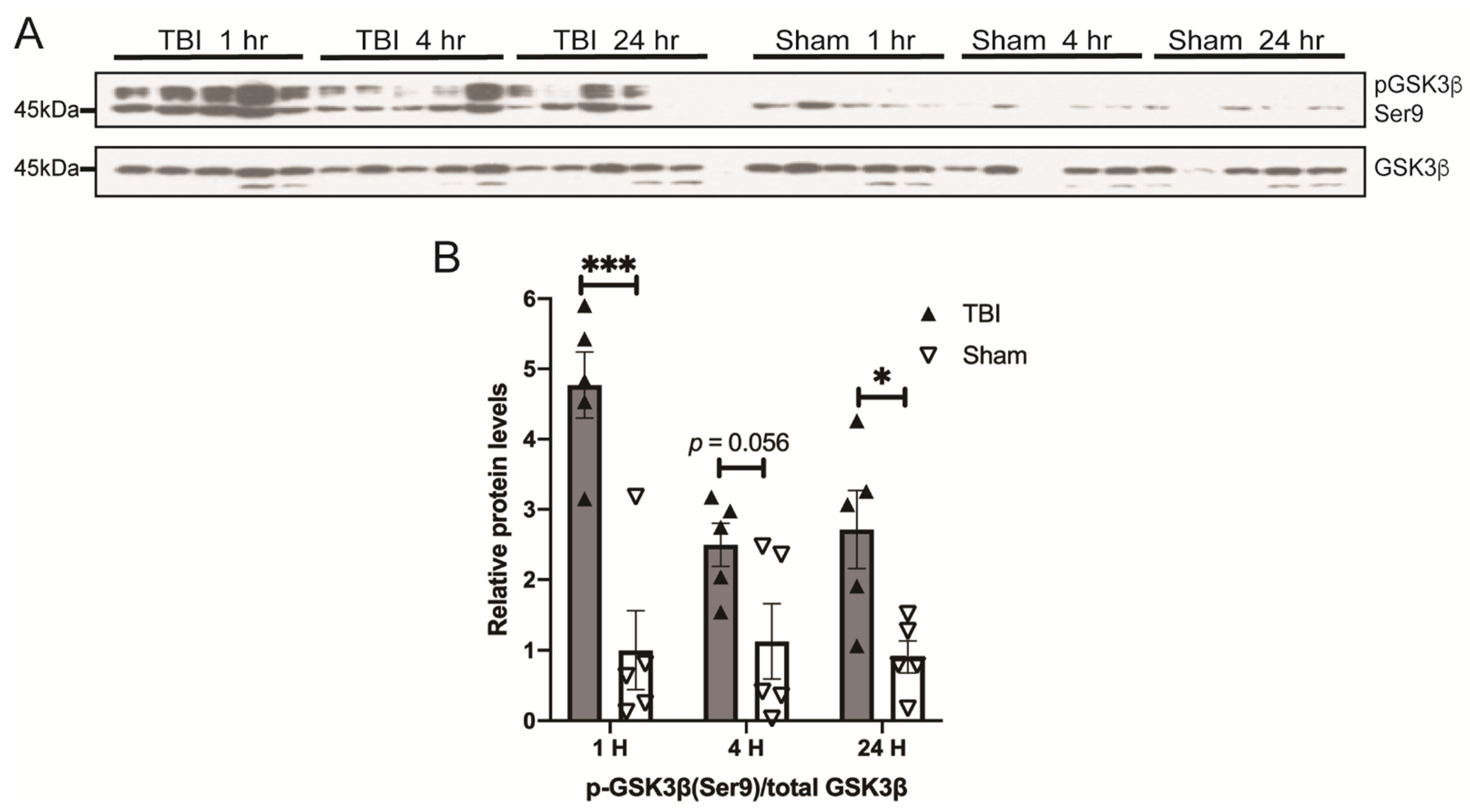
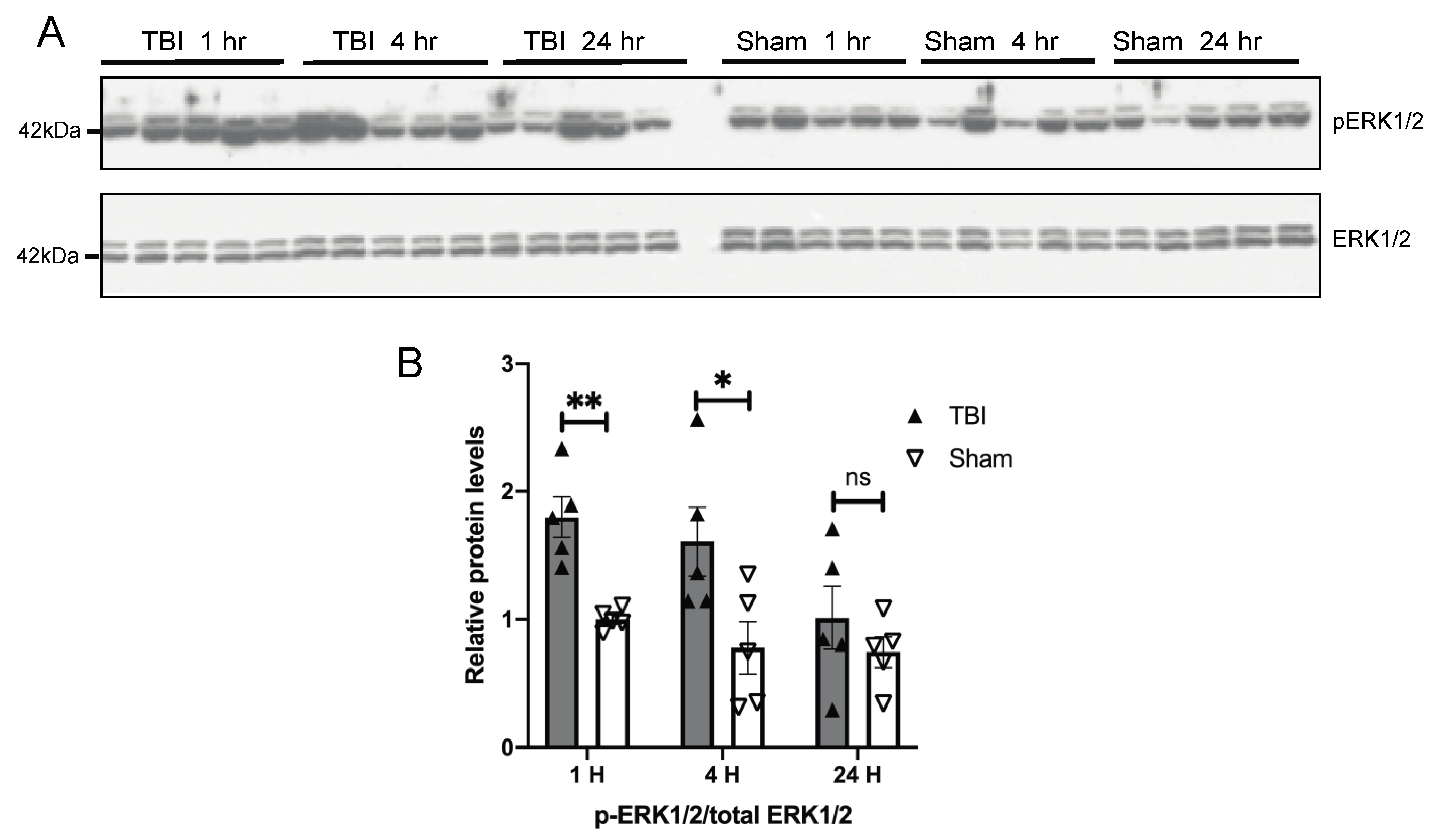
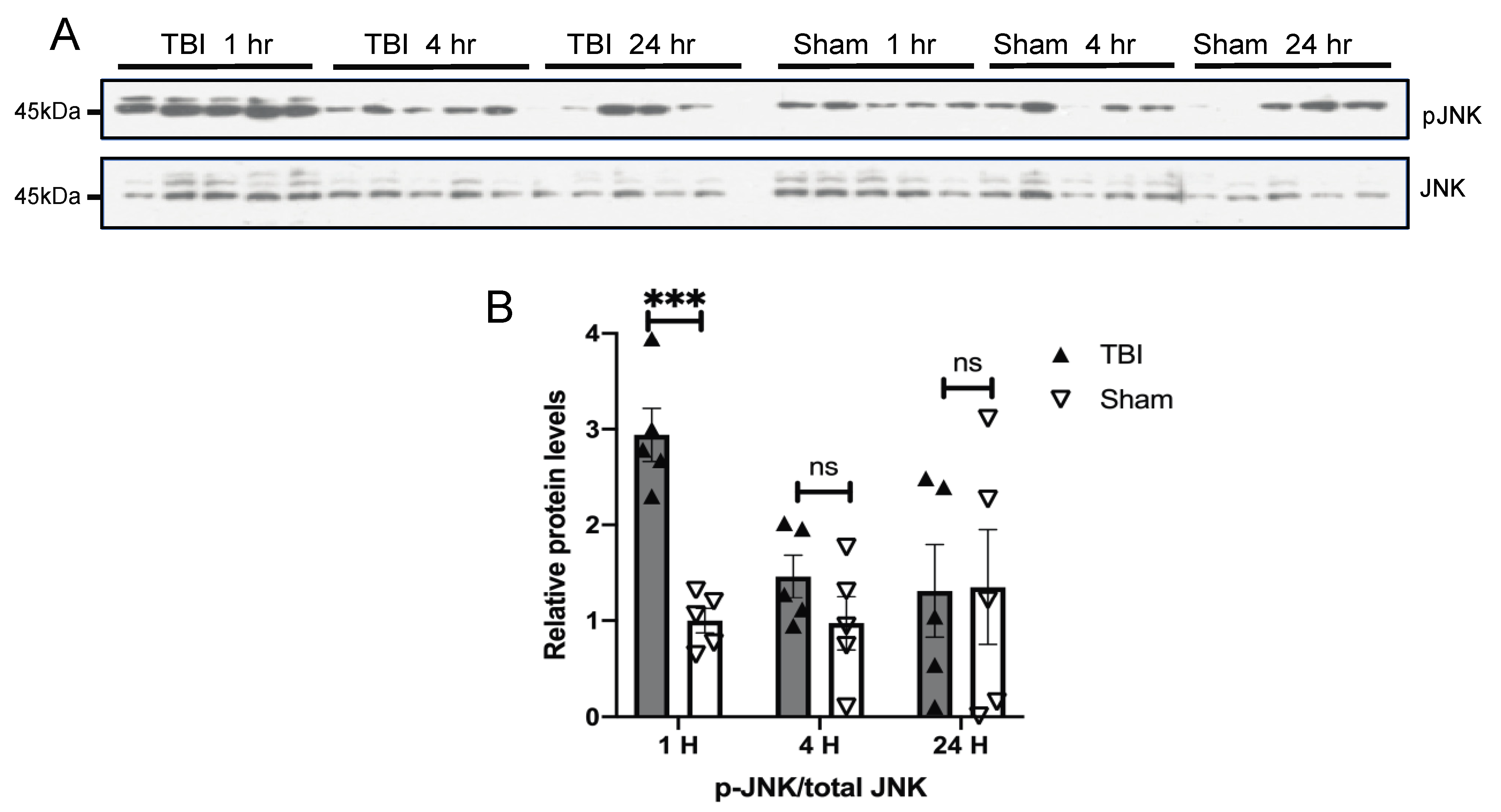
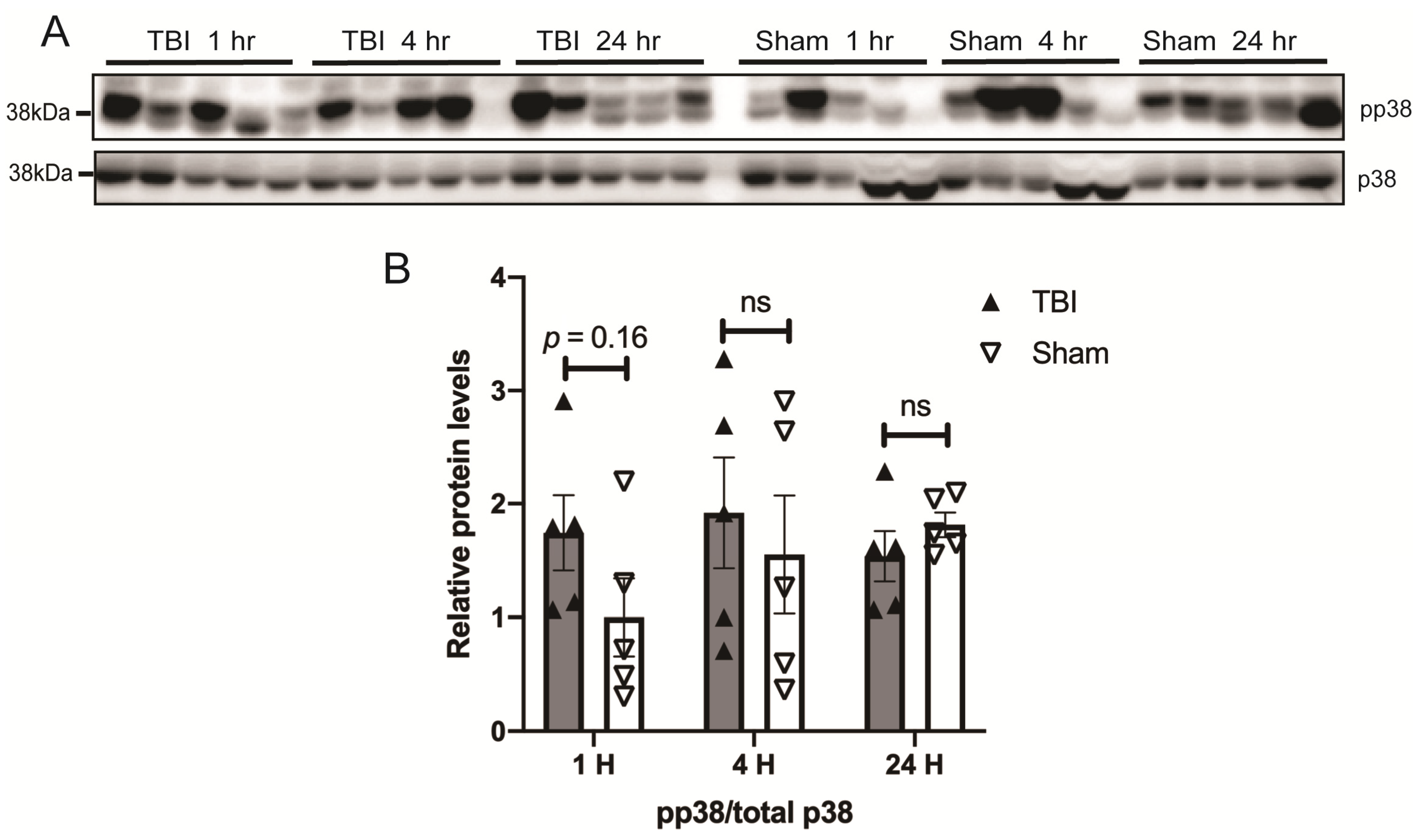
3.3. Effects of a Single Mild BOP Exposure on Tau Kinases
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Elder, G.A.; Mitsis, E.M.; Ahlers, S.T.; Cristian, A. Blast-induced mild traumatic brain injury. Psychiatr. Clin. N. Am. 2010, 33, 757–781. [Google Scholar] [CrossRef]
- Vincent, A.S.; Roebuck-Spencer, T.M.; Cernich, A. Cognitive changes and dementia risk after traumatic brain injury: Implications for aging military personnel. Alzheimers Dement. 2014, 10, S174–S187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lange, R.T.; Brickell, T.A.; Ivins, B.; Vanderploeg, R.D.; French, L.M. Variable, not always persistent, postconcussion symptoms after mild TBI in U.S. military service members: A five-year cross-sectional outcome study. J. Neurotrauma 2013, 30, 958–969. [Google Scholar] [CrossRef] [PubMed]
- Mac Donald, C.L.; Barber, J.; Jordan, M.; Johnson, A.M.; Dikmen, S.; Fann, J.R.; Temkin, N. Early clinical predictors of 5-year outcome after concussive blast traumatic brain injury. JAMA Neurol. 2017, 74, 821–829. [Google Scholar] [CrossRef] [Green Version]
- Kornblith, E.; Peltz, C.B.; Xia, F.; Plassman, B.; Novakovic-Apopain, T.; Yaffe, K. Sex, race, and risk of dementia diagnosis after traumatic brain injury among older veterans. Neurology 2020, 95, e1768–e1775. [Google Scholar] [CrossRef]
