A Peptide Inhibitor of Peroxiredoxin 6 Phospholipase A2 Activity Significantly Protects against Lung Injury in a Mouse Model of Ventilator Induced Lung Injury (VILI)


Abstract
:
1. Introduction
2. Materials and Methods
2.1. Mice
2.2. PIP-2
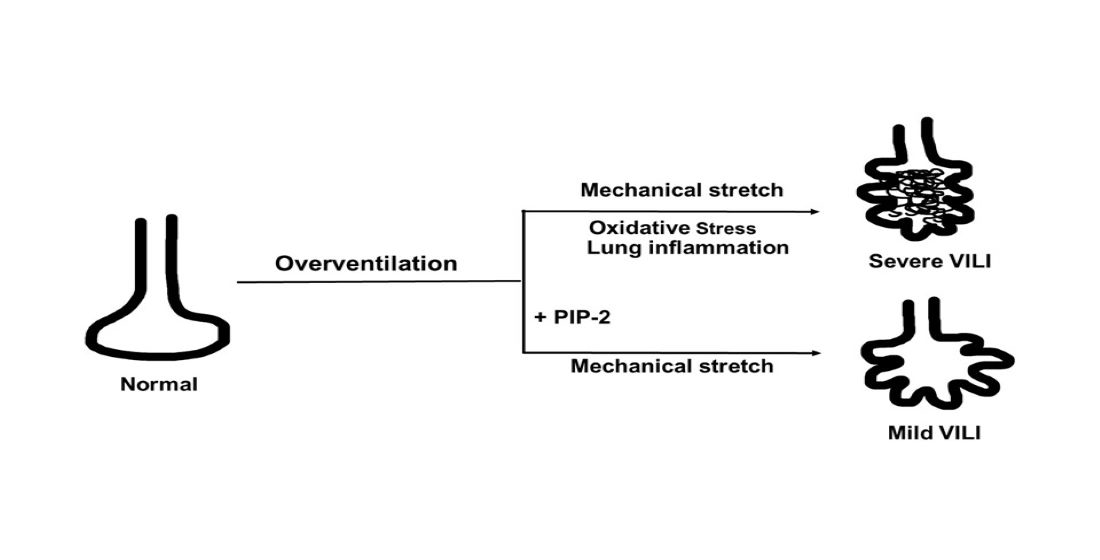
2.3. Ventilator Induced Lung Injury (VILI)
2.4. Indices of Lung Injury and Oxidative Stress
2.5. Measurement of ROS in HL60 Cells
2.6. Statistical Analysis
3. Results
3.1. Liposome Encapsulation Is Essential for Cellular Uptake of PIP-2
3.2. PIP-2 Administered Intratracheally Abrogates Oxidative Stress Arising from High Tidal Volume-Induced VILI
3.3. Intravenous vs. Intratracheal Administration of PIP-2
3.4. Male vs. Female Mice
3.5. Effect of PIP-2 on ROS Production in Human Cells
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Patent Pending
References
- Bernard, G.R.; Artigas, A.; Brigham, K.L.; Carlet, J.; Falke, K.; Hudson, L.; Lamy, M.; Legall, J.R.; Morris, A.; Spragg, R. The american-european consensus conference on ARDS. Definitions, mechanisms, relevant outcomes, and clinical trial coordination. Am. J. Respir. Crit. Care Med. 1994, 149, 818–824. [Google Scholar] [CrossRef]
- Chow, C.W.; Herrera Abreu, M.T.; Suzuki, T.; Downey, G.P. Oxidative stress and acute lung injury. Am. J. Respir. Cell Mol. Biol. 2003, 29, 427–431. [Google Scholar] [CrossRef] [PubMed]
- Johnson, E.R.; Matthay, M.A. Acute lung injury: Epidemiology, pathogenesis, and treatment. J. Aerosol Med. Pulm. Drug Deliv. 2010, 23, 243–252. [Google Scholar] [CrossRef]
- Allardet-Servent, J.; Sicard, G.; Metz, V.; Chiche, L. Benefits and risks of oxygen therapy during acute medical illness: Just a matter of dose! Rev. Med. Interne 2019, 40, 670–676. [Google Scholar] [CrossRef]
- Mayoralas-Alises, S.; Carratala, J.M.; Diaz-Lobato, S. New perspectives in oxygen therapy titration: Is automatic titration the future? Arch. Bronconeumol. 2019, 55, 319–327. [Google Scholar] [CrossRef] [PubMed]
- Dreyfuss, D.; Saumon, G. Ventilator-induced lung injury: Lessons from experimental studies. Am. J. Respir. Crit. Care Med. 1998, 157, 294–323. [Google Scholar] [CrossRef]
- Beitler, J.R.; Malhotra, A.; Thompson, B.T. Ventilator-induced lung injury. Clin. Chest Med. 2016, 37, 633–646. [Google Scholar] [CrossRef] [PubMed]
- Brower, R.G.; Matthay, M.A.; Morris, A.; Schoenfeld, D.; Thompson, B.T.; Wheeler, A. Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. N. Engl. J. Med. 2000, 342, 1301–1308. [Google Scholar] [CrossRef] [PubMed]
- Nieman, G.F.; Satalin, J.; Andrews, P.; Aiash, H.; Habashi, N.M.; Gatto, L.A. Personalizing mechanical ventilation according to physiologic parameters to stabilize alveoli and minimize ventilator induced lung injury (VILI). Intensive Care Med. Exp. 2017, 5, 8. [Google Scholar] [CrossRef] [Green Version]
