
Melatonin Successfully Rescues the Hippocampal Molecular Machinery and Enhances Anti-oxidative Activity Following Early-Life Sleep Deprivation Injury

 ,
, 
Abstract
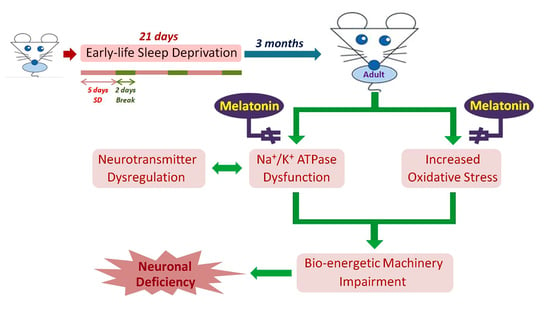
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Experimental Animals
2.2. Sleep Deprivation Procedure
2.3. Perfusion and Tissue Preparation
2.4. Immunohistochemistry
2.5. Nissl Staining
2.6. Quantitative Image Analysis for Light Microscopic (LM) Study
2.7. Western Blotting
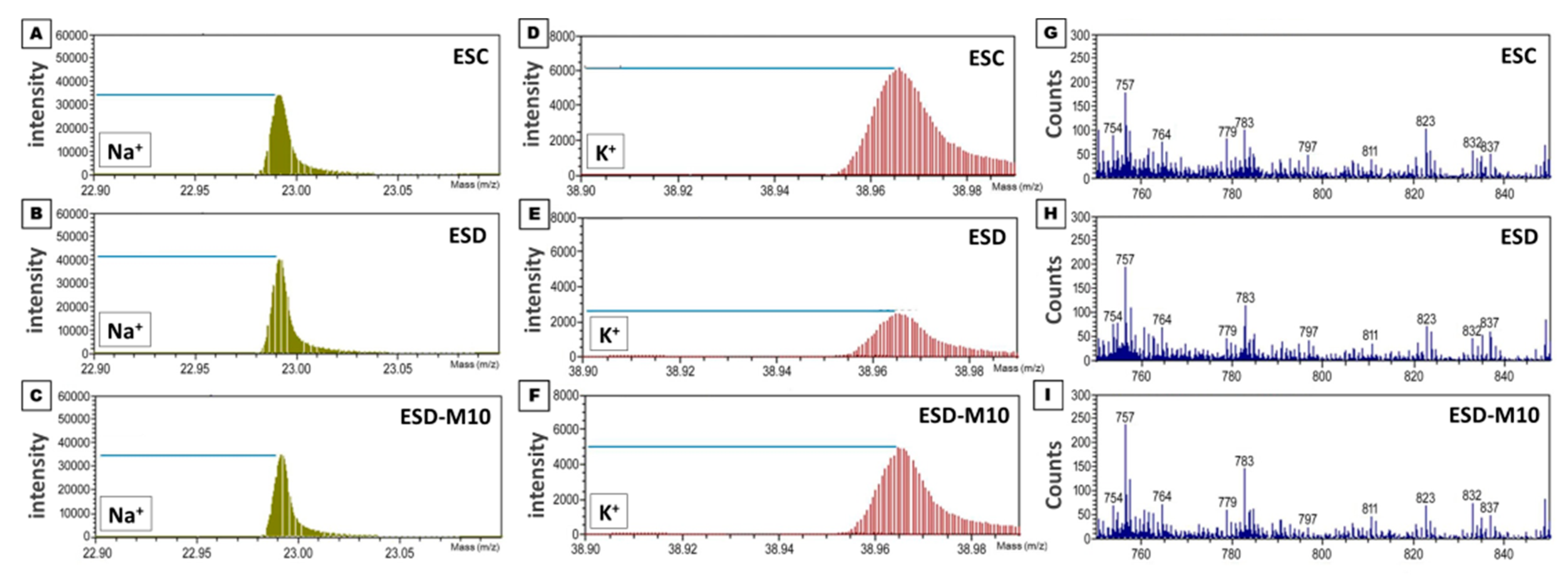
2.8. TOF-SIMS Analysis
2.9. Statistical Analysis
3. Results
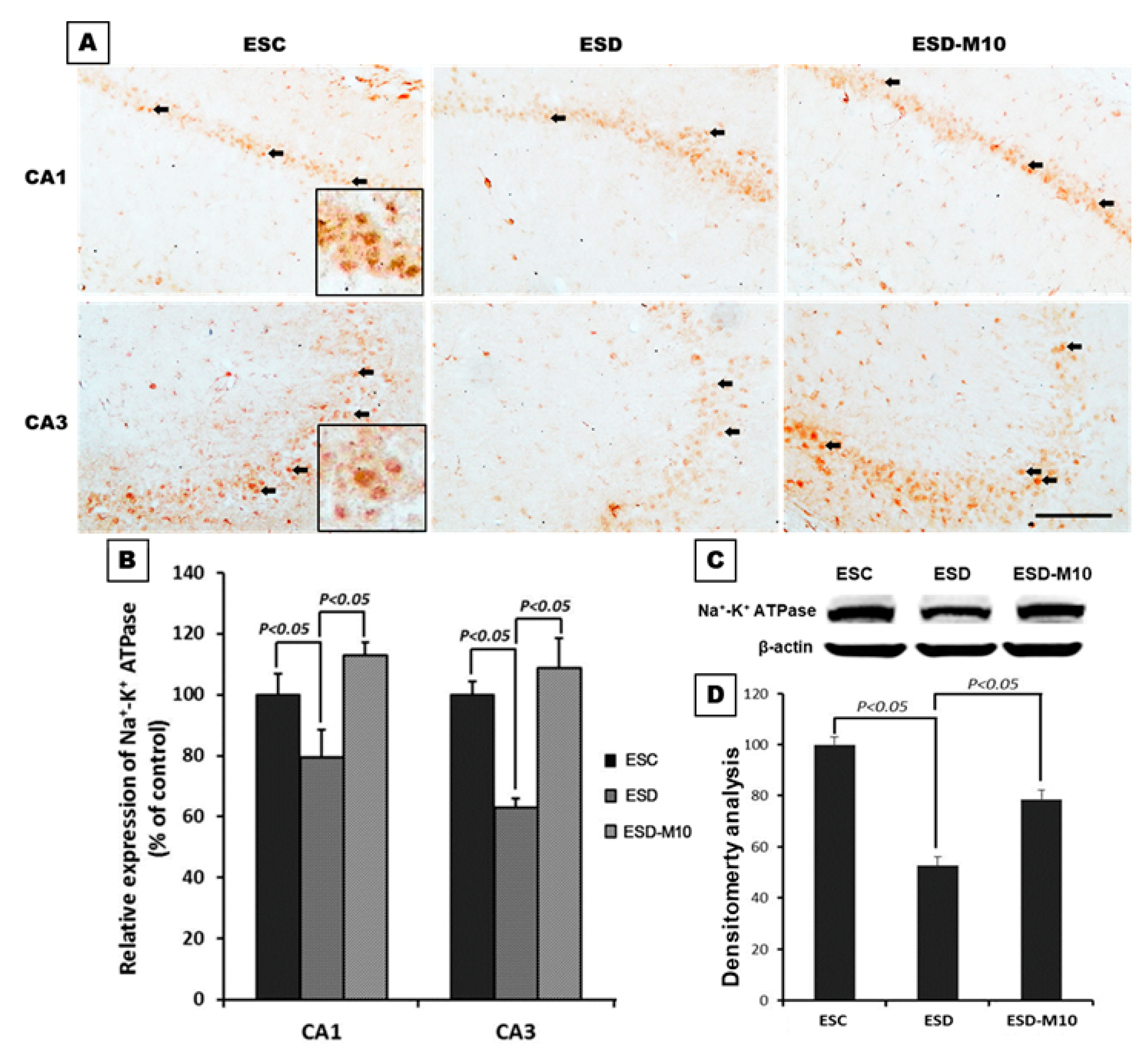
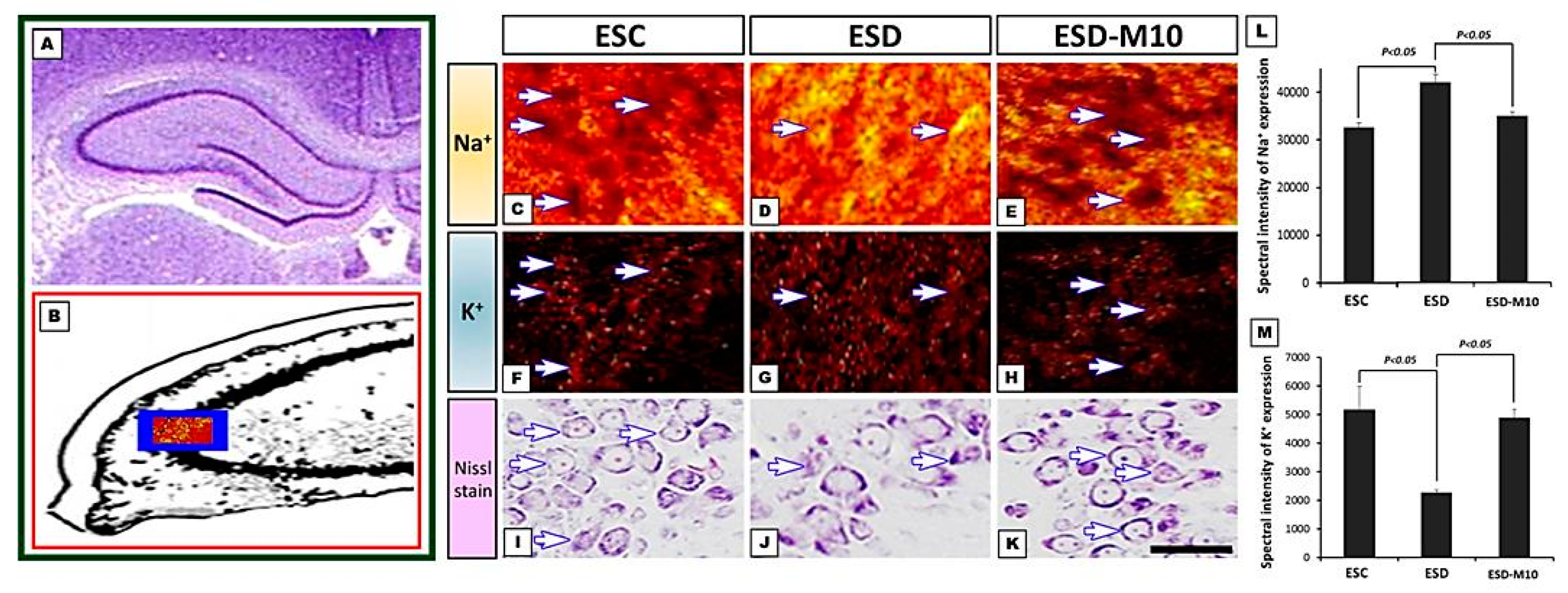
3.1. Melatonin Preserves Hippocampus Ion Homeostasis
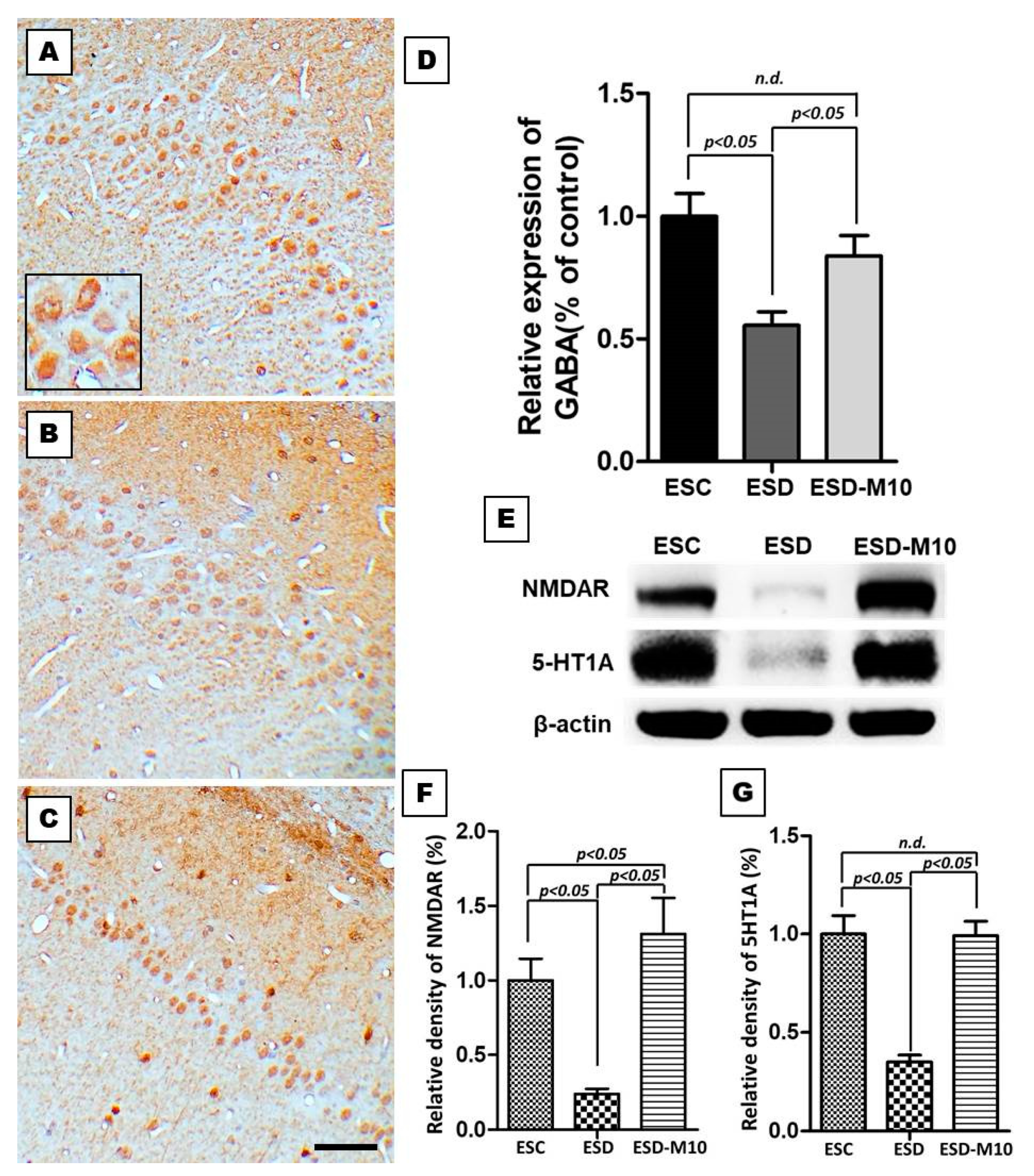
3.2. Melatonin Effectively Enhances the Expressions of GABA, NMDAR, and 5-HT1 A in the Hippocampus
