
Free Radicals and Neonatal Brain Injury: From Underlying Pathophysiology to Antioxidant Treatment Perspectives

 ,
,
Abstract
:1. Introduction
2. Pathophysiological Mechanisms of Oxidative Brain Damage
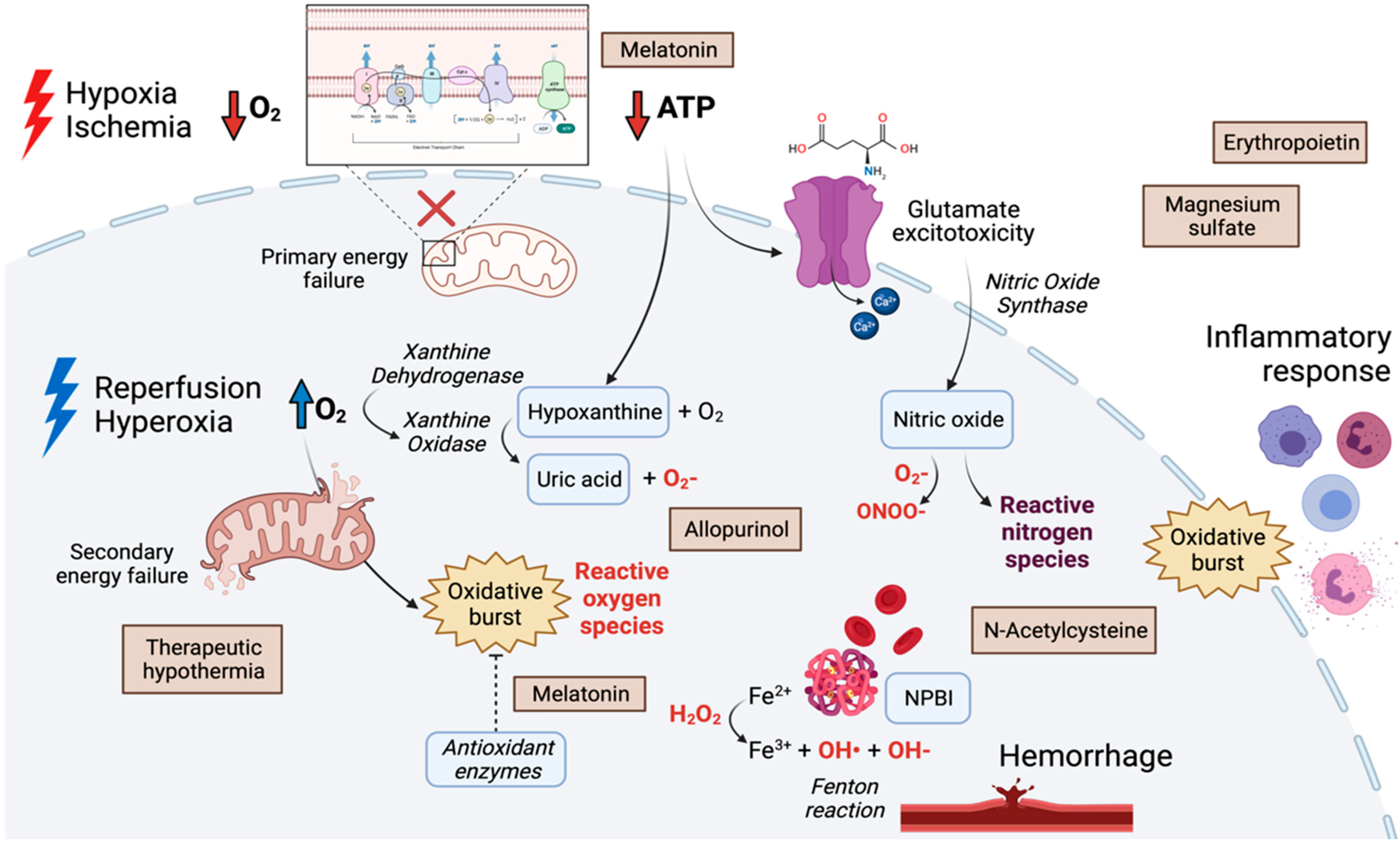
2.1. Hypoxia-Ischemia-Reperfusion
2.2. Intermittent Hypoxia
2.3. Inflammation
2.4. Hemorrhage (Hemoglobin-Induced Oxidative Damage)
3. Free Radical Biomarkers in Neonatal Brain Injury
3.1. Lipid Peroxidation Biomarkers
3.2. Protein Oxidation Markers
3.3. Nucleic Acid Oxidation Markers
3.4. Antioxidant Enzymes
3.5. Non-Protein-Bound Iron (NPBI)
3.6. Uric Acid
3.7. Nitric Oxide
3.8. Other Free Radical Biomarkers
4. Antioxidant Neuroprotective Treatments
4.1. Therapeutic Hypothermia
4.2. Erythropoietin
4.3. Melatonin
4.4. Allopurinol
4.5. N-Acetylcysteine
4.6. Magnesium Sulfate
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Lushchak, V.I. Free radicals, reactive oxygen species, oxidative stress and its classification. Chem. Biol. Interact. 2014, 224, 164–175. [Google Scholar] [CrossRef] [PubMed]
- Cobb, C.A.; Cole, M.P. Oxidative and nitrative stress in neurodegeneration. Neurobiol. Dis. 2015, 84, 4–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Folkerth, R.D.; Haynes, R.L.; Borenstein, N.S.; Belliveau, R.A.; Trachtenberg, F.; Rosenberg, P.A.; Volpe, J.J.; Kinney, H.C. Developmental lag in superoxide dismutases relative to other antioxidant enzymes in premyelinated human telencephalic white matter. J. Neuropathol. Exp. Neurol. 2004, 63, 990–999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lushchak, V.I. Glutathione homeostasis and functions: Potential targets for medical interventions. J. Amino Acids 2012, 2012, 736837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.D.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef]
- Thibeault, D.W. The precarious antioxidant defenses of the preterm infant. Am. J. Perinatol. 2000, 17, 167–181. [Google Scholar] [CrossRef]
- Khwaja, O.; Volpe, J.J. Pathogenesis of cerebral white matter injury of prematurity. Arch. Dis. Child. Fetal Neonatal Ed. 2008, 93, F153–F161. [Google Scholar] [CrossRef]
- Volpe, J.J. Brain injury in premature infants: A complex amalgam of destructive and developmental disturbances. Lancet Neurol. 2009, 8, 110–124. [Google Scholar] [CrossRef] [Green Version]
- Brekke, E.; Berger, H.R.; Widerøe, M.; Sonnewald, U.; Morken, T.S. Glucose and Intermediary Metabolism and Astrocyte–Neuron Interactions Following Neonatal Hypoxia–Ischemia in Rat. Neurochem. Res. 2017, 42, 115–132. [Google Scholar] [CrossRef]
- Iadecola, C. Bright and dark sides of nitric oxide in ischemic brain injury. Trends Neurosci. 1997, 20, 132–139. [Google Scholar] [CrossRef]
- Ferriero, D.M. Neonatal brain injury. N. Engl. J. Med. 2004, 351, 1985–1995. [Google Scholar] [CrossRef]
- Hope, P.L.; Cady, E.B.; Chu, A.; Delpy, D.T.; Gardiner, R.M.; Reynolds, E.O. Brain metabolism and intracellular pH during ischaemia and hypoxia: An in vivo 31P and 1H nuclear magnetic resonance study in the lamb. J. Neurochem. 1987, 49, 75–82. [Google Scholar] [CrossRef] [PubMed]
- McCord, J.M. Oxygen-Derived Free Radicals in Postischemic Tissue Injury. N. Engl. J. Med. 1985, 312, 159–163. [Google Scholar]
- Amaya, Y.; Yamazaki, K.; Sato, M.; Noda, K.; Nishino, T.; Nishino, T. Proteolytic conversion of xanthine dehydrogenase from the NAD-dependent type to the O2-dependent type. Amino acid sequence of rat liver xanthine dehydrogenase and identification of the cleavage sites of the enzyme protein during irreversible conversion by trypsin. J. Biol. Chem. 1990, 265, 14170–14175. [Google Scholar]
- Chung, H.Y.; Baek, B.S.; Song, S.H.; Kim, M.S.; Huh, J.I.; Shim, K.H.; Kim, K.W.; Lee, K.H. Xanthine dehydrogenase/xanthine oxidase and oxidative stress. Age 1997, 20, 127–140. [Google Scholar] [CrossRef] [Green Version]
- Hagberg, H.; Mallard, C.; Rousset, C.I.; Thornton, C. Mitochondria: Hub of injury responses in the developing brain. Lancet Neurol. 2014, 13, 217–232. [Google Scholar] [CrossRef]
