
A Synthetic SOD/Catalase Mimic Compound for the Treatment of ALS


Abstract
:
1. Introduction
2. Materials and Methods
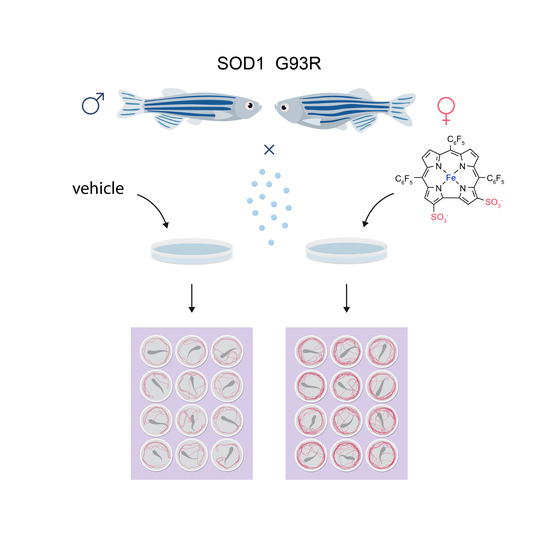
2.1. Zebrafish
2.2. Drug Administration Protocol
2.3. Toxicity Evaluation
2.4. Motor Performance of SOD1 G93R Larvae
2.5. Statistics
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.-X.; et al. Mutations in Cu/Zn Superoxide Dismutase Gene Are Associated with Familial Amyotrophic Lateral Sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef] [PubMed]
- Masrori, P.; Van Damme, P. Amyotrophic Lateral Sclerosis: A Clinical Review. Eur. J. Neurol. 2020, 27, 1918–1929. [Google Scholar] [CrossRef] [PubMed]
- Huai, J.; Zhang, Z. Structural Properties and Interaction Partners of Familial ALS-Associated SOD1 Mutants. Front. Neurol. 2019, 527. [Google Scholar] [CrossRef] [Green Version]
- Sathasivam, S.; Ince, P.G.; Shaw, P.J. Apoptosis in Amyotrophic Lateral Sclerosis: A Review of the Evidence. Neuropathol. Appl. Neurobiol. 2001, 27, 257–274. [Google Scholar] [CrossRef]
- Obrador, E.; Salvador, R.; López-Blanch, R.; Jihad-Jebbar, A.; Vallés, S.L.; Estrela, J.M. Oxidative Stress, Neuroinflammation and Mitochondria in the Pathophysiology of Amyotrophic Lateral Sclerosis. Antioxidants 2020, 9, 901. [Google Scholar] [CrossRef] [PubMed]
- Cassina, P.; Cassina, A.; Pehar, M.; Castellanos, R.; Gandelman, M.; de León, A.; Robinson, K.M.; Mason, R.P.; Beckman, J.S.; Barbeito, L.; et al. Mitochondrial Dysfunction in SOD1G93A-Bearing Astrocytes Promotes Motor Neuron Degeneration: Prevention by Mitochondrial-Targeted Antioxidants. J. Neurosci. 2008, 28, 4115–4122. [Google Scholar] [CrossRef] [Green Version]
- Burch, P.M.; Heintz, N.H. Redox Regulation of Cell-Cycle Re-Entry: Cyclin D1 as a Primary Target for the Mitogenic Effects of Reactive Oxygen and Nitrogen Species. Antioxid. Redox Signal. 2005, 7, 741–751. [Google Scholar] [CrossRef]
- Oktyabrsky, O.N.; Smirnova, G.V. Redox Regulation of Cellular Functions. Biochem. 2007, 72, 132–145. [Google Scholar] [CrossRef]
- Byrne, D.P.; Shrestha, S.; Galler, M.; Cao, M.; Daly, L.A.; Campbell, A.E.; Eyers, C.E.; Veal, E.A.; Kannan, N.; Eyers, P.A. Aurora A Regulation by Reversible Cysteine Oxidation Reveals Evolutionarily Conserved Redox Control of Ser/Thr Protein Kinase Activity. Sci. Signal. 2020, 13, eaax2713. [Google Scholar] [CrossRef]
- Shrestha, S.; Katiyar, S.; Sanz-Rodriguez, C.E.; Kemppinen, N.R.; Kim, H.W.; Kadirvelraj, R.; Panagos, C.; Keyhaninejad, N.; Colonna, M.; Chopra, P.; et al. A Redox-Active Switch in Fructosamine-3-Kinases Expands the Regulatory Repertoire of the Protein Kinase Superfamily. Sci. Signal. 2020, 13, eaax6313. [Google Scholar] [CrossRef]
- Pryor, W.A.; Squadrito, G.L. The Chemistry of Peroxynitrite: A Product from the Reaction of Nitric Oxide with Superoxide. Am. J. Physiol. 1995, 268, L699–L722. [Google Scholar] [CrossRef]