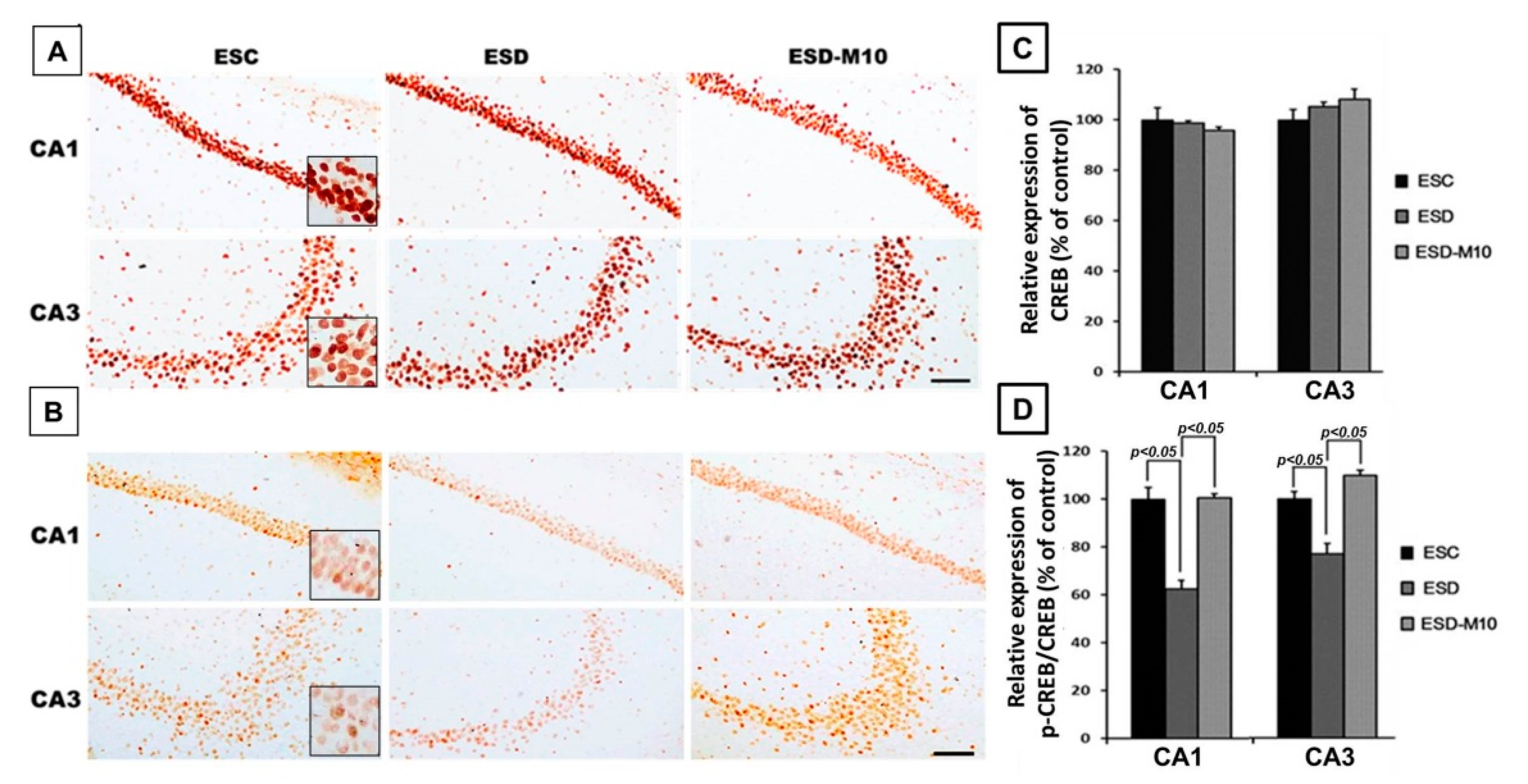
3.3. Melatonin Increases p-CREB Protein Levels in Hippocampal Neurons in ESD Rats
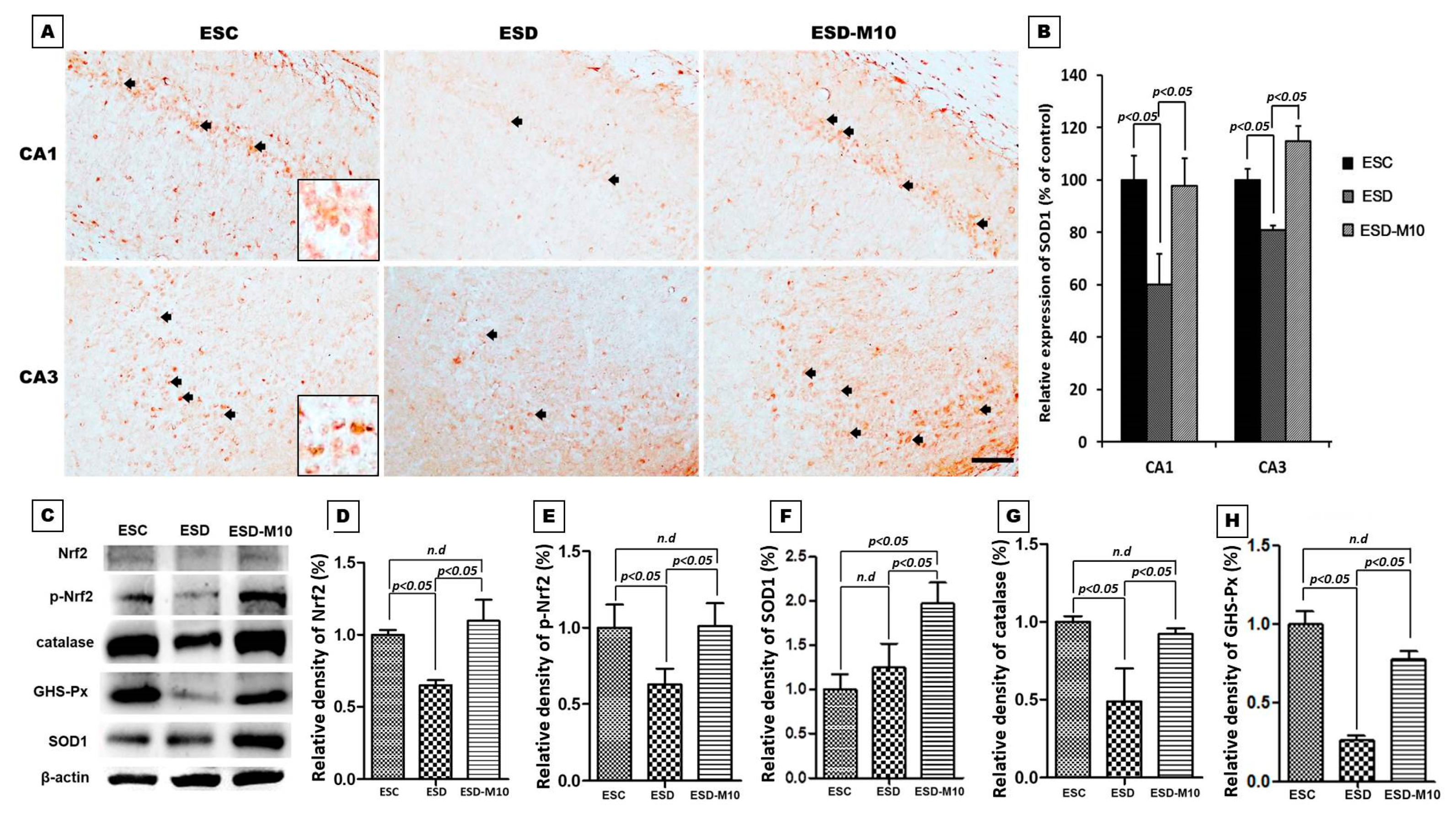
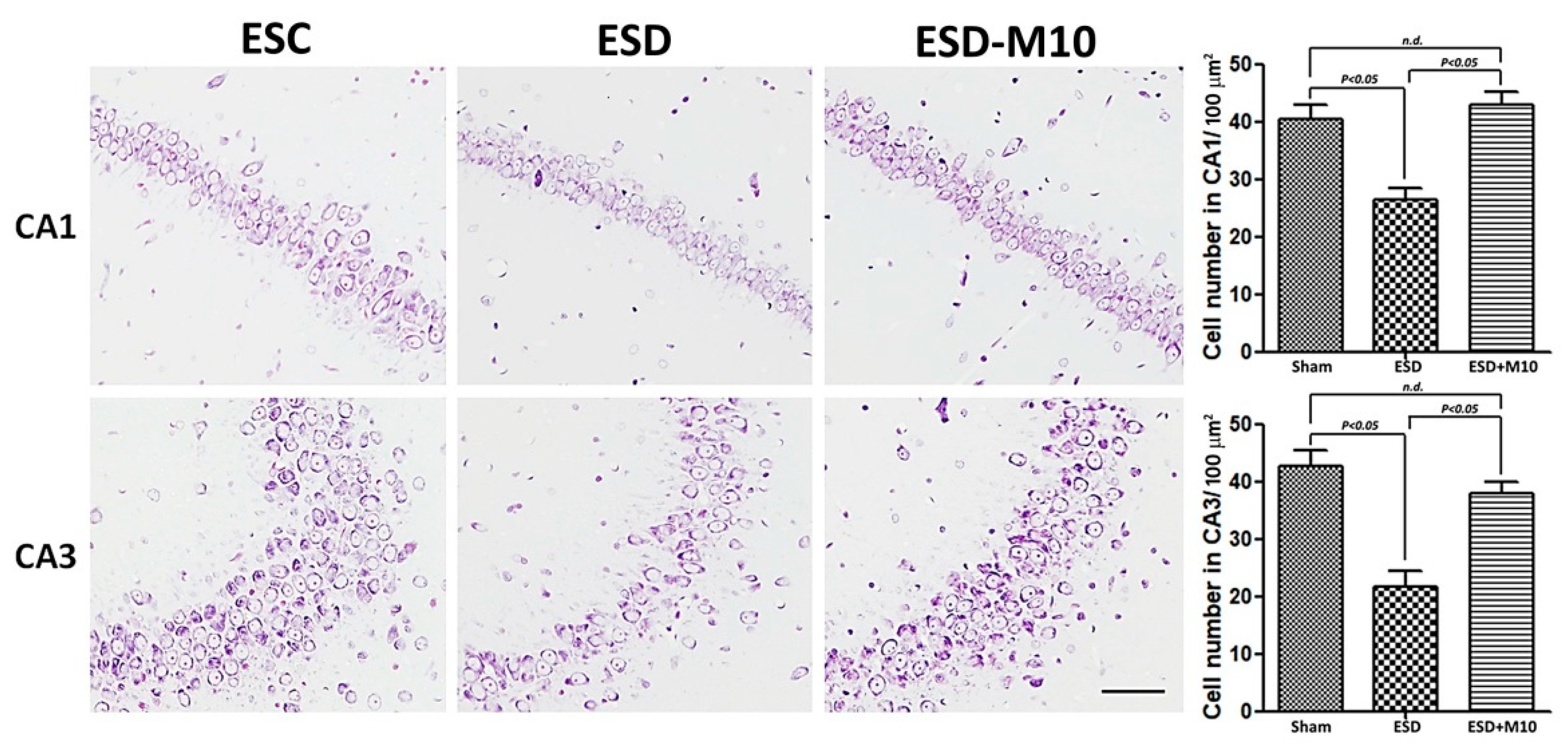
3.4. Melatonin Effectively Promotes p-Nrf2, Anti-oxidative Enzyme Activities, and Neuronal Cell Survival in the Hippocampus
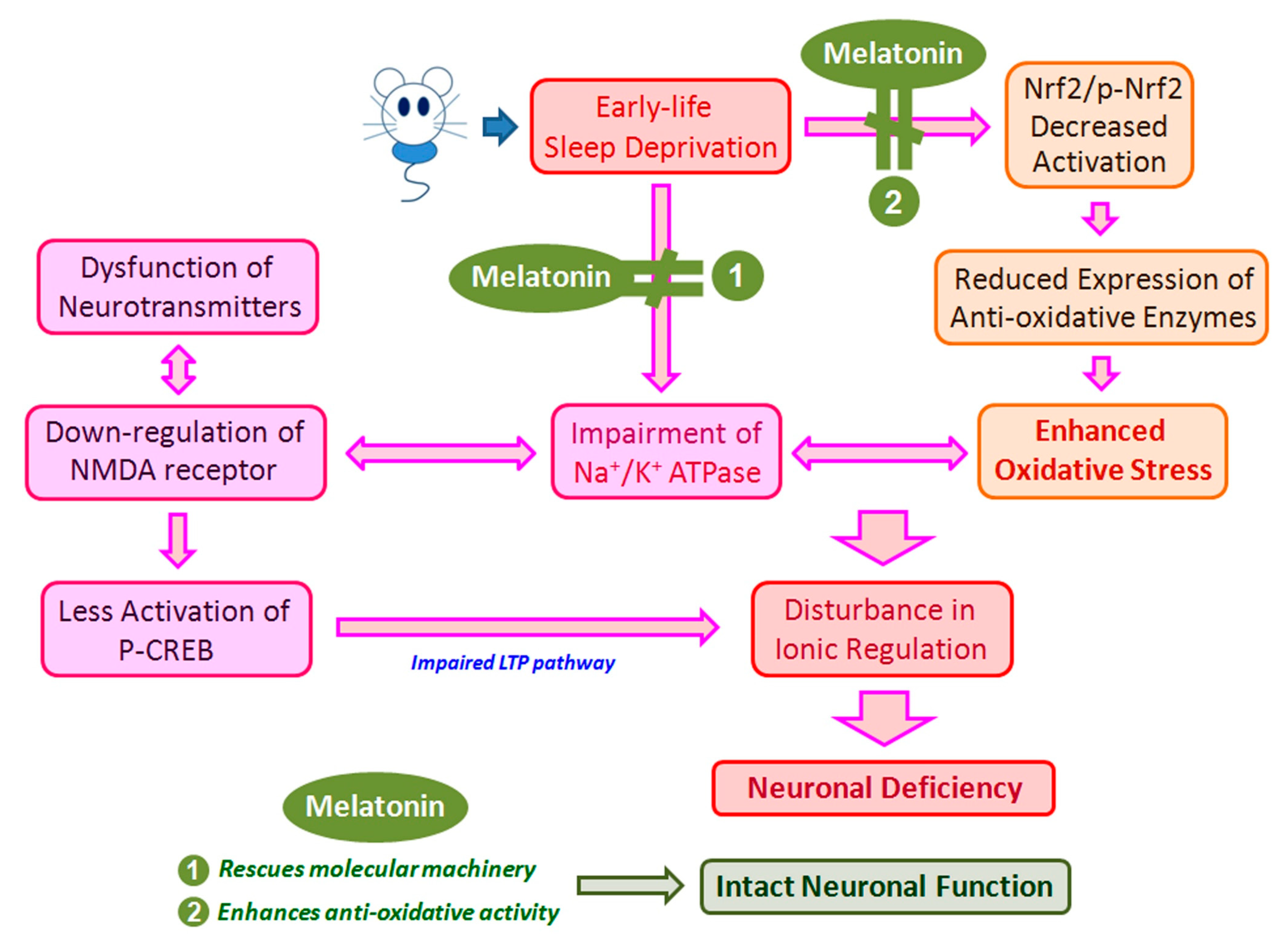
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CREB | cAMP response element binding protein |
| ESD | early-life sleep deprivation |
| GABA | γ-aminobutyric acid |
| GSH-Px | glutathione peroxidase |
| LTP | long-term potentiation |
| NMDA | N-methyl-d-aspartate |
| Nrf2 | nuclear factor E2-related factor 2 |
| ROS | reactive oxygen species |
| SOD1 | superoxide dismutase |
| TOF-SIMS | time-of-flight secondary ion mass spectrometry |
References
- Palagini, L.; Rosenlicht, N. Sleep, dreaming, and mental health: A review of historical and neurobiological perspectives. Sleep Med. Rev. 2011, 15, 179–186. [Google Scholar] [CrossRef]
- Grandner, M.A. Sleep, health, and society. Sleep Med. Clin. 2020, 15, 319–340. [Google Scholar] [CrossRef]
- Sleep in America Poll-Children and Sleep. National Sleep Fundation. 2004. Available online: http://www.sleepfoundation.org/article/sleep-america-polls/2004-children-and-sleep (accessed on 14 February 2021).