- Douglas-Escobar, M.; Weiss, M.D. Hypoxic-ischemic encephalopathy: A review for the clinician. JAMA Pediatr. 2015, 169, 397–403. [Google Scholar] [CrossRef] [PubMed]
- Ferriero, D.M.; Arcavi, L.J.; Simon, R.P. Ontogeny of excitotoxic injury to nicotinamide adenine dinucleotide phosphate diaphorase reactive neurons in the neonatal rat striatum. Neuroscience 1990, 36, 417–424. [Google Scholar] [CrossRef]
- Barkhuizen, M.; Van de Berg, W.D.J.; De Vente, J.; Blanco, C.E.; Gavilanes, A.W.D.; Steinbusch, H.W.M. Nitric Oxide Production in the Striatum and Cerebellum of a Rat Model of Preterm Global Perinatal Asphyxia. Neurotox. Res. 2017, 31, 400–409. [Google Scholar] [CrossRef] [Green Version]
- Laptook, A.R. Birth Asphyxia and Hypoxic-Ischemic Brain Injury in the Preterm Infant. Clin. Perinatol. 2016, 43, 529–545. [Google Scholar] [CrossRef]
- Soul, J.S.; Hammer, P.E.; Tsuji, M.; Saul, J.P.; Bassan, H.; Limperopoulos, C.; Disalvo, D.N.; Moore, M.; Akins, P.; Ringer, S.; et al. Fluctuating pressure-passivity is common in the cerebral circulation of sick premature infants. Pediatr. Res. 2007, 61, 467–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Fiore, J.M.; Vento, M. Intermittent hypoxemia and oxidative stress in preterm infants. Respir. Physiol. Neurobiol. 2019, 266, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Martini, S.; Frabboni, G.; Rucci, P.; Czosnyka, M.; Smielewski, P.; Galletti, S.; Cimatti, A.G.; Faldella, G.; Corvaglia, L.; Austin, T. Cardiovascular and cerebrovascular responses to cardio-respiratory events in preterm infants during the transitional period. J. Physiol. 2020, 598, 4107–4119. [Google Scholar] [CrossRef] [PubMed]
- Fabian, R.H.; Perez-Polo, J.R.; Kent, T.A. Extracellular superoxide concentration increases following cerebral hypoxia but does not affect cerebral blood flow. Int. J. Dev. Neurosci. 2004, 22, 225–230. [Google Scholar] [CrossRef]
- Di Fiore, J.M.; Kaffashi, F.; Loparo, K.; Sattar, A.; Schluchter, M.; Foglyano, R.; Martin, R.J.; Wilson, C.G. The relationship between patterns of intermittent hypoxia and retinopathy of prematurity in preterm infants. Pediatr. Res. 2012, 72, 606–612. [Google Scholar] [CrossRef] [Green Version]
- Poets, C.F.; Roberts, R.S.; Schmidt, B.; Whyte, R.K.; Asztalos, E.V.; Bader, D.; Bairam, A.; Moddemann, D.; Peliowski, A.; Rabi, Y.; et al. Association between intermittent hypoxemia or bradycardia and late death or disability in extremely preterm infants. JAMA J. Am. Med. Assoc. 2015, 314, 595–603. [Google Scholar] [CrossRef]
- Darnall, R.A.; Chen, X.; Nemani, K.V.; Sirieix, C.M.; Gimi, B.; Knoblach, S.; McEntire, B.L.; Hunt, C.E. Early postnatal exposure to intermittent hypoxia in rodents is proinflammatory, impairs white matter integrity, and alters brain metabolism. Pediatr. Res. 2017, 82, 164–172. [Google Scholar] [CrossRef]
- Juliano, C.; Sosunov, S.; Niatsetskaya, Z.; Isler, J.A.; Utkina-Sosunova, I.; Jang, I.; Ratner, V.; Ten, V. Mild intermittent hypoxemia in neonatal mice causes permanent neurofunctional deficit and white matter hypomyelination. Exp. Neurol. 2015, 264, 33–42. [Google Scholar] [CrossRef]
- French, H.M.; Reid, M.; Mamontov, P.; Simmons, R.A.; Grinspan, J.B. Oxidative stress disrupts oligodendrocyte maturation. J. Neurosci. Res. 2009, 87, 3076–3087. [Google Scholar] [CrossRef] [Green Version]
- Back, S.A.; Craig, A.; Luo, N.L.; Ren, J.; Akundi, R.S.; Ribeiro, I.; Rivkees, S.A. Protective effects of caffeine on chronic hypoxia-induced perinatal white matter injury. Ann. Neurol. 2006, 60, 696–705. [Google Scholar] [CrossRef]
- Liu, S.; Zhang, X.; Liu, Y.; Yuan, X.; Yang, L.; Zhang, R.; Zhang, X.; Wang, X.; Xu, F.; Zhu, C. Early application of caffeine improves white matter development in very preterm infants. Respir. Physiol. Neurobiol. 2020, 281, 103495. [Google Scholar] [CrossRef] [PubMed]
- Jaganjac, M.; Cipak, A.; Schaur, R.J.; Zarkovic, N. Pathophysiology of neutrophil-mediated extracellular redox reactions. Front. Biosci. -Landmark 2016, 21, 839–855. [Google Scholar] [CrossRef] [Green Version]