- Yang, S.; Lian, G. ROS and Diseases: Role in Metabolism and Energy Supply. Mol. Cell. Biochem. 2020, 467, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Casas, A.I.; Dao, V.T.-V.; Daiber, A.; Maghzal, G.J.; Di Lisa, F.; Kaludercic, N.; Leach, S.; Cuadrado, A.; Jaquet, V.; Seredenina, T.; et al. Reactive Oxygen-Related Diseases: Therapeutic Targets and Emerging Clinical Indications. Antioxid. Redox Signal. 2015, 23, 1171–1185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abe, K.; Pan, L.H.; Watanabe, M.; Konno, H.; Kato, T.; Itoyama, Y. Upregulation of Protein-Tyrosine Nitration in the Anterior Horn Cells of Amyotrophic Lateral Sclerosis. Neurol. Res. 1997, 19, 124–128. [Google Scholar] [CrossRef] [PubMed]
- Beal, M.F.; Ferrante, R.J.; Browne, S.E.; Matthews, R.T.; Kowall, N.W.; Brown, R.H.J. Increased 3-Nitrotyrosine in Both Sporadic and Familial Amyotrophic Lateral Sclerosis. Ann. Neurol. 1997, 42, 644–654. [Google Scholar] [CrossRef]
- Radi, R. Peroxynitrite, a Stealthy Biological Oxidant. J. Biol. Chem. 2013, 288, 26464–26472. [Google Scholar] [CrossRef] [Green Version]
- Richeson, C.E.; Mulder, P.; Bowry, V.W.; Ingold, K.U. The Complex Chemistry of Peroxynitrite Decomposition: New Insights1. J. Am. Chem. Soc. 1998, 120, 7211–7219. [Google Scholar] [CrossRef]
- Calingasan, N.Y.; Chen, J.; Kiaei, M.; Beal, M.F. Beta-Amyloid 42 Accumulation in the Lumbar Spinal Cord Motor Neurons of Amyotrophic Lateral Sclerosis Patients. Neurobiol. Dis. 2005, 19, 340–347. [Google Scholar] [CrossRef]
- Shaw, P.J.; Ince, P.G.; Falkous, G.; Mantle, D. Oxidative Damage to Protein in Sporadic Motor Neuron Disease Spinal Cord. Ann. Neurol. 1995, 38, 691–695. [Google Scholar] [CrossRef]
- Ferrante, R.J.; Browne, S.E.; Shinobu, L.A.; Bowling, A.C.; Baik, M.J.; MacGarvey, U.; Kowall, N.W.; Brown, R.H.J.; Beal, M.F. Evidence of Increased Oxidative Damage in Both Sporadic and Familial Amyotrophic Lateral Sclerosis. J. Neurochem. 1997, 69, 2064–2074. [Google Scholar] [CrossRef]
- Smith, R.G.; Henry, Y.K.; Mattson, M.P.; Appel, S.H. Presence of 4-Hydroxynonenal in Cerebrospinal Fluid of Patients with Sporadic Amyotrophic Lateral Sclerosis. Ann. Neurol. 1998, 44, 696–699. [Google Scholar] [CrossRef] [PubMed]
- Simpson, E.P.; Henry, Y.K.; Henkel, J.S.; Smith, R.G.; Appel, S.H. Increased Lipid Peroxidation in Sera of ALS Patients: A Potential Biomarker of Disease Burden. Neurology 2004, 62, 1758–1765. [Google Scholar] [CrossRef] [PubMed]
- Ihara, Y.; Nobukuni, K.; Takata, H.; Hayabara, T. Oxidative Stress and Metal Content in Blood and Cerebrospinal Fluid of Amyotrophic Lateral Sclerosis Patients with and without a Cu, Zn-Superoxide Dismutase Mutation. Neurol. Res. 2005, 27, 105–108. [Google Scholar] [CrossRef] [PubMed]
- Tohgi, H.; Abe, T.; Yamazaki, K.; Murata, T.; Ishizaki, E.; Isobe, C. Remarkable Increase in Cerebrospinal Fluid 3-Nitrotyrosine in Patients with Sporadic Amyotrophic Lateral Sclerosis. Ann. Neurol. 1999, 46, 129–131. [Google Scholar] [CrossRef]
- Weishaupt, J.H.; Bartels, C.; Pölking, E.; Dietrich, J.; Rohde, G.; Poeggeler, B.; Mertens, N.; Sperling, S.; Bohn, M.; Hüther, G.; et al. Reduced Oxidative Damage in ALS by High-Dose Enteral Melatonin Treatment. J. Pineal Res. 2006, 41, 313–323. [Google Scholar] [CrossRef]
- Mitsumoto, H.; Santella, R.M.; Liu, X.; Bogdanov, M.; Zipprich, J.; Wu, H.-C.; Mahata, J.; Kilty, M.; Bednarz, K.; Bell, D.; et al. Oxidative Stress Biomarkers in Sporadic ALS. Amyotroph. lateral Scler. Off. Publ. World Fed. Neurol. Res. Gr. Mot. Neuron Dis. 2008, 9, 177–183. [Google Scholar] [CrossRef]