- Aldag, M.; Armstrong, R.C.; Bandak, F.; Bellgowan, P.S.F.; Bentley, T.; Biggerstaff, S.; Caravelli, K.; Cmarik, J.; Crowder, A.; DeGraba, T.J.; et al. The biological basis of chronic traumatic encephalopathy following blast injury: A literature review. J. Neurotrauma 2017, 34, S26–S43. [Google Scholar] [CrossRef]
- Dickstein, D.L.; De Gasperi, R.; Gama Sosa, M.A.; Perez-Garcia, G.; Short, J.A.; Sosa, H.; Perez, G.M.; Tschiffely, A.E.; Dams-O’Connor, K.; Pullman, M.Y.; et al. Brain and blood biomarkers of tauopathy and neuronal injury in humans and rats with neurobehavioral syndromes following blast exposure. Mol. Psychiatry 2020. [Google Scholar] [CrossRef] [Green Version]
- McKee, A.C. The neuropathology of chronic traumatic encephalopathy: The status of the literature. Semin. Neurol. 2020, 40, 359–369. [Google Scholar] [CrossRef]
- Katz, D.I.; Bernick, C.; Dodick, D.W.; Mez, J.; Mariani, M.L.; Adler, C.H.; Alosco, M.L.; Balcer, L.J.; Banks, S.J.; Barr, W.B.; et al. National Institute of Neurological Disorders and Stroke Consensus Diagnostic Criteria for Traumatic Encephalopathy Syndrome. Neurology 2021, 96. [Google Scholar] [CrossRef] [PubMed]
- Tripathy, A.; Shade, A.; Erskine, B.; Bailey, K.; Grande, A.; deLong, J.J.; Perry, G.; Castellani, R.J. No evidence of increased chronic traumatic encephalopathy pathology or neurodegenerative proteinopathy in former military service members: A preliminary study. J. Alzheimers Dis. 2019, 67, 1277–1289. [Google Scholar] [CrossRef] [PubMed]
- Shively, S.B.; Horkayne-Szakaly, I.; Jones, R.V.; Kelly, J.P.; Armstrong, R.C.; Perl, D.P. Characterisation of interface astroglial scarring in the human brain after blast exposure: A post-mortem case series. Lancet Neurol. 2016, 15, 944–953. [Google Scholar] [CrossRef] [Green Version]