- Folkerth, R.D.; Keefe, R.J.; Haynes, R.L.; Trachtenberg, F.L.; Volpe, J.J.; Kinney, H.C. Interferon-γ expression in periventricular leukomalacia in the human brain. Brain Pathol. 2004, 14, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Kadhim, H.; Tabarki, B.; De Prez, C.; Rona, A.M.; Sébire, G. Interleukin-2 in the pathogenesis of perinatal white matter damage. Neurology 2002, 58, 1125–1128. [Google Scholar] [CrossRef]
- Kadhim, H.; Tabarki, B.; De Prez, C.; Sébire, G. Cytokine immunoreactivity in cortical and subcortical neurons in periventricular leukomalacia: Are cytokines implicated in neuronal dysfunction in cerebral palsy? Acta Neuropathol. 2003, 105, 209–216. [Google Scholar] [CrossRef]
- Kadhim, H.; Tabarki, B.; Verellen, G.; De Prez, C.; Rona, A.M.; Sébire, G. Inflammatory cytokines in the pathogenesis of periventricular leukomalacia. Neurology 2001, 56, 1278–1284. [Google Scholar] [CrossRef]
- Yoon, B.H.; Romero, R.; Kim, C.J.; Koo, J.N.; Choe, G.; Syn, H.C.; Chi, J.G. High expression of tumor necrosis factor-α and interleukin-6 in periventricular leukomalacia. Am. J. Obstet. Gynecol. 1997, 177, 406–411. [Google Scholar] [CrossRef]
- Haynes, R.L.; Folkerth, R.D.; Keefe, R.J.; Sung, I.; Swzeda, L.I.; Rosenberg, P.A.; Volpe, J.J.; Kinney, H.C. Nitrosative and oxidative injury to premyelinating oligodendrocytes in periventricular leukomalacia. J. Neuropathol. Exp. Neurol. 2003, 62, 441–450. [Google Scholar] [CrossRef]
- Perrone, S.; Tataranno, M.L.; Negro, S.; Longini, M.; Toti, M.S.; Alagna, M.G.; Proietti, F.; Bazzini, F.; Toti, P.; Buonocore, G. Placental histological examination and the relationship with oxidative stress in preterm infants. Placenta 2016, 46, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Musilova, I.; Tothova, L.; Menon, R.; Vlkova, B.; Celec, P.; Hornychova, H.; Kutova, R.; Andrys, C.; Stepan, M.; Kacerovsky, M. Umbilical cord blood markers of oxidative stress in pregnancies complicated by preterm prelabor rupture of membranes. J. Matern. Neonatal Med. 2016, 29, 1900–1910. [Google Scholar] [CrossRef]
- Kacerovsky, M.; Tothova, L.; Menon, R.; Vlkova, B.; Musilova, I.; Hornychova, H.; Prochazka, M.; Celec, P. Amniotic fluid markers of oxidative stress in pregnancies complicated by preterm prelabor rupture of membranes. J. Matern. Neonatal Med. 2015, 28, 1250–1259. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Zhang, Q.; Wang, Q.; Lu, J. Contribution of Histologic Chorioamnionitis and Fetal Inflammatory Response Syndrome to Increased Risk of Brain Injury in Infants with Preterm Premature Rupture of Membranes. Pediatr. Neurol. 2016, 61, 94–98.e1. [Google Scholar] [CrossRef] [PubMed]
- Duggan, P.J.; Maalouf, E.F.; Watts, T.L.; Sullivan, M.H.F.; Counsell, S.J.; Allsop, J.; Al-Nakib, L.; Rutherford, M.A.; Battin, M.; Roberts, I.; et al. Intrauterine T-cell activation and increased proinflammatory cytokine concentrations in preterm infants with cerebral lesions. Lancet 2001, 358, 1699–1700. [Google Scholar] [CrossRef]
- Kakita, H.; Hussein, M.H.; Yamada, Y.; Henmi, H.; Kato, S.; Kobayashi, S.; Ito, T.; Kato, I.; Fukuda, S.; Suzuki, S.; et al. High postnatal oxidative stress in neonatal cystic periventricular leukomalacia. Brain Dev. 2009, 31, 641–648. [Google Scholar] [CrossRef] [PubMed]
- Maleki, Z.; Bailis, A.; Argani, C.H.; Askin, F.B.; Graham, E.M. Periventricular leukomalacia and placental histopathologic abnormalities. Obstet. Gynecol. 2009, 114, 1115–1120. [Google Scholar] [CrossRef] [PubMed]
- Anblagan, D.; Pataky, R.; Evans, M.J.; Telford, E.J.; Serag, A.; Sparrow, S.; Piyasena, C.; Semple, S.I.; Wilkinson, A.G.; Bastin, M.E.; et al. Association between preterm brain injury and exposure to chorioamnionitis during fetal life. Sci. Rep. 2016, 6, 37932. [Google Scholar] [CrossRef] [PubMed]
- Procianoy, R.S.; Silveira, R.C. Association between high cytokine levels with white matter injury in preterm infants with sepsis. Pediatr. Crit. Care Med. 2012, 13, 183–187. [Google Scholar] [CrossRef]