- Nagase, M.; Yamamoto, Y.; Miyazaki, Y.; Yoshino, H. Increased Oxidative Stress in Patients with Amyotrophic Lateral Sclerosis and the Effect of Edaravone Administration. Redox Rep. 2016, 21, 104–112. [Google Scholar] [CrossRef]
- Mahammed, A.; Gross, Z. Highly Efficient Catalase Activity of Metallocorroles. Chem. Commun. 2010, 46, 7040–7042. [Google Scholar] [CrossRef]
- Eckshtain, M.; Zilbermann, I.; Mahammed, A.; Saltsman, I.; Okun, Z.; Maimon, E.; Cohen, H.; Meyerstein, D.; Gross, Z. Superoxide Dismutase Activity of Corrole Metal Complexes. Dalt. Trans. 2009, 38, 7879–7882. [Google Scholar] [CrossRef]
- Mahammed, A.; Gross, Z. Iron and Manganese Corroles Are Potent Catalysts for the Decomposition of Peroxynitrite. Angew. Chem. Int. Ed. Engl. 2006, 45, 6544–6547. [Google Scholar] [CrossRef]
- Haber, A.; Gross, Z. Catalytic Antioxidant Therapy by Metallodrugs: Lessons from Metallocorroles. Chem. Commun. 2015, 51, 5812–5827. [Google Scholar] [CrossRef]
- Okun, Z.; Kupershmidt, L.; Amit, T.; Mandel, S.; Bar-Am, O.; Youdim, M.B.H.; Gross, Z. Manganese Corroles Prevent Intracellular Nitration and Subsequent Death of Insulin-Producing Cells. ACS Chem. Biol. 2009, 4, 910–914. [Google Scholar] [CrossRef] [PubMed]
- Haber, A. Metallocorroles for Attenuation of Atherosclerosis; Springer: Berlin/Heidelberg, Germany, 2012. [Google Scholar]
- Haber, A.; Angel, I.; Mahammed, A.; Gross, Z. Combating Diabetes Complications by 1-Fe, a Corrole-Based Catalytic Antioxidant. J. Diabetes Complicat. 2013, 27, 316–321. [Google Scholar] [CrossRef]
- Haber, A.; Gross, Z. Protecting the Beneficial Functionality of Lipoproteins by 1-Fe, a Corrole-Based Catalytic Antioxidant. Chem. Sci. 2011, 2, 295–302. [Google Scholar] [CrossRef]
- Haber, A.; Mahammed, A.; Fuhrman, B.; Volkova, N.; Coleman, R.; Hayek, T.; Aviram, M.; Gross, Z. Amphiphilic/Bipolar Metallocorroles That Catalyze the Decomposition of Reactive Oxygen and Nitrogen Species, Rescue Lipoproteins from Oxidative Damage, and Attenuate Atherosclerosis in Mice. Angew. Chem. Int. Ed. Engl. 2008, 47, 7896–7900. [Google Scholar] [CrossRef] [PubMed]
- Kupershmidt, L.; Okun, Z.; Amit, T.; Mandel, S.; Saltsman, I.; Mahammed, A.; Bar-Am, O.; Gross, Z.; Youdim, M.B.H. Metallocorroles as Cytoprotective Agents against Oxidative and Nitrative Stress in Cellular Models of Neurodegeneration. J. Neurochem. 2010, 113, 363–373. [Google Scholar] [CrossRef]
- Soll, M.; Bar am, O.; Mahammed, A.; Saltsman, I.; Mandel, S.; Youdim, M.B.H.; Gross, Z. Neurorescue by a ROS Decomposition Catalyst. ACS Chem. Neurosci. 2016, 7, 1374–1382. [Google Scholar] [CrossRef]
- Kanamori, A.; Catrinescu, M.-M.; Mahammed, A.; Gross, Z.; Levin, L.A. Neuroprotection against Superoxide Anion Radical by Metallocorroles in Cellular and Murine Models of Optic Neuropathy. J. Neurochem. 2010, 114, 488–498. [Google Scholar] [CrossRef] [Green Version]