- Huber, B.R.; Meabon, J.S.; Martin, T.J.; Mourad, P.D.; Bennett, R.; Kraemer, B.C.; Cernak, I.; Petrie, E.C.; Emery, M.J.; Swenson, E.R.; et al. Blast exposure causes early and persistent aberrant phospho- and cleaved-tau expression in a murine model of mild blast-induced traumatic brain injury. J. Alzheimers Dis. 2013, 37, 309–323. [Google Scholar] [CrossRef] [Green Version]
- Abdul-Muneer, P.M.; Schuetz, H.; Wang, F.; Skotak, M.; Jones, J.; Gorantla, S.; Zimmerman, M.C.; Chandra, N.; Haorah, J. Induction of oxidative and nitrosative damage leads to cerebrovascular inflammation in an animal model of mild traumatic brain injury induced by primary blast. Free Radic. Biol. Med. 2013, 60, 282–291. [Google Scholar] [CrossRef] [PubMed]
- Cernak, I.; Savic, V.J.; Kotur, J.; Prokic, V.; Veljovic, M.; Grbovic, D. Characterization of plasma magnesium concentration and oxidative stress following graded traumatic brain injury in humans. J. Neurotrauma 2000, 17, 53–68. [Google Scholar] [CrossRef]
- Johnson, V.E.; Stewart, J.E.; Begbie, F.D.; Trojanowski, J.Q.; Smith, D.H.; Stewart, W. Inflammation and white matter degeneration persist for years after a single traumatic brain injury. Brain 2013, 136, 28–42. [Google Scholar] [CrossRef] [Green Version]