- Shah, D.K.; Doyle, L.W.; Anderson, P.J.; Bear, M.; Daley, A.J.; Hunt, R.W.; Inder, T.E. Adverse Neurodevelopment in Preterm Infants with Postnatal Sepsis or Necrotizing Enterocolitis is Mediated by White Matter Abnormalities on Magnetic Resonance Imaging at Term. J. Pediatr. 2008, 153, 170–175.e1. [Google Scholar] [CrossRef]
- Gagliardi, L.; Bellù, R.; Zanini, R.; Dammann, O. Bronchopulmonary dysplasia and brain white matter damage in the preterm infant: A complex relationship. Paediatr. Perinat. Epidemiol. 2009, 23, 582–590. [Google Scholar] [CrossRef] [PubMed]
- Barron, A.; McCarthy, C.M.; O’Keeffe, G.W. Preeclampsia and Neurodevelopmental Outcomes: Potential Pathogenic Roles for Inflammation and Oxidative Stress? Mol. Neurobiol. 2021, 58, 2734–2756. [Google Scholar] [CrossRef]
- Reeder, B.J. The redox activity of hemoglobins: From physiologic functions to pathologic mechanisms. Antioxid. Redox Signal. 2010, 13, 1087–1123. [Google Scholar] [CrossRef] [PubMed]
- Sadrzadeh, S.M.H.; Graf, E.; Panter, S.S.; Hallaway, P.E.; Eaton, J.W. Hemoglobin. A biologic Fenton reagent. J. Biol. Chem. 1984, 259, 14354–14356. [Google Scholar] [CrossRef]
- Wan, J.; Ren, H.; Wang, J. Iron toxicity, lipid peroxidation and ferroptosis after intracerebral haemorrhage. Stroke Vasc. Neurol. 2019, 4, 93–95. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Cao, F.; Yin, H.; Huang, Z.; Lin, Z.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, present and future. Cell Death Dis. 2020, 11, 88. [Google Scholar] [CrossRef] [PubMed]
- Adler, I.; Batton, D.; Betz, B.; Bezinque, S.; Ecklund, K.; Junewick, J.; McCauley, R.; Miller, C.; Seibert, J.; Specter, B.; et al. Mechanisms of injury to white matter adjacent to a large intraventricular hemorrhage in the preterm brain. J. Clin. Ultrasound 2010, 38, 254–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romantsik, O.; Bruschettini, M.; Ley, D. Intraventricular hemorrhage and white matter injury in preclinical and clinical studies. Neoreviews 2019, 20, e636–e652. [Google Scholar] [CrossRef]
- Ozawa, H.; Nishida, A.; Mito, T.; Takashima, S. Development of ferritin-containing cells in the pons and cerebellum of the human brain. Brain Dev. 1994, 16, 92–95. [Google Scholar] [CrossRef]
- Zia, M.T.; Csiszar, A.; Labinskyy, N.; Hu, F.; Vinukonda, G.; Lagamma, E.F.; Ungvari, Z.; Ballabh, P. Oxidative-nitrosative stress in a rabbit pup model of germinal matrix hemorrhage: Role of nad(p)h oxidase. Stroke 2009, 40, 2191–2198. [Google Scholar] [CrossRef] [Green Version]
- Goulding, D.S.; Vogel, R.C.; Gensel, J.C.; Morganti, J.M.; Stromberg, A.J.; Miller, B.A. Acute brain inflammation, white matter oxidative stress, and myelin deficiency in a model of neonatal intraventricular hemorrhage. J. Neurosurg. Pediatr. 2020, 26, 613–623. [Google Scholar] [CrossRef]
- Pandya, C.D.; Vekaria, H.; Joseph, B.; Slone, S.A.; Gensel, J.C.; Sullivan, P.G.; Miller, B.A. Hemoglobin induces oxidative stress and mitochondrial dysfunction in oligodendrocyte progenitor cells. Transl. Res. 2021, 231, 13–23. [Google Scholar] [CrossRef]
- Torres-Cuevas, I.; Parra-Llorca, A.; Sánchez-Illana, A.; Nuñez-Ramiro, A.; Kuligowski, J.; Cháfer-Pericás, C.; Cernada, M.; Escobar, J.; Vento, M. Oxygen and oxidative stress in the perinatal period. Redox Biol. 2017, 12, 674–681. [Google Scholar] [CrossRef]
- Banupriya, C.; Doureradjou, P.; Mondal, N.; Vishnu, B.; Koner, B.C. Can urinary excretion rate of malondialdehyde, uric acid and protein predict the severity and impending death in perinatal asphyxia? Clin. Biochem. 2008, 41, 968–973. [Google Scholar] [CrossRef] [PubMed]
- El Bana, S.M.; Maher, S.E.; Gaber, A.F.; Aly, S.S. Serum and Urinary Malondialdehyde (MDA), Uric acid, and Protein as markers of perinatal asphyxia. Electron. Physician 2016, 8, 2614–2619. [Google Scholar] [CrossRef] [Green Version]
- Siciarz, A.; Weinberger, B.; Witz, G.; Hiatt, M.; Hegyi, T. Urinary thiobarbituric acid-reacting substances as potential biomarkers of intrauterine hypoxia. Arch. Pediatr. Adolesc. Med. 2001, 155, 718–722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuligowski, J.; Escobar, J.; Quintás, G.; Lliso, I.; Torres-Cuevas, I.; Nuñez, A.; Cubells, E.; Rook, D.; van Goudoever, J.B.; Vento, M. Analysis of lipid peroxidation biomarkers in extremely low gestational age neonate urines by UPLC-MS/MS. Anal. Bioanal. Chem. 2014, 406, 4345–4356. [Google Scholar] [CrossRef]