- Banks, S.; Dinges, D.F. Behavioral and physiological consequences of sleep restriction. J. Clin. Sleep Med. 2007, 3, 519–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ford, D.E.; Kamerow, D.B. Epidemiologic study of sleep disturbances and psychiatric disorders. An opportunity for prevention? JAMA 1989, 262, 1479–1484. [Google Scholar] [CrossRef] [PubMed]
- Walker, M.P. Cognitive consequences of sleep and sleep loss. Sleep Med. 2008, 9 (Suppl. S1), S29–S34. [Google Scholar] [CrossRef]
- Sawangjit, A.; Oyanedel, C.N.; Niethard, N.; Salazar, C.; Born, J.; Inostroza, M. The hippocampus is crucial for forming non-hippocampal long-term memory during Sleep. Nature 2018, 564, 109–113. [Google Scholar] [CrossRef]
- Malerba, P.; Bazhenov, M. Circuit mechanisms of hippocampal reactivation during Sleep. Neurobiol. Learn. Mem. 2019, 160, 98–107. [Google Scholar] [CrossRef]
- Havekes, R.; Vecsey, C.G.; Abel, T. The impact of sleep deprivation on neuronal and glial signaling pathways important for memory and synaptic plasticity. Cell. Signal. 2012, 24, 1251–1260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Havekes, R.; Park, A.J.; Tudor, J.C.; Luczak, V.G.; Hansen, R.T.; Ferri, S.L.; Bruinenberg, V.M.; Poplawski, S.G.; Day, J.P.; Aton, S.J.; et al. Sleep deprivation causes memory deficits by negatively impacting neuronal connectivity in hippocampal area CA1. Elife 2016, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez-Burgos, I.; Feria-Velasco, A. Serotonin/dopamine interaction in memory formation. Prog. Brain Res. 2008, 172, 603–623. [Google Scholar] [CrossRef] [PubMed]
- Martinez, J.L., Jr.; Derrick, B.E. Long-term potentiation and learning. Annu. Rev. Psychol. 1996, 47, 173–203. [Google Scholar] [CrossRef] [Green Version]
- Lynch, M.A. Long-term potentiation and memory. Physiol. Rev. 2004, 84, 87–136. [Google Scholar] [CrossRef] [PubMed]
- Murphy, D.D.; Segal, M. Morphological plasticity of dendritic spines in central neurons is mediated by activation of cAMP response element binding protein. Proc. Natl. Acad. Sci. USA 1997, 94, 1482–1487. [Google Scholar] [CrossRef] [Green Version]
- Miyamoto, E. Molecular mechanism of neuronal plasticity: Induction and maintenance of long-term potentiation in the hippocampus. J. Pharmacol. Sci. 2006, 100, 433–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, H.M.; Liao, W.C.; Sheu, J.N.; Chang, C.C.; Lan, C.T.; Mai, F.D. Sleep deprivation impairs Ca2+ expression in the hippocampus: Ionic imaging analysis for cognitive deficiency with TOF-SIMS. Microsc. Microanal. 2012, 18, 425–435. [Google Scholar] [CrossRef]
- Reynolds, A.; Laurie, C.; Mosley, R.L.; Gendelman, H.E. Oxidative stress and the pathogenesis of neurodegenerative disorders. Int. Rev. Neurobiol. 2007, 82, 297–325. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; O, W.; Li, W.; Jiang, Z.G.; Ghanbari, H.A. Oxidative stress and neurodegenerative disorders. Int. J. Mol. Sci. 2013, 14, 24438–24475. [Google Scholar] [CrossRef] [Green Version]
- Silva, R.H.; Abilio, V.C.; Takatsu, A.L.; Kameda, S.R.; Grassl, C.; Chehin, A.B.; Medrano, W.A.; Calzavara, M.B.; Registro, S.; Andersen, M.L.; et al. Role of hippocampal oxidative stress in memory deficits induced by sleep deprivation in mice. Neuropharmacology 2004, 46, 895–903. [Google Scholar] [CrossRef]
- D’Almeida, V.; Lobo, L.L.; Hipolide, D.C.; de Oliveira, A.C.; Nobrega, J.N.; Tufik, S. Sleep deprivation induces brain region-specific decreases in glutathione levels. Neuroreport 1998, 9, 2853–2856. [Google Scholar] [CrossRef]
- Wu, L.J.; Kim, S.S.; Zhuo, M. Molecular targets of anxiety: From membrane to nucleus. Neurochem. Res. 2008, 33, 1925–1932. [Google Scholar] [CrossRef]
- Davis, M.; Rainnie, D.; Cassell, M. Neurotransmission in the rat amygdala related to fear and anxiety. Trends Neurosci. 1994, 17, 208–214. [Google Scholar] [CrossRef]
- Chen, L.Y.; Tiong, C.; Tsai, C.H.; Liao, W.C.; Yang, S.F.; Youn, S.C.; Mai, F.D.; Chang, H.M. Early-life sleep deprivation persistently depresses melatonin production and bio-energetics of the pineal gland: Potential implications for the development of metabolic deficiency. Brain Struct. Funct. 2015, 220, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Garcia, J.J.; Lopez-Pingarron, L.; Almeida-Souza, P.; Tres, A.; Escudero, P.; Garcia-Gil, F.A.; Tan, D.X.; Reiter, R.J.; Ramirez, J.M.; Bernal-Perez, M. Protective effects of melatonin in reducing oxidative stress and in preserving the fluidity of biological membranes: A review. J. Pineal Res. 2014, 56, 225–237. [Google Scholar] [CrossRef] [PubMed]
- Litvinenko, G.I.; Shurlygina, A.V.; Gritsyk, O.B.; Mel’nikova, E.V.; Tenditnik, M.V.; Avrorov, P.A.; Trufakin, V.A. Effects of melatonin on morphological and functional parameters of the pineal gland and organs of immune system in rats during natural light cycle and constant illumination. Bull. Exp. Biol. Med. 2015, 159, 732–735. [Google Scholar] [CrossRef] [PubMed]
- Reiter, R.J.; Mayo, J.C.; Tan, D.X.; Sainz, R.M.; Alatorre-Jimenez, M.; Qin, L. Melatonin as an antioxidant: Under promises but over delivers. J. Pineal Res. 2016, 61, 253–278. [Google Scholar] [CrossRef]
- Favero, G.; Bonomini, F.; Franco, C.; Rezzani, R. Mitochondrial dysfunction in skeletal muscle of a fibromyalgia model: The potential benefits of melatonin. Int. J. Mol. Sci. 2019, 20, 765. [Google Scholar] [CrossRef] [Green Version]
- Tan, D.X.; Hardeland, R. Targeting host defense system and rescuing compromised mitochondria to increase tolerance against pathogens by melatonin may impact outcome of deadly virus infection pertinent to COVID-19. Molecules 2020, 25, 4410. [Google Scholar] [CrossRef]