- Mac Donald, C.L.; Johnson, A.M.; Cooper, D.; Nelson, E.C.; Werner, N.J.; Shimony, J.S.; Snyder, A.Z.; Raichle, M.E.; Witherow, J.R.; Fang, R.; et al. Detection of blast-related traumatic brain injury in U.S. military personnel. N. Engl. J. Med. 2011, 364, 2091–2100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lipton, M.L.; Gulko, E.; Zimmerman, M.E.; Friedman, B.W.; Kim, M.; Gellella, E.; Gold, T.; Shifteh, K.; Ardekani, B.A.; Branch, C.A. Diffusion-tensor imaging implicates prefrontal axonal injury in executive function impairment following very mild traumatic brain injury. Radiology 2009, 252, 816–824. [Google Scholar] [CrossRef]
- McKee, A.C.; Robinson, M.E. Military-related traumatic brain injury and neurodegeneration. Alzheimers Dement. 2014, 10, S242–S253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldstein, L.E.; Fisher, A.M.; Tagge, C.A.; Zhang, X.L.; Velisek, L.; Sullivan, J.A.; Upreti, C.; Kracht, J.M.; Ericsson, M.; Wojnarowicz, M.W.; et al. Chronic traumatic encephalopathy in blast-exposed military veterans and a blast neurotrauma mouse model. Sci. Transl. Med. 2012, 4, 134ra160. [Google Scholar] [CrossRef] [Green Version]
- Omalu, B.; Hammers, J.L.; Bailes, J.; Hamilton, R.L.; Kamboh, M.I.; Webster, G.; Fitzsimmons, R.P. Chronic traumatic encephalopathy in an Iraqi war veteran with posttraumatic stress disorder who committed suicide. Neurosurg. Focus 2011, 31, E3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryu, J.; Horkayne-Szakaly, I.; Xu, L.; Pletnikova, O.; Leri, F.; Eberhart, C.; Troncoso, J.C.; Koliatsos, V.E. The problem of axonal injury in the brains of veterans with histories of blast exposure. Acta Neuropathol. Commun. 2014, 2, 153. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.T.; Siddarth, P.; Merrill, D.A.; Martinez, J.; Emerson, N.D.; Liu, J.; Wong, K.P.; Satyamurthy, N.; Giza, C.C.; Huang, S.C.; et al. FDDNP-PET tau brain protein binding patterns in military personnel with suspected chronic traumatic encephalopathy. J. Alzheimers Dis. 2018, 65, 79–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, M.E.; McKee, A.C.; Salat, D.H.; Rasmusson, A.M.; Radigan, L.J.; Catana, C.; Milberg, W.P.; McGlinchey, R.E. Positron emission tomography of tau in Iraq and Afghanistan Veterans with blast neurotrauma. Neuroimage Clin. 2019, 21, 101651. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Song, H.; Cui, J.; Johnson, C.E.; Hubler, G.K.; DePalma, R.G.; Gu, Z.; Xia, W. Proteomic profiling of mouse brains exposed to blast-induced mild traumatic brain injury reveals changes in axonal proteins and phosphorylated tau. J. Alzheimers Dis. 2018, 66, 751–773. [Google Scholar] [CrossRef] [PubMed]
- Lucke-Wold, B.P.; Turner, R.C.; Logsdon, A.F.; Nguyen, L.; Bailes, J.E.; Lee, J.M.; Robson, M.J.; Omalu, B.I.; Huber, J.D.; Rosen, C.L. Endoplasmic reticulum stress implicated in chronic traumatic encephalopathy. J. Neurosurg. 2016, 124, 687–702. [Google Scholar] [CrossRef] [Green Version]
- Murphy, E.K.; Iacono, D.; Pan, H.; Grimes, J.B.; Parks, S.; Raiciulescu, S.; Leonessa, F.; Perl, D.P. Explosive-driven double-blast exposure: Molecular, histopathological, and behavioral consequences. Sci. Rep. 2020, 10, 17446. [Google Scholar] [CrossRef]