- Derwall, M.; Martin, L.; Rossaint, R. The acute respiratory distress syndrome: Pathophysiology, current clinical practice, and emerging therapies. Expert Rev. Respir. Med. 2018, 12, 1021–1029. [Google Scholar] [CrossRef] [PubMed]
- Hoegl, S.; Burns, N.; Angulo, M.; Francis, D.; Osborne, C.M.; Mills, T.W.; Blackburn, M.R.; Eltzschig, H.K.; Vohwinkel, C.U. Capturing the multifactorial nature of ARDS—“Two-hit” approach to model murine acute lung injury. Physiol. Rep. 2018, 6, e13648. [Google Scholar] [CrossRef]
- Cho, W.H.; Kim, Y.H.; Heo, H.J.; Kim, D.; Kwak, T.W.; Kim, K.H.; Yeo, H.J. Ginsenoside ameliorated ventilator-induced lung injury in rats. J. Intensive Care 2020, 8, 89. [Google Scholar] [CrossRef]
- Kuethe, D.O.; Filipczak, P.T.; Hix, J.M.; Gigliotti, A.P.; Estepar, R.S.; Washko, G.R.; Baron, R.M.; Fredenburgh, L.E. Magnetic resonance imaging provides sensitive in vivo assessment of experimental ventilator-induced lung injury. Am. J. Physiol. Lung Cell Mol. Physiol. 2016, 311, L208–L218. [Google Scholar] [CrossRef] [Green Version]
- Nieman, G.F.; Al-Khalisy, H.; Kollisch-Singule, M.; Satalin, J.; Blair, S.; Trikha, G.; Andrews, P.; Madden, M.; Gatto, L.A.; Habashi, N.M. A physiologically informed strategy to effectively open, stabilize, and protect the acutely injured lung. Front. Physiol. 2020, 11, 227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caironi, P.; Langer, T.; Carlesso, E.; Protti, A.; Gattinoni, L. Time to generate ventilator-induced lung injury among mammals with healthy lungs: A unifying hypothesis. Intensive Care Med. 2011, 37, 1913–1920. [Google Scholar] [CrossRef] [Green Version]
- Letsiou, E.; Sammani, S.; Zhang, W.; Zhou, T.; Quijada, H.; Moreno-Vinasco, L.; Dudek, S.M.; Garcia, J.G. Pathologic mechanical stress and endotoxin exposure increases lung endothelial microparticle shedding. Am. J. Respir. Cell Mol. Biol. 2015, 52, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Gross, C.; Desai, A.A.; Zemskov, E.; Wu, X.; Garcia, A.N.; Jacobson, J.R.; Yuan, J.X.; Garcia, J.G.; Black, S.M. Endothelial cell signaling and ventilator-induced lung injury: Molecular mechanisms, genomic analyses, and therapeutic targets. Am. J. Physiol. Lung Cell Mol. Physiol. 2017, 312, L452–L476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, H.; He, J.; Liu, J.; Zhang, X.; Yang, F.; Liu, P.; Wang, S. Alpha 1-antitrypsin ameliorates ventilator-induced lung injury in rats by inhibiting inflammatory responses and apoptosis. Exp. Biol. Med. 2018, 243, 87–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huppert, L.A.; Matthay, M.A.; Ware, L.B. Pathogenesis of acute respiratory distress syndrome. Semin. Respir. Crit Care Med. 2019, 40, 31–39. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, S.; Feinstein, S.I.; Dodia, C.; Sorokina, E.; Lien, Y.C.; Nguyen, S.; Debolt, K.; Speicher, D.; Fisher, A.B. Peroxiredoxin 6 phosphorylation and subsequent phospholipase A2 activity are required for agonist-mediated activation of NADPH oxidase in mouse pulmonary microvascular endothelium and alveolar macrophages. J. Biol. Chem. 2011, 286, 11696–11706. [Google Scholar] [CrossRef] [Green Version]