- Gomes, L.M.F.; Mahammed, A.; Prosser, K.E.; Smith, J.R.; Silverman, M.A.; Walsby, C.J.; Gross, Z.; Storr, T. A Catalytic Antioxidant for Limiting Amyloid-Beta Peptide Aggregation and Reactive Oxygen Species Generation. Chem. Sci. 2018, 10, 1634–1643. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, G.A.B.; Drapeau, P. Calcium Channel Agonists Protect against Neuromuscular Dysfunction in a Genetic Model of TDP-43 Mutation in ALS. J. Neurosci. 2013, 33, 1741. [Google Scholar] [CrossRef] [Green Version]
- Ramesh, T.; Lyon, A.N.; Pineda, R.H.; Wang, C.; Janssen, P.M.L.; Canan, B.D.; Burghes, A.H.M.; Beattie, C.E. A Genetic Model of Amyotrophic Lateral Sclerosis in Zebrafish Displays Phenotypic Hallmarks of Motoneuron Disease. Dis. Model. Mech. 2010, 3, 652–662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldshtein, H.; Muhire, A.; Petel Légaré, V.; Pushett, A.; Rotkopf, R.; Shefner, J.M.; Peterson, R.T.; Armstrong, G.A.B.; Russek- Blum, N. Efficacy of Ciprofloxacin/Celecoxib Combination in Zebrafish Models of Amyotrophic Lateral Sclerosis. Ann. Clin. Transl. Neurol. 2020, 7, 1883–1897. [Google Scholar] [CrossRef]
- Parng, C.; Seng, W.L.; Semino, C.; McGrath, P. Zebrafish: A Preclinical Model for Drug Screening. Assay Drug Dev. Technol. 2002, 1, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Wiley, D.S.; Redfield, S.E.; Zon, L.I. Chemical Screening in Zebrafish for Novel Biological and Therapeutic Discovery. Methods Cell Biol. 2017, 138, 651–679. [Google Scholar]
- Westerfield, M. The Zebrafish Book: A Guide for the Laboratory Use of Zebrafish; Institute of Neuroscience, University of Oregon Press: Eugene, OR, USA.
- Hill, A.J.; Teraoka, H.; Heideman, W.; Peterson, R.E. Zebrafish as a Model Vertebrate for Investigating Chemical Toxicity. Toxicol. Sci. 2005, 86, 6–19. [Google Scholar] [CrossRef] [Green Version]
- Haynes, C.M.; Titus, E.A.; Cooper, A.A. Degradation of Misfolded Proteins Prevents ER-Derived Oxidative Stress and Cell Death. Mol. Cell 2004, 15, 767–776. [Google Scholar] [CrossRef] [PubMed]
- Uddin, M.S.; Tewari, D.; Sharma, G.; Kabir, M.T.; Barreto, G.E.; Bin-Jumah, M.N.; Perveen, A.; Abdel-Daim, M.M.; Ashraf, G.M. Molecular Mechanisms of ER Stress and UPR in the Pathogenesis of Alzheimer’s Disease. Mol. Neurobiol. 2020, 57, 2902–2919. [Google Scholar] [CrossRef]
- Hetz, C.; Zhang, K.; Kaufman, R.J. Mechanisms, Regulation and Functions of the Unfolded Protein Response. Nat. Rev. Mol. Cell Biol. 2020, 21, 421–438. [Google Scholar] [CrossRef]
- Polymenidou, M.; Cleveland, D.W. The Seeds of Neurodegeneration: Prion-like Spreading in ALS. Cell 2011, 147, 498–508. [Google Scholar] [CrossRef] [Green Version]
- D’Amico, E.; Factor-Litvak, P.; Santella, R.M.; Mitsumoto, H. Clinical Perspective on Oxidative Stress in Sporadic Amyotrophic Lateral Sclerosis. Free Radic. Biol. Med. 2013, 65, 509–527. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Li, C.; Chen, X.; Li, S.; Shang, H. Abnormal Serum Iron-Status Indicator Changes in Amyotrophic Lateral Sclerosis (ALS) Patients: A Meta-Analysis. Front. Neurol. 2020, 380. [Google Scholar] [CrossRef] [PubMed]
- Kwan, J.Y.; Jeong, S.Y.; Van Gelderen, P.; Deng, H.-X.; Quezado, M.M.; Danielian, L.E.; Butman, J.A.; Chen, L.; Bayat, E.; Russell, J.; et al. Iron Accumulation in Deep Cortical Layers Accounts for MRI Signal Abnormalities in ALS: Correlating 7 Tesla MRI and Pathology. PLoS ONE 2012, 7, e35241. [Google Scholar] [CrossRef] [PubMed]