- Shi, Q.X.; Chen, B.; Nie, C.; Zhao, Z.P.; Zhang, J.H.; Si, S.Y.; Cui, S.J.; Gu, J.W. A novel model of blast induced traumatic brain injury caused by compressed gas produced sustained cognitive deficits in rats: Involvement of phosphorylation of tau at the Thr205 epitope. Brain Res. Bull. 2020, 157, 149–161. [Google Scholar] [CrossRef] [PubMed]
- Shi, Q.X.; Chen, B.; Nie, C.; Zhao, Z.P.; Zhang, J.H.; Si, S.Y.; Cui, S.J.; Gu, J.W. Improvement in cognitive dysfunction following blast induced traumatic brain injury by thymosin alpha1 in rats: Involvement of inhibition of tau phosphorylation at the Thr205 epitope. Brain Res. 2020, 1747, 147038. [Google Scholar] [CrossRef]
- Peng, J.H.; Kung, F.T.; Peng, W.; Parker, J.C., Jr. Increased ALZ-50 immunoreactivity in CSF of pseudotumor cerebri patients. Ann. Clin. Lab. Sci. 2006, 36, 151–156. [Google Scholar]
- Miller, B.E. Comparison of A68 levels in Alzheimer diseased and non-Alzheimer’s diseased brain by two ALZ50 based methods. Life Sci. 1999, 65, 2215–2222. [Google Scholar] [CrossRef]
- Jicha, G.A.; Weaver, C.; Lane, E.; Vianna, C.; Kress, Y.; Rockwood, J.; Davies, P. cAMP-dependent protein kinase phosphorylations on tau in Alzheimer’s disease. J. Neurosci. 1999, 19, 7486–7494. [Google Scholar] [CrossRef]
- Rana, P.; Rama Rao, K.V.; Ravula, A.; Trivedi, R.; D’Souza, M.; Singh, A.K.; Gupta, R.K.; Chandra, N. Oxidative stress contributes to cerebral metabolomic profile changes in animal model of blast-induced traumatic brain injury. Metabolomics 2020, 16, 39. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Yin, Y.L.; Liu, X.Z.; Shen, P.; Zheng, Y.G.; Lan, X.R.; Lu, C.B.; Wang, J.Z. Current understanding of metal ions in the pathogenesis of Alzheimer’s disease. Transl. Neurodegener. 