- Cháfer-Pericás, C.; Rahkonen, L.; Sánchez-Illana, A.; Kuligowski, J.; Torres-Cuevas, I.; Cernada, M.; Cubells, E.; Nuñez-Ramiro, A.; Andersson, S.; Vento, M.; et al. Ultra high performance liquid chromatography coupled to tandem mass spectrometry determination of lipid peroxidation biomarkers in newborn serum samples. Anal. Chim. Acta 2015, 886, 214–220. [Google Scholar] [CrossRef] [PubMed]
- Mondal, N.; Bhat, B.V.; Banupriya, C.; Koner, B.C. Oxidative stress in perinatal asphyxia in relation to outcome. Indian J. Pediatr. 2010, 77, 515–517. [Google Scholar] [CrossRef] [PubMed]
- Mutlu, M.; Sarıaydın, M.; Aslan, Y.; Kader, Ş.; Dereci, S.; Kart, C.; Yaman, S.Ö.; Kural, B. Status of vitamin D, antioxidant enzymes, and antioxidant substances in neonates with neonatal hypoxic-ischemic encephalopathy. J. Matern. Neonatal Med. 2015, 29, 2259–2263. [Google Scholar] [CrossRef] [PubMed]
- Shouman, B.O.; Mesbah, A.; Aly, H. Iron metabolism and lipid peroxidation products in infants with hypoxic ischemic encephalopathy. J. Perinatol. 2008, 28, 487–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, A.; Mittal, R.; Khanna, H.D.; Basu, S.; Van de Bor, M.; Van Bel, F. Free radical injury and blood-brain barrier permeability in hypoxic-ischemic encephalopathy. Pediatrics 2008, 122, e722–e727. [Google Scholar] [CrossRef]
- Thorat, V.N.; Suryakar, A.N.; Sardeshmukh, A.S.; Sarawade, S.S. Oxidants and antioxidants in hypoxic ischaemic encephalopathy. Indian J. Clin. Biochem. 2004, 19, 32–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, S.K.; Dua, T.; Tandon, A.; Kumari, S.; Ray, G.; Batra, S. Status of lipid peroxidation and antioxidant enzymes in hypoxic ischemic encephalopathy. Indian Pediatr. 1999, 36, 561–566. [Google Scholar]
- Wan, Z.T.; Wang, W.X.; Lu, H.Y.; Zhou, Y.X. Relationship between serum MDA level and brain CT value in neonatal hypoxic ischemic encephalopathy (HIE). Chin. J. Child. Health Care 2001, 9, 3–4. [Google Scholar]
- Yu, T.; Kui, L.Q.; Ming, Q.Z. Effect of asphyxia on non-protein-bound iron and lipid peroxidation in newborn infants. Dev. Med. Child. Neurol. 2003, 45, 24–27. [Google Scholar] [CrossRef]
- Sarnat, H.B.; Sarnat, M.S. Neonatal encephalopathy following fetal distress. A clinical and electroencephalographic study. Arch. Neurol. 1976, 33, 696–705. [Google Scholar] [CrossRef]
- Krediet, T.G.; Kavelaars, A.; Vreman, H.J.; Heijnen, C.J.; van Bel, F. Respiratory distress syndrome-associated inflammation is related to early but not late peri/intraventricular hemorrhage in preterm infants. J. Pediatr. 2006, 148, 740–746. [Google Scholar] [CrossRef] [PubMed]
- Weinberger, B.; Anwar, M.; Henien, S.; Sosnovsky, A.; Hiatt, M.; Jochnowitz, N.; Witz, G.; Hegyi, T. Association of lipid peroxidation with antenatal betamethasone and oxygen radical disorders in preterm infants. Biol. Neonate 2004, 85, 121–127. [Google Scholar] [CrossRef]
- Katti, K.; Ayasolla, K.R.; Iurcotta, T.; Potak, D.; Codipilly, C.; Weinberger, B. Lipid peroxidation products as predictors of oxidant-mediated disease in preterm infants. J. Matern. Neonatal Med. 2021. [Google Scholar] [CrossRef]
- Chafer-Pericas, C.; Cernada, M.; Rahkonen, L.; Stefanovic, V.; Andersson, S.; Vento, M. Preliminary case control study to establish the correlation between novel peroxidation biomarkers in cord serum and the severity of hypoxic ischemic encephalopathy. Free Radic. Biol. Med. 2016, 97, 244–249. [Google Scholar] [CrossRef] [Green Version]
- Negro, S.; Benders, M.J.N.L.; Tataranno, M.L.; Coviello, C.; De Vries, L.S.; Van Bel, F.; Groenendaal, F.; Longini, M.; Proietti, F.; Belvisi, E.; et al. Early prediction of hypoxic-ischemic brain injury by a new panel of biomarkers in a population of term newborns. Oxid. Med. Cell. Longev. 2018, 2018, 7608108. [Google Scholar] [CrossRef]