- Gehrmann, J.; Matsumoto, Y.; Kreutzberg, G.W. Microglia: Intrinsic Immuneffector Cell of the Brain. Brain Res. Rev. 1995, 20, 269–287. [Google Scholar] [CrossRef]
- Saha, P.P.; Vishwanathan, V.; Bankapalli, K.; D’Silva, P. Iron-Sulfur Protein Assembly in Human Cells BT - Reviews of Physiology, Biochemistry and Pharmacology Vol. 174; Nilius, B., de Tombe, P., Gudermann, T., Jahn, R., Lill, R., Petersen, O.H., Eds.; Springer International Publishing: Cham, Switzerland, 2018. [Google Scholar]
- Comporti, M.; Signorini, C.; Buonocore, G.; Ciccoli, L. Iron Release, Oxidative Stress and Erythrocyte Ageing. Free Radic. Biol. Med. 2002, 32, 568–576. [Google Scholar] [CrossRef]
- Trotti, D.; Danbolt, N.C.; Volterra, A. Glutamate Transporters Are Oxidant-Vulnerable: A Molecular Link between Oxidative and Excitotoxic Neurodegeneration? Trends Pharmacol. Sci. 1998, 19, 328–334. [Google Scholar] [CrossRef]
- Rojas, F.; Gonzalez, D.; Cortes, N.; Ampuero, E.; Hernández, D.E.; Fritz, E.; Abarzua, S.; Martinez, A.; Elorza, A.A.; Alvarez, A.; et al. Reactive Oxygen Species Trigger Motoneuron Death in Non-Cell-Autonomous Models of ALS through Activation of c-Abl Signaling. Front. Cell. Neurosci. 2015, 203. [Google Scholar] [CrossRef] [Green Version]
- D’Ambrosi, N.; Cozzolino, M.; Carrì, M.T. Neuroinflammation in Amyotrophic Lateral Sclerosis: Role of Redox (Dys)Regulation. Antioxid. Redox Signal. 2017, 29, 15–36. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Material | Concentration | Survival % (I treatment; t = 0) | Survival % (I treatment; t = 24 h) | Survival % (II treatment; t = 72 h) | Toxicity | ||||
|---|---|---|---|---|---|---|---|---|---|
| WT | mSod1 | WT | mSod1 | WT | mSod1 | WT | mSod1 | ||
| 1-Fe | 100 μM | 100 | 100 | 0 | 0 | 0 | 0 | death | death |
| 10 μM | 100 | 100 | 100 | 100 | 40 | 45 | decreased heart rate, severe behavioral toxicity with significantly slower and weaker movements and death | decreased heart rate, severe behavioral toxicity with significantly slower and weaker movements and death | |
| 5 μM | 100 | 100 | 100 | 100 | 100 | 100 | mild behavioral toxicity | no obvious drug induced effects | |
| 1 μM | 100 | 100 | 100 | 100 | 100 | 100 | no obvious drug induced effects | no obvious drug induced effects | |
| 0.05 μM | 100 | 100 | 100 | 100 | 100 | 100 | no obvious drug induced effects | no obvious drug induced effects | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Soll, M.; Goldshtein, H.; Rotkopf, R.; Russek-Blum, N.; Gross, Z. A Synthetic SOD/Catalase Mimic Compound for the Treatment of ALS. Antioxidants 2021, 10, 827. https://doi.org/10.3390/antiox10060827
Soll M, Goldshtein H, Rotkopf R, Russek-Blum N, Gross Z. A Synthetic SOD/Catalase Mimic Compound for the Treatment of ALS. Antioxidants. 2021; 10(6):827. https://doi.org/10.3390/antiox10060827
Chicago/Turabian StyleSoll, Matan, Hagit Goldshtein, Ron Rotkopf, Niva Russek-Blum, and Zeev Gross. 2021. "A Synthetic SOD/Catalase Mimic Compound for the Treatment of ALS" Antioxidants 10, no. 6: 827. https://doi.org/10.3390/antiox10060827