- Vazquez-Medina, J.P.; Dodia, C.; Weng, L.; Mesaros, C.; Blair, I.A.; Feinstein, S.I.; Chatterjee, S.; Fisher, A.B. The phospholipase A2 activity of peroxiredoxin 6 modulates NADPH oxidase 2 activation via lysophosphatidic acid receptor signaling in the pulmonary endothelium and alveolar macrophages. FASEB J. 2016, 30, 2885–2898. [Google Scholar] [CrossRef] [Green Version]
- Lee, I.; Dodia, C.; Chatterjee, S.; Zagorski, J.; Mesaros, C.; Blair, I.A.; Feinstein, S.I.; Jain, M.; Fisher, A.B. A novel nontoxic inhibitor of the activation of NADPH oxidase reduces reactive oxygen species production in mouse lung. J. Pharmacol. Exp. Ther. 2013, 345, 284–296. [Google Scholar] [CrossRef] [Green Version]
- Lee, I.; Dodia, C.; Chatterjee, S.; Feinstein, S.I.; Fisher, A.B. Protection against LPS-induced acute lung injury by a mechanism-based inhibitor of NADPH oxidase (type 2). Am. J. Physiol. Lung Cell Mol. Physiol. 2014, 306, L635–L644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benipal, B.; Feinstein, S.I.; Chatterjee, S.; Dodia, C.; Fisher, A.B. Inhibition of the phospholipase A2 activity of peroxiredoxin 6 prevents lung damage with exposure to hyperoxia. Redox Biol. 2015, 4, 321–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vazquez-Medina, J.P.; Tao, J.Q.; Patel, P.; Bannitz-Fernandes, R.; Dodia, C.; Sorokina, E.M.; Feinstein, S.I.; Chatterjee, S.; Fisher, A.B. Genetic inactivation of the phospholipase A2 activity of peroxiredoxin 6 in mice protects against LPS-induced acute lung injury. Am. J. Physiol. Lung Cell Mol. Physiol. 2019, 316, L656–L668. [Google Scholar] [CrossRef]
- Fisher, A.B. The serpentine path to a novel mechanism-based inhibitor of acute inflammatory lung injury. J. Appl Physiol. (1985) 2014, 116, 1521–1530. [Google Scholar] [CrossRef] [Green Version]
- Fisher, A.B.; Dodia, C.; Chander, A. Inhibition of lung calcium-independent phospholipase A2 by surfactant protein A. Am. J. Physiol. 1994, 267, L335–L341. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.Z.; Manevich, Y.; Baldwin, J.L.; Dodia, C.; Yu, K.; Feinstein, S.I.; Fisher, A.B. Interaction of surfactant protein A with peroxiredoxin 6 regulates phospholipase A2 activity. J. Biol. Chem. 2006, 281, 7515–7525. [Google Scholar] [CrossRef] [Green Version]
- Krishnaiah, S.Y.; Dodia, C.; Sorokina, E.M.; Li, H.; Feinstein, S.I.; Fisher, A.B. Binding sites for interaction of peroxiredoxin 6 with surfactant protein A. Biochim. Biophys. Acta 2016, 1864, 419–425. [Google Scholar] [CrossRef] [Green Version]