- Coviello, C.; Perrone, S.; Buonocore, G.; Negro, S.; Longini, M.; Dani, C.; de Vries, L.S.; Groenendaal, F.; Vijlbrief, D.C.; Benders, M.J.N.L.; et al. Isoprostanes as Biomarker for White Matter Injury in Extremely Preterm Infants. Front. Pediatr. 2020, 8, 618622. [Google Scholar] [CrossRef]
- Ahola, T.; Fellman, V.; Kjellmer, I.; Raivio, K.O.; Lapatto, R. Plasma 8-isoprostane is increased in preterm infants who develop bronchopulmonary dysplasia or periventricular leukomalacia. Pediatr. Res. 2004, 56, 88–93. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Ramakrishna, S.V.K.; Basu, S.; Rao, G.R.K. Oxidative stress in perinatal asphyxia. Pediatr. Neurol. 2008, 38, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Inder, T.; Mocatta, T.; Darlow, B.; Spencer, C.; Volpe, J.J.; Winterbourn, C. Elevated free radical products in the cerebrospinal fluid of VLBW infants with cerebral white matter injury. Pediatr. Res. 2002, 52, 213–218. [Google Scholar] [CrossRef] [PubMed]
- Perrone, S.; Tataranno, M.L.; Stazzoni, G.; Buonocore, G. Biomarkers of oxidative stress in fetal and neonatal diseases. J. Matern. Neonatal Med. 2012, 25, 2575–2578. [Google Scholar] [CrossRef]
- Buonocore, G.; Perrone, S.; Longini, M.; Terzuoli, L.; Bracci, R. Total hydroperoxide and advanced oxidation protein products in preterm hypoxic babies. Pediatr. Res. 2000, 47, 221–224. [Google Scholar] [CrossRef] [Green Version]
- Buonocore, G.; Perrone, S.; Longini, M.; Vezzosi, P.; Marzocchi, B.; Paffetti, P.; Bracci, R. Oxidative stress in preterm neonates at birth and on the seventh day of life. Pediatr. Res. 2002, 52, 46–49. [Google Scholar] [CrossRef] [PubMed]
- Perrone, S.; Tataranno, M.L.; Negro, S.; Longini, M.; Marzocchi, B.; Proietti, F.; Iacoponi, F.; Capitani, S.; Buonocore Giuseppe, G. Early identification of the risk for free radical-related diseases in preterm newborns. Early Hum. Dev. 2010, 86, 241–244. [Google Scholar] [CrossRef] [PubMed]
- Shoji, H.; Ikeda, N.; Hosozawa, M.; Ohkawa, N.; Matsunaga, N.; Suganuma, H.; Hisata, K.; Tanaka, K.; Shimizu, T. Oxidative stress early in infancy and neurodevelopmental outcome in very low-birthweight infants. Pediatr. Int. 2014, 56, 709–713. [Google Scholar] [CrossRef]
- Nassi, N.; Ponziani, V.; Becatti, M.; Galvan, P.; Donzelli, G. Anti-oxidant enzymes and related elements in term and preterm newborns. Pediatr. Int. 2009, 51, 183–187. [Google Scholar] [CrossRef]
- Matsubasa, T.; Uchino, T.; Karashima, S.; Tanimura, M.; Endo, F. Oxidative stress in very low birth weight infants as measured by urinary 8-OHdG. Free Radic. Res. 2002, 36, 189–193. [Google Scholar] [CrossRef]
- Fukuda, M.; Yamauchi, H.; Yamamoto, H.; Aminaka, M.; Murakami, H.; Kamiyama, N.; Miyamoto, Y.; Koitabashi, Y. The evaluation of oxidative DNA damage in children with brain damage using 8-hydroxydeoxyguanosine levels. Brain Dev. 2008, 30, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Gulcan, H.; Ozturk, I.C.; Arslan, S. Alterations in Antioxidant Enzyme Activities in Cerebrospinal Fluid Related with Severity of Hypoxic Ischemic Encephalopathy in Newborns. Neonatology 2005, 88, 87–91. [Google Scholar] [CrossRef]
- Tsukimori, K.; Komatsu, H.; Yoshimura, T.; Hikino, S.; Hara, T.; Wake, N.; Nakano, H. Increased inflammatory markers are associated with early periventricular leukomalacia. Dev. Med. Child. Neurol. 2007, 49, 587–590. [Google Scholar] [CrossRef]
- Signorini, C.; Perrone, S.; Sgherri, C.; Ciccoli, L.; Buonocore, G.; Leoncini, S.; Rossi, V.; Vecchio, D.; Comporti, M. Plasma Esterified F2-Isoprostanes and Oxidative Stress in Newborns: Role of Nonprotein-Bound Iron. Pediatr. Res. 2008, 63, 287–291. [Google Scholar] [CrossRef] [Green Version]
- Marzocchi, B.; Perrone, S.; Paffetti, P.; Magi, B.; Bini, L.; Tani, C.; Longini, M.; Buonocore, G. Nonprotein-bound iron and plasma protein oxidative stress at birth. Pediatr. Res. 2005, 58, 1295–1299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buonocore, G.; Perrone, S.; Longini, M.; Paffetti, P.; Vezzosi, P.; Gatti, M.G.; Bracci, R. Non protein bound iron as early predictive marker of neonatal brain damage. Brain 2003, 126, 1224–1230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorrepaal, C.A.; Berger, H.M.; Benders, M.J.; van Zoeren-Grobben, D.; Van de Bor, M.; Van Bel, F. Nonprotein-bound iron in postasphyxial reperfusion injury of the newborn. Pediatrics 1996, 98, 883–889. [Google Scholar] [CrossRef] [PubMed]