2020, 9, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, X.; Lee, H.G.; Raina, A.K.; Perry, G.; Smith, M.A. The role of mitogen-activated protein kinase pathways in Alzheimer’s disease. Neurosignals 2002, 11, 270–281. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Wang, J.Z. Nature of tau-associated neurodegeneration and the molecular mechanisms. J. Alzheimers Dis. 2018, 62, 1305–1317. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Smith, M.A.; Avila, J.; DeBernardis, J.; Kansal, M.; Takeda, A.; Zhu, X.; Nunomura, A.; Honda, K.; Moreira, P.I.; et al. Alzheimer-specific epitopes of tau represent lipid peroxidation-induced conformations. Free Radic. Biol. Med. 2005, 38, 746–754. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Lee, H.G.; Casadesus, G.; Avila, J.; Drew, K.; Perry, G.; Smith, M.A. Oxidative imbalance in Alzheimer’s disease. Mol. Neurobiol. 2005, 31, 205–217. [Google Scholar] [CrossRef]
- Wang, W.; Zhao, F.; Ma, X.; Perry, G.; Zhu, X. Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: Recent advances. Mol. Neurodegener. 2020, 15, 30. [Google Scholar] [CrossRef]
- Takeda, A.; Smith, M.A.; Avila, J.; Nunomura, A.; Siedlak, S.L.; Zhu, X.; Perry, G.; Sayre, L.M. In Alzheimer’s disease, heme oxygenase is coincident with Alz50, an epitope of tau induced by 4-hydroxy-2-nonenal modification. J. Neurochem. 2000, 75, 1234–1241. [Google Scholar] [CrossRef]
- Rashedinia, M.; Lari, P.; Abnous, K.; Hosseinzadeh, H. Protective effect of crocin on acrolein-induced tau phosphorylation in the rat brain. Acta Neurobiol. Exp. 2015, 75, 208–219. [Google Scholar]