- Fisher, A.B.; Dodia, C.; Feinstein, S.I. Identification of small peptides that inhibit NADPH oxidase (Nox2) activation. Antioxidants 2018, 7, 181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fisher, A.B.; Dodia, C.; Chatterjee, S.; Feinstein, S.I. A peptide inhibitor of NADPH oxidase (NOX2) activation markedly decreases mouse lung injury and mortality following administration of lipopolysaccharide (LPS). Int. J. Mol. Sci. 2019, 20, 2395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, H.C.; Hellwig, K.; Rosseau, S.; Tschernig, T.; Schmiedl, A.; Gutbier, B.; Schmeck, B.; Hippenstiel, S.; Peters, H.; Morawietz, L.; et al. Simvastatin attenuates ventilator-induced lung injury in mice. Crit. Care 2010, 14, R143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, G.; Feinstein, S.I.; Wang, Y.; Dodia, C.; Fisher, D.; Yu, K.; Ho, Y.S.; Fisher, A.B. Comparison of glutathione peroxidase 1 and peroxiredoxin 6 in protection against oxidative stress in the mouse lung. Free Radic. Biol. Med. 2010, 49, 1172–1181. [Google Scholar] [CrossRef] [Green Version]
- Millius, A.; Weiner, O.D. Manipulation of neutrophil-like HL-60 cells for the study of directed cell migration. Methods Mol. Biol. 2010, 591, 147–158. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, S.; Tao, J.Q.; Johncola, A.; Guo, W.; Caporale, A.; Langham, M.C.; Wehrli, F.W. Acute exposure to e-cigarettes causes inflammation and pulmonary endothelial oxidative stress in nonsmoking, healthy young subjects. Am. J. Physiol. Lung Cell Mol. Physiol. 2019, 317, L155–L166. [Google Scholar] [CrossRef] [PubMed]
- Pietrofesa, R.A.; Woodruff, P.; Hwang, W.T.; Patel, P.; Chatterjee, S.; Albelda, S.M.; Christofidou-Solomidou, M. The synthetic lignan secoisolariciresinol diglucoside prevents asbestos-induced NLRP3 inflammasome activation in murine macrophages. Oxidative. Med. Cell Longev. 2017, 2017, 7395238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poelma, D.L.; Lachmann, B.; Haitsma, J.J.; Zimmermann, L.J.; van Iwaarden, J.F. Influence of phosphatidylglycerol on the uptake of liposomes by alveolar cells and on lung function. J. Appl. Physiol. (1985) 2005, 98, 1784–1791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, S.; Lear, T.B.; Jerome, J.A.; Rajbhandari, S.; Snavely, C.A.; Gulick, D.L.; Gibson, K.F.; Zou, C.; Chen, B.B.; Mallampalli, R.K. Lipopolysaccharide primes the NALP3 inflammasome by inhibiting its ubiquitination and degradation mediated by the SCFFBXL2 E3 Ligase. J. Biol. Chem. 2015, 290, 18124–18133. [Google Scholar] [CrossRef] [Green Version]