- Savman, K.; Nilsson, U.A.; Blennow, M.; Kjellmer, I.; Whitelaw, A. Non-protein-bound iron is elevated in cerebrospinal fluid from preterm infants with posthemorrhagic ventricular dilatation. Pediatr. Res. 2001, 49, 208–212. [Google Scholar] [CrossRef] [Green Version]
- Basu, P.; Som, S.; Choudhuri, N.; Das, H. Correlation between Apgar score and urinary uric acid to creatinine ratio in perinatal asphyxia. Indian J. Clin. Biochem. 2008, 23, 361–364. [Google Scholar] [CrossRef] [Green Version]
- Bhongir, A.V.; Yakama, A.V.V.; Saha, S.; Radia, S.B.; Pabbati, J. The Urinary Uric Acid/Creatinine Ratio is An Adjuvant Marker for Perinatal Asphyxia. Eur. J. Pharm. Med. Res. 2015, 2, 520–528. [Google Scholar]
- Chen, H.J.; Yau, K.I.; Tsai, K.S. Urinary uric acid/creatinine ratio as an additional marker of perinatal asphyxia. J. Formos. Med. Assoc. 2000, 99, 771–774. [Google Scholar]
- Patel, K.P.; Makadia, M.G.; Patel, V.I.; Nilayangode, H.N.; Nimbalkar, S.M. Urinary Uric Acid/Creatinine Ratio—A Marker for Perinatal Asphyxia. J. Clin. Diagn. Res. 2017, 11, SC08–SC10. [Google Scholar] [CrossRef]
- Akisü, M.; Kültürsay, N. Value of the urinary uric acid to creatinine ratio in term infants with perinatal asphyxia. Acta Paediatr. Jpn. Overseas Ed. 1998, 40, 78–81. [Google Scholar] [CrossRef]
- Perlman, J.M.; Risser, R.R. Relationship of uric acid concentrations and severe intraventricular hemorrhage/leukomalacia in the premature infant. J. Pediatr. 1998, 132, 436–439. [Google Scholar] [CrossRef]
- Sysyn, G.D.; Rozycki, H.J. Lack of prognostic significance of early elevated serum uric acid levels in low birthweight infants. Biol. Neonate 2003, 83, 253–257. [Google Scholar] [CrossRef] [PubMed]
- Aliefendioǧlu, D.; Gürsoy, T.; Hayran, K.M.; Aslan, A.T. Can cerebrospinal fluid uric acid levels differentiate intraventricular hemorrhage from traumatic tap? Biol. Neonate 2006, 90, 268–272. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Pan, F.; Li, H.; Pan, J.; Qin, S.; Shen, C. Role of carbon monoxide and nitric oxide in newborn infants with postasphyxial hypoxic-ischemic encephalopathy. Pediatrics 2000, 106, 1447–1451. [Google Scholar] [CrossRef]
- Gunes, T.; Ozturk, M.A.; Koklu, E.; Kose, K.; Gunes, I. Effect of allopurinol supplementation on nitric oxide levels in asphyxiated newborns. Pediatr. Neurol. 2007, 36, 17–24. [Google Scholar] [CrossRef]
- Yan, C.; Zhang, B. Clinical significance of detecting serum melatonin and SBDPs in brain injury in preterm infants. Pediatr. Neonatol. 2019, 60, 435–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martini, S.; Austin, T.; Aceti, A.; Faldella, G.; Corvaglia, L. Free radicals and neonatal encephalopathy: Mechanisms of injury, biomarkers, and antioxidant treatment perspectives. Pediatr. Res. 2020, 87, 823–833. [Google Scholar] [CrossRef] [PubMed]
- Martinello, K.; Hart, A.R.; Yap, S.; Mitra, S.; Robertson, N.J. Management and investigation of neonatal encephalopathy: 2017 update. Arch. Dis. Child. Fetal Neonatal Ed. 2017, 102, F346–F358. [Google Scholar] [CrossRef]
- Thoresen, M.; Penrice, J.; Lorek, A.; Cady, E.B.; Wylezinska, M.; Kirkbride, V.; Cooper, C.E.; Brown, G.C.; Edwards, A.D.; Wyatt, J.S.; et al. Mild Hypothermia after Severe Transient Hypoxia-Ischemia Ameliorates Delayed Cerebral Energy Failure in the Newborn Piglet. Pediatr. Res. 1995, 37, 667–670. [Google Scholar] [CrossRef] [Green Version]
- Edwards, A.D.; Yue, X.; Squier, M.V.; Thoresen, M.; Cady, E.B.; Penrice, J.; Cooper, C.E.; Wyatt, J.S.; Reynolds, E.O.; Mehmet, H. Specific inhibition of apoptosis after cerebral hypoxia-ischaemia by moderate post-insult hypothermia. Biochem. Biophys. Res. Commun. 1995, 217, 1193–1199. [Google Scholar] [CrossRef]