- Su, B.; Wang, X.; Lee, H.G.; Tabaton, M.; Perry, G.; Smith, M.A.; Zhu, X. Chronic oxidative stress causes increased tau phosphorylation in M17 neuroblastoma cells. Neurosci. Lett. 2010, 468, 267–271. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, K.; Liu, F.; Gong, C.X. Tau and neurodegenerative disease: The story so far. Nat. Rev. Neurol. 2016, 12, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Sawyer, T.W.; Tse, Y.C.; Fan, C.; Hennes, G.; Barnes, J.; Josey, T.; Weiss, T.; Nelson, P.; Wong, T.P. Primary blast-induced changes in Akt and GSK3beta phosphorylation in rat hippocampus. Front. Neurol. 2017, 8, 413. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, C.; Shao, C.; Zhang, L.; Siedlak, S.L.; Meabon, J.S.; Peskind, E.R.; Lu, Y.; Wang, W.; Perry, G.; Cook, D.G.; et al. Oxidative Stress Signaling in Blast TBI-Induced Tau Phosphorylation. Antioxidants 2021, 10, 955. https://doi.org/10.3390/antiox10060955
Wang C, Shao C, Zhang L, Siedlak SL, Meabon JS, Peskind ER, Lu Y, Wang W, Perry G, Cook DG, et al. Oxidative Stress Signaling in Blast TBI-Induced Tau Phosphorylation. Antioxidants. 2021; 10(6):955. https://doi.org/10.3390/antiox10060955
Chicago/Turabian StyleWang, Chunyu, Changjuan Shao, Li Zhang, Sandra L. Siedlak, James S. Meabon, Elaine R. Peskind, Yubing Lu, Wenzhang Wang, George Perry, David G. Cook, and et al. 2021. "Oxidative Stress Signaling in Blast TBI-Induced Tau Phosphorylation" Antioxidants 10, no. 6: 955. https://doi.org/10.3390/antiox10060955