- Herrero, R.; Sanchez, G.; Lorente, J.A. New insights into the mechanisms of pulmonary edema in acute lung injury. Ann. Transl. Med. 2018, 6, 32. [Google Scholar] [CrossRef]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- Orndorff, R.L.; Hong, N.; Yu, K.; Feinstein, S.I.; Zern, B.J.; Fisher, A.B.; Muzykantov, V.R.; Chatterjee, S. NOX2 in lung inflammation: Quantum dot based in situ imaging of NOX2-mediated expression of vascular cell adhesion molecule-1. Am. J. Physiol. Lung Cell Mol. Physiol. 2014, 306, L260–L268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diebold, B.A.; Smith, S.M.; Li, Y.; Lambeth, J.D. NOX2 As a target for drug development: Indications, possible complications, and progress. Antioxid. Redox Signal. 2015, 23, 375–405. [Google Scholar] [CrossRef] [Green Version]
- Damodarasamy, M.; Zhang, M.; Dienger, K.; McCormack, F.X. Two rat surfactant protein A isoforms arise by a novel mechanism that includes alternative translation initiation. Biochemistry 2000, 39, 10189–10195. [Google Scholar] [CrossRef] [PubMed]
- Ambruso, D.R.; Ellison, M.A.; Thurman, G.W.; Leto, T.L. Peroxiredoxin 6 translocates to the plasma membrane during neutrophil activation and is required for optimal NADPH oxidase activity. Biochim. Biophys. Acta 2011, 1823, 306–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leavey, P.J.; Gonzalez-Aller, C.; Thurman, G.; Kleinberg, M.; Rinckel, L.; Ambruso, D.W.; Freeman, S.; Kuypers, F.A.; Ambruso, D.R. A 29-kDa protein associated with p67phox expresses both peroxiredoxin and phospholipase A2 activity and enhances superoxide anion production by a cell-free system of NADPH oxidase activity. J. Biol. Chem. 2002, 277, 45181–45187. [Google Scholar] [CrossRef] [Green Version]
- Ellison, M.A.; Thurman, G.W.; Ambruso, D.R. Phox activity of differentiated PLB-985 cells is enhanced, in an agonist specific manner, by the PLA2 activity of Prdx6-PLA2. Eur. J. Immunol. 2012, 42, 1609–1617. [Google Scholar] [CrossRef] [PubMed]
- Dipaolo, B.C.; Davidovich, N.; Kazanietz, M.G.; Margulies, S.S. Rac1 pathway mediates stretch response in pulmonary alveolar epithelial cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2013, 305, L141–L153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiumello, D.; Brioni, M. Severe hypoxemia: Which strategy to choose. Crit. Care 2016, 20, 132. [Google Scholar] [CrossRef] [Green Version]
- Fisher, A.B.; Beers, M.F. Hyperoxia and acute lung injury (letter). Am. J. Physiol. Lung Cell Mol. Physiol. 2008, 295, L1066. [Google Scholar] [CrossRef]
- Budinger, G.R.S.; Mutlu, G.M. Balancing the risks and benefits of oxygen therapy in critically III adults. Chest 2013, 143, 1151–1162. [Google Scholar] [CrossRef] [Green Version]
- Kallet, R.H.; Matthay, M.A. Hyperoxic acute lung injury. Respir. Care 2013, 58, 123–141. [Google Scholar] [CrossRef] [Green Version]