- Fernández-Palanca, P.; Méndez-Blanco, C.; Fondevila, F.; Tuñón, M.J.; Reiter, R.J.; Mauriz, J.L.; González-Gallego, J. Melatonin as an antitumor agent against liver cancer: An updated systematic review. Antioxidants 2021, 10, 103. [Google Scholar] [CrossRef]
- Antolin, I.; Rodriguez, C.; Sainz, R.M.; Mayo, J.C.; Uria, H.; Kotler, M.L.; Rodriguez-Colunga, M.J.; Tolivia, D.; Menendez-Pelaez, A. Neurohormone melatonin prevents cell damage: Effect on gene expression for antioxidant enzymes. FASEB J. 1996, 10, 882–890. [Google Scholar] [CrossRef]
- Tan, D.X.; Manchester, L.C.; Terron, M.P.; Flores, L.J.; Reiter, R.J. One molecule, many derivatives: A never-ending interaction of melatonin with reactive oxygen and nitrogen species? J. Pineal Res. 2007, 42, 28–42. [Google Scholar] [CrossRef]
- Reiter, R.J.; Tan, D.X.; Jou, M.J.; Korkmaz, A.; Manchester, L.C.; Paredes, S.D. Biogenic amines in the reduction of oxidative stress: Melatonin and its metabolites. Neuroendocrinol. Lett. 2008, 29, 391–398. [Google Scholar]
- Chang, H.M.; Wu, U.I.; Lan, C.T. Melatonin preserves longevity protein (sirtuin 1) expression in the hippocampus of total sleep-deprived rats. J. Pineal Res. 2009, 47, 211–220. [Google Scholar] [CrossRef]
- Banke, I.S.; Folorunsho, A.S.; Mohammed, B.; Musa, S.M.; Charles, O.; Olusegun, A.J. Effects of melatonin on changes in cognitive performances and brain malondialdehyde concentration induced by sub-chronic co-administration of chlorpyrifos and cypermethrin in male Wister rats. Asian Pac. J. Trop. Biomed. 2014, 4, 318–323. [Google Scholar] [CrossRef] [Green Version]
- Dilek, M.; Naziroglu, M.; Baha Oral, H.; Suat Ovey, I.; Kucukayaz, M.; Mungan, M.T.; Kara, H.Y.; Sutcu, R. Melatonin modulates hippocampus NMDA receptors, blood and brain oxidative stress levels in ovariectomized rats. J. Membr. Biol. 2010, 233, 135–142. [Google Scholar] [CrossRef] [PubMed]
- El-Sherif, Y.; Tesoriero, J.; Hogan, M.V.; Wieraszko, A. Melatonin regulates neuronal plasticity in the hippocampus. J. Neurosci. Res. 2003, 72, 454–460. [Google Scholar] [CrossRef]
- Everson, C.A.; Bergmann, B.M.; Rechtschaffen, A. Sleep deprivation in the rat: III. Total sleep deprivation. Sleep 1989, 12, 13–21. [Google Scholar] [CrossRef]
- Rechtschaffen, A.; Bergmann, B.M. Sleep deprivation in the rat by the disk-over-water method. Behav. Brain Res. 1995, 69, 55–63. [Google Scholar] [CrossRef]
- Bergmann, B.M.; Kushida, C.A.; Everson, C.A.; Gilliland, M.A.; Obermeyer, W.; Rechtschaffen, A. Sleep deprivation in the rat: II. Methodology. Sleep 1989, 12, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Shaywitz, A.J.; Greenberg, M.E. CREB: A stimulus-induced transcription factor activated by a diverse array of extracellular signals. Annu. Rev. Biochem. 1999, 68, 821–861. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, C.; Mayo, J.C.; Sainz, R.M.; Antolin, I.; Herrera, F.; Martin, V.; Reiter, R.J. Regulation of antioxidant enzymes: A significant role for melatonin. J. Pineal Res. 2004, 36, 1–9. [Google Scholar] [CrossRef]
- Chen, L.Y.; Renn, T.Y.; Liao, W.C.; Mai, F.D.; Ho, Y.J.; Hsiao, G.; Lee, A.W.; Chang, H.M. Melatonin successfully rescues hippocampal bioenergetics and improves cognitive function following drug intoxication by promoting Nrf2-ARE signaling activity. J. Pineal Res. 2017, 63. [Google Scholar] [CrossRef]
- Famularo, R.; Kinscherff, R.; Fenton, T. Psychiatric diagnoses of maltreated children: Preliminary findings. J. Am. Acad. Child Adolesc. Psychiatry 1992, 31, 863–867. [Google Scholar] [CrossRef]
- Heim, C.; Nemeroff, C.B. The role of childhood trauma in the neurobiology of mood and anxiety disorders: Preclinical and clinical studies. Biol. Psychiatry 2001, 49, 1023–1039. [Google Scholar] [CrossRef] [Green Version]
- Taylor, D.J.; Lichstein, K.L.; Durrence, H.H.; Reidel, B.W.; Bush, A.J. Epidemiology of insomnia, depression, and anxiety. Sleep 2005, 28, 1457–1464. [Google Scholar] [CrossRef]
- Harvey, A.G. The adverse consequences of sleep disturbance in pediatric bipolar disorder: Implications for intervention. Child Adolesc. Psychiatr. Clin. N. Am. 2009, 18, 321–338. [Google Scholar] [CrossRef] [PubMed]
- Sterpenich, V.; Albouy, G.; Darsaud, A.; Schmidt, C.; Vandewalle, G.; Dang Vu, T.T.; Desseilles, M.; Phillips, C.; Degueldre, C.; Balteau, E.; et al. Sleep promotes the neural reorganization of remote emotional memory. J. Neurosci. 2009, 29, 5143–5152. [Google Scholar] [CrossRef] [PubMed]
- Dos Reis, E.A.; de Oliveira, L.S.; Lamers, M.L.; Netto, C.A.; Wyse, A.T. Arginine administration inhibits hippocampal Na(+),K(+)-ATPase activity and impairs retention of an inhibitory avoidance task in rats. Brain Res. 2002, 951, 151–157. [Google Scholar] [CrossRef]
- Amaral, A.U.; Seminotti, B.; Cecatto, C.; Fernandes, C.G.; Busanello, E.N.; Zanatta, A.; Kist, L.W.; Bogo, M.R.; de Souza, D.O.; Woontner, M.; et al. Reduction of Na+, K+-ATPase activity and expression in cerebral cortex of glutaryl-CoA dehydrogenase deficient mice: A possible mechanism for brain injury in glutaric aciduria type I. Mol. Genet. Metab. 2012, 107, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Mandal, J.; Chakraborty, A.; Chandra, A.K. Altered acetylcholinesterase and Na+-K+ATPase activities in different areas of brain in relation to thyroid gland function and morphology under the influence of excess iodine. Int. J. Pharm. Clin. Res. 2016, 8, 9. [Google Scholar]
- Goldstein, I.; Levy, T.; Galili, D.; Ovadia, H.; Yirmiya, R.; Rosen, H.; Lichtstein, D. Involvement of Na(+), K(+)-ATPase and endogenous digitalis-like compounds in depressive disorders. Biol. Psychiatry 2006, 60, 491–499. [Google Scholar] [CrossRef] [PubMed]