- Fisher, A.B.; Dodia, C.; Tan, Z.T.; Ayene, I.; Eckenhoff, R.G. Oxygen-dependent lipid peroxidation during lung ischemia. J. Clin. Investig. 1991, 88, 674–679. [Google Scholar] [CrossRef]
- Nin, N.; Muriel, A.; Penuelas, O.; Brochard, L.; Lorente, J.A.; Ferguson, N.D.; Raymondos, K.; Rios, F.; Violi, D.A.; Thille, A.W.; et al. Severe hypercapnia and outcome of mechanically ventilated patients with moderate or severe acute respiratory distress syndrome. Intensive Care Med. 2017, 43, 200–208. [Google Scholar] [CrossRef]
- Strosing, K.M.; Faller, S.; Gyllenram, V.; Engelstaedter, H.; Buerkle, H.; Spassov, S.; Hoetzel, A. Inhaled Anesthetics Exert Different protective properties in a mouse model of ventilator-induced lung injury. Anesth. Analg. 2016, 123, 143–151. [Google Scholar] [CrossRef]
- Tao, S.; de la Vega, M.R.; Quijada, H.; Wondrak, G.T.; Wang, T.; Garcia, J.G.; Zhang, D.D. Bixin protects mice against ventilation-induced lung injury in an NRF2-dependent manner. Sci. Rep. 2016, 6, 18760. [Google Scholar] [CrossRef] [Green Version]
- Hoegl, S.; Zwissler, B. Preventing ventilator-induced lung injury-what does the evidence say? J. Thorac. Dis. 2017, 9, 2259–2263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, X.; Sun, J.; Fei, A.; Gao, C.; Pan, S.; Wu, Z. Hydrogen sulfide treatment alleviated ventilator-induced lung injury through regulation of autophagy and endoplasmic reticulum stress. Int. J. Biol. Sci. 2019, 15, 2872–2884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bobba, C.M.; Fei, Q.; Shukla, V.; Lee, H.; Patel, P.; Putman, R.K.; Spitzer, C.; Tsai, M.; Wewers, M.D.; Lee, R.J.; et al. Nanoparticle delivery of microRNA-146a regulates mechanotransduction in lung macrophages and mitigates injury during mechanical ventilation. Nat. Commun. 2021, 12, 289. [Google Scholar] [CrossRef]
- Zhu, C.H.; Yu, J.; Wang, B.Q.; Nie, Y.; Wang, L.; Shan, S.Q. Dexmedetomidine reduces ventilator-induced lung injury via ERK1/2 pathway activation. Mol. Med. Rep. 2020, 22, 5378–5384. [Google Scholar] [CrossRef]
- Wang, Q.; Xu, G.X.; Tai, Q.H.; Wang, Y. Lipoxin A4 reduces ventilator-induced lung injury in rats with large-volume mechanical ventilation. Mediat. Inflamm. 2020, 2020, 6705985. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| aiPLA2 Activity nmol/hr/mg Protein | |
|---|---|
| Control (Liposomes only) | 8.81 ± 0.42 |
| PIP-2 (Encapsulated in Liposomes) | 1.55 ± 0.03 * |
| PIP-2 (No Liposomes) | 8.43 ± 0.40 |
| Composition * DPPC: PC: PG | PIP-2 | n | Activity ϯ nmol/mg prot/hr | Inhibition % |
|---|---|---|---|---|
| 50/25/10 | NO | 2 | 8.83 ± 0.10 | ----- |
| 50/25/10 | YES | 3 | 1.64 ± 0.07 | 81.4 |
| 0/75/10 | YES | 3 | 1.72 ± 0.10 | 80.5 |
| 75/0/10 | YES | 2 | 1.84 ± 0.02 | 79.2 |
| 55/30/10 | YES | 2 | 2.51 ± 0.12 | 71.7 |
| Condition | # of cells in BALf (×104/g body wt) | Total Protein in BALf (mg/g Body wt | Wet/Dry Weight Ratio of Lung | TBARS pmol/mg prot. | 8—Isoprotanes pmol/mg prot | Protein Carbonyls nmol/mg prot |
|---|---|---|---|---|---|---|
| Control (non-ventilated) | 0.97 ± 0.12 | 76.9 ± 4.2 | 5.61 ± 0.04 | 74.6 ± 4.6 | 33.1 ± 6.0 | 5.63 ± 0.48 |
| Low tidal volume | 1.03 ± 0.06 | 80.1 ± 4.4 | 6.02 ± 0.26 | 81.7 ± 3.0 | 37.8 ± 4.2 | 5.90 ± 0.46 |
| Low tidal volume + PIP-2 | 1.01 ± 0.08 | 78.8 ± 3.6 | 5.98 ± 0.22 | 79.9 ± 4.2 | 36.2 ± 2.4 | 5.82 ± 0.48 |
| High tidal volume (VILI) * | 19.6 ± 2.84 | 235 ± 20 | 10.8 ± 0.42 | 530 ± 14.4 | 174 ± 9.8 | 22.3 ± 1.56 |
| VILI + PIP-2 (2 µg/g) ϯ | 8.4 ± 0.62 | 102 ± 16.2 | 8.14 ± 0.22 | 206 ± 24 | 75.9 ± 2.0 | 8.85 ± 0.84 |
| VILI + PIP-2 (10 µg/g) ϯ | 8.1 ± 0.46 | 107 ± 16.2 | 7.62 ± 0.36 | 202 ± 12.4 | 76.5 ± 2.4 | 8.12 ± 0.34 |
| Condition | # of Cells in BALf (×104/g body wt) | Total Protein in BALf (mg/g Body wt | Wet/Dry Weight Ratio of Lung | TBARS pmol/mg prot. | 8—Isoprostanes pmol/mg prot | Protein Carbonyls nmol/mg prot |
|---|---|---|---|---|---|---|
| Control (non-ventilated) | 1.04 ± 0.14 | 82.4 ± 10.0 | 5.94 ± 0.50 | 80.5 ± 3.2 | 31.5 ± 5.4 | 5.48 ± 0.58 |
| VILI * | 18.9 ± 1.16 | 218 ± 19 | 10.4 ± 0.70 | 512 ± 26 | 178 ± 34 | 21.1 ± 1.32 |
| VILI + PIP-2 ϯ | 8.17 ± 0.28 | 103 ± 5.6 | 7.4 ± 0.24 | 186 ± 36 | 81.3 ± 7.2 | 8.44 ± 1.38 |
| Condition | # of Cells in BALf (×104/g Body wt) | Total Protein in BALf (mg/g Body wt | Wet/Dry Weight Ratio of Lung | TBARS pmol/mg prot. | 8—Isoprostanes pmol/mg prot | Protein Carbonyls nmol/mg prot |
|---|---|---|---|---|---|---|
| Control (non-ventilated) | 1.01 ± 0.06 | 81.7 ± 10.6 | 5.72 ± 0.24 | 81.7 ± 2.0 | 39.6 ± 1.82 | 5.83 ± 0.24 |
| VILI (no PIP-2) * | 17.7± 0.46 | 222 ± 16 | 9.85 ± 0.54 | 479 ± 28 | 183 ± 22 | 20.6 ± 2.8 |
| VILI + PIP-2 ϯ | 7.67 ± 0.58 | 93.6 ± 7.0 | 7.49 ± 0.34 | 180 ± 36 | 85.2 ± 5.6 | 8.87 ± 0.20 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fisher, A.B.; Dodia, C.; Chatterjee, S. A Peptide Inhibitor of Peroxiredoxin 6 Phospholipase A2 Activity Significantly Protects against Lung Injury in a Mouse Model of Ventilator Induced Lung Injury (VILI). Antioxidants 2021, 10, 925. https://doi.org/10.3390/antiox10060925
Fisher AB, Dodia C, Chatterjee S. A Peptide Inhibitor of Peroxiredoxin 6 Phospholipase A2 Activity Significantly Protects against Lung Injury in a Mouse Model of Ventilator Induced Lung Injury (VILI). Antioxidants. 2021; 10(6):925. https://doi.org/10.3390/antiox10060925
Chicago/Turabian StyleFisher, Aron B., Chandra Dodia, and Shampa Chatterjee. 2021. "A Peptide Inhibitor of Peroxiredoxin 6 Phospholipase A2 Activity Significantly Protects against Lung Injury in a Mouse Model of Ventilator Induced Lung Injury (VILI)" Antioxidants 10, no. 6: 925. https://doi.org/10.3390/antiox10060925