- Pivovarov, A.S.; Calahorro, F.; Walker, R.J. Na(+)/K(+)-pump and neurotransmitter membrane receptors. Invert. Neurosci. 2018, 19, 1. [Google Scholar] [CrossRef]
- Tremblay, R.; Lee, S.; Rudy, B. GABAergic interneurons in the neocortex: From cellular properties to circuits. Neuron 2016, 91, 260–292. [Google Scholar] [CrossRef] [Green Version]
- Taylor, S.F.; Tso, I.F. GABA abnormalities in schizophrenia: A methodological review of in vivo studies. Schizophr. Res. 2015, 167, 84–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akkuratov, E.E.; Lopacheva, O.M.; Kruusmagi, M.; Lopachev, A.V.; Shah, Z.A.; Boldyrev, A.A.; Liu, L. Functional interaction between Na/K-ATPase and NMDA receptor in cerebellar neurons. Mol. Neurobiol. 2015, 52, 1726–1734. [Google Scholar] [CrossRef]
- Lakhan, S.E.; Caro, M.; Hadzimichalis, N. NMDA receptor activity in neuropsychiatric disorders. Front. Psychiatry 2013, 4, 52. [Google Scholar] [CrossRef] [Green Version]
- Zhong, P.; Yuen, E.Y.; Yan, Z. Modulation of neuronal excitability by serotonin-NMDA interactions in prefrontal cortex. Mol. Cell Neurosci. 2008, 38, 290–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasefi, M.S.; Yang, K.; Li, J.; Kruk, J.S.; Heikkila, J.J.; Jackson, M.F.; MacDonald, J.F.; Beazely, M.A. Acute 5-HT7 receptor activation increases NMDA-evoked currents and differentially alters NMDA receptor subunit phosphorylation and trafficking in hippocampal neurons. Mol. Brain 2013, 6, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sala, C.; Rudolph-Correia, S.; Sheng, M. Developmentally regulated NMDA receptor-dependent dephosphorylation of cAMP response element-binding protein (CREB) in hippocampal neurons. J. Neurosci. 2000, 20, 3529–3536. [Google Scholar] [CrossRef]
- Atrooz, F.; Salim, S. Sleep deprivation, oxidative stress and inflammation. Adv. Protein Chem. Struct. Biol. 2020, 119, 309–336. [Google Scholar] [CrossRef]
- Nabaee, E.; Kesmati, M.; Shahriari, A.; Khajehpour, L.; Torabi, M. Cognitive and hippocampus biochemical changes following sleep deprivation in the adult male rat. Biomed. Pharmacother. 2018, 104, 69–76. [Google Scholar] [CrossRef]
- Wang, X.Q.; Xiao, A.Y.; Sheline, C.; Hyrc, K.; Yang, A.; Goldberg, M.P.; Choi, D.W.; Yu, S.P. Apoptotic insults impair Na+, K+-ATPase activity as a mechanism of neuronal death mediated by concurrent ATP deficiency and oxidant stress. J. Cell Sci. 2003, 116, 2099–2110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, B.; Cao, R.; Choi, Y.S.; Cho, H.Y.; Rhee, A.D.; Hah, C.K.; Hoyt, K.R.; Obrietan, K. The CREB/CRE transcriptional pathway: Protection against oxidative stress-mediated neuronal cell death. J. Neurochem. 2009, 108, 1251–1265. [Google Scholar] [CrossRef] [Green Version]
- Reiter, R.J.; Acuna-Castroviejo, D.; Tan, D.X.; Burkhardt, S. Free radical-mediated molecular damage. Mechanisms for the protective actions of melatonin in the central nervous system. Ann. N. Y. Acad. Sci. 2001, 939, 200–215. [Google Scholar] [CrossRef]
- Reiter, R.J.; Tan, D.X.; Manchester, L.C.; Lopez-Burillo, S.; Sainz, R.M.; Mayo, J.C. Melatonin: Detoxification of oxygen and nitrogen-based toxic reactants. Adv. Exp. Med. Biol. 2003, 527, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, M.A.; Contini Mdel, C.; Millen, N.; Mahieu, S.T. Role of melatonin in the oxidative damage prevention at different times of hepatic regeneration. Cell Biochem. Funct. 2012, 30, 701–708. [Google Scholar] [CrossRef]
- Wang, Z.; Ma, C.; Meng, C.J.; Zhu, G.Q.; Sun, X.B.; Huo, L.; Zhang, J.; Liu, H.X.; He, W.C.; Shen, X.M.; et al. Melatonin activates the Nrf2-ARE pathway when it protects against early brain injury in a subarachnoid hemorrhage model. J. Pineal Res. 2012, 53, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Ritter, L.; Kleemann, D.; Hickmann, F.H.; Amaral, A.U.; Sitta, A.; Wajner, M.; Ribeiro, C.A. Disturbance of energy and redox homeostasis and reduction of Na+,K+-ATPase activity provoked by in vivo intracerebral administration of ethylmalonic acid to young rats. Biochim. Biophys. Acta 2015, 1852, 759–767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliveira-Abreu, K.; Silva-Dos-Santos, N.M.; Coelho-de-Souza, A.N.; Ferreira-da-Silva, F.W.; Silva-Alves, K.S.D.; Cardoso-Teixeira, A.C.; Cipolla-Neto, J.; Leal-Cardoso, J.H. Melatonin reduces excitability in dorsal root ganglia neurons with inflection on the repolarization phase of the action potential. Int. J. Mol. Sci. 2019, 20, 2611. [Google Scholar] [CrossRef] [Green Version]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chang, H.-M.; Lin, H.-C.; Cheng, H.-L.; Liao, C.-K.; Tseng, T.-J.; Renn, T.-Y.; Lan, C.-T.; Chen, L.-Y. Melatonin Successfully Rescues the Hippocampal Molecular Machinery and Enhances Anti-oxidative Activity Following Early-Life Sleep Deprivation Injury. Antioxidants 2021, 10, 774. https://doi.org/10.3390/antiox10050774
Chang H-M, Lin H-C, Cheng H-L, Liao C-K, Tseng T-J, Renn T-Y, Lan C-T, Chen L-Y. Melatonin Successfully Rescues the Hippocampal Molecular Machinery and Enhances Anti-oxidative Activity Following Early-Life Sleep Deprivation Injury. Antioxidants. 2021; 10(5):774. https://doi.org/10.3390/antiox10050774
Chicago/Turabian StyleChang, Hung-Ming, Hsing-Chun Lin, Hsin-Lin Cheng, Chih-Kai Liao, To-Jung Tseng, Ting-Yi Renn, Chyn-Tair Lan, and Li-You Chen. 2021. "Melatonin Successfully Rescues the Hippocampal Molecular Machinery and Enhances Anti-oxidative Activity Following Early-Life Sleep Deprivation Injury" Antioxidants 10, no. 5: 774. https://doi.org/10.3390/antiox10050774