- Schumacker, P.T.; Rowland, J.; Saltz, S.; Nelson, D.P.; Wood, L.D.H. Effects of hyperthermia and hypothermia on oxygen extraction by tissues during hypovolemia. J. Appl. Physiol. 1987, 63, 1246–1252. [Google Scholar] [CrossRef] [PubMed]
- Thoresen, M.; Wyatt, J. Keeping a cool head, post-hypoxic hypothermia—An old idea revisited. Acta Paediatr. 1997, 86, 1029–1033. [Google Scholar] [CrossRef]
- Thoresen, M.; Satas, S.; Puka-Sundvall, M.; Whitelaw, A.; Hallström, A.; Løberg, E.M.; Ungerstedt, U.; Steen, P.A.; Hagberg, H. Post-hypoxic hypothermia reduces cerebrocortical release of NO and excitotoxins. Neuroreport 1997, 8, 3359–3362. [Google Scholar] [CrossRef] [PubMed]
- Han, H.S.; Karabiyikoglu, M.; Kelly, S.; Sobel, R.A.; Yenari, M.A. Mild hypothermia inhibits nuclear factor-κB translocation in experimental stroke. J. Cereb. Blood Flow Metab. 2003, 23, 589–598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loidl, C.F.; De Vente, J.; van Ittersum, M.M.; van Dijk, E.H.; Vles, J.S.; Steinbusch, H.W.; Blanco, C.E. Hypothermia during or after severe perinatal asphyxia prevents increase in cyclic GMP-related nitric oxide levels in the newborn rat striatum. Brain Res. 1998, 791, 303–307. [Google Scholar] [CrossRef]
- Kader, A.; Frazzini, V.I.; Baker, C.J.; Solomon, R.A.; Trifiletti, R.R. Effect of mild hypothermia on nitric oxide synthesis during focal cerebral ischemia. Neurosurgery 1994, 35, 272–277. [Google Scholar] [CrossRef]
- Xiong, M.; Li, J.; Ma, S.M.; Yang, Y.; Zhou, W.H. Effects of hypothermia on oligodendrocyte precursor cell proliferation, differentiation and maturation following hypoxia ischemia in vivo and in vitro. Exp. Neurol. 2013, 247, 720–729. [Google Scholar] [CrossRef]
- Rutherford, M.A.; Azzopardi, D.; Whitelaw, A.; Cowan, F.; Renowden, S.; Edwards, A.D.; Thoresen, M. Mild hypothermia and the distribution of cerebral lesions in neonates with hypoxic-ischemic encephalopathy. Pediatrics 2005, 116, 1001–1006. [Google Scholar] [CrossRef]
- Rutherford, M.; Ramenghi, L.A.; Edwards, A.D.; Brocklehurst, P.; Halliday, H.; Levene, M.; Strohm, B.; Thoresen, M.; Whitelaw, A.; Azzopardi, D. Assessment of brain tissue injury after moderate hypothermia in neonates with hypoxic-ischaemic encephalopathy: A nested substudy of a randomised controlled trial. Lancet Neurol. 2010, 9, 39–45. [Google Scholar] [CrossRef] [Green Version]
- Jacobs, S.E.; Berg, M.; Hunt, R.; Tarnow-Mordi, W.O.; Inder, T.E.; Davis, P.G. Cooling for newborns with hypoxic ischaemic encephalopathy. In Cochrane Database of Systematic Reviews; Jacobs, S.E., Ed.; John Wiley & Sons, Ltd.: Chichester, UK, 2013; p. CD003311. [Google Scholar]
- Edwards, A.D.; Brocklehurst, P.; Gunn, A.J.; Halliday, H.; Juszczak, E.; Levene, M.; Strohm, B.; Thoresen, M.; Whitelaw, A.; Azzopardi, D. Neurological outcomes at 18 months of age after moderate hypothermia for perinatal hypoxic ischaemic encephalopathy: Synthesis and meta-analysis of trial data. BMJ 2010, 340, c363. [Google Scholar] [CrossRef] [Green Version]
- Simbruner, G.; Mittal, R.A.; Rohlmann, F.; Muche, R.; neo.nEURO.network Trial Participants. Systemic Hypothermia after Neonatal Encephalopathy: Outcomes of neo.nEURO.network RCT. Pediatrics 2010, 126, e771–e778. [Google Scholar] [CrossRef]
- Azzopardi, D.; Strohm, B.; Marlow, N.; Brocklehurst, P.; Deierl, A.; Eddama, O.; Goodwin, J.; Halliday, H.L.; Juszczak, E.; Kapellou, O.; et al. Effects of Hypothermia for Perinatal Asphyxia on Childhood Outcomes. N. Engl. J. Med. 2014, 371, 140–149. [Google Scholar] [CrossRef] [Green Version]
- Rao, R.; Trivedi, S.; Vesoulis, Z.; Liao, S.M.; Smyser, C.D.; Mathur, A.M. Safety and Short-Term Outcomes of Therapeutic Hypothermia in Preterm Neonates 34–35 Weeks Gestational Age with Hypoxic-Ischemic Encephalopathy. J. Pediatr. 2017, 183, 37–42. [Google Scholar] [CrossRef] [Green Version]
- Herrera, T.I.; Edwards, L.; Malcolm, W.F.; Smith, P.B.; Fisher, K.A.; Pizoli, C.; Gustafson, K.E.; Goldstein, R.F.; Cotten, C.M.; Goldberg, R.N.; et al. Outcomes of preterm infants treated with hypothermia for hypoxic-ischemic encephalopathy. Early Hum. Dev. 2018, 125, 1–7. [Google Scholar] [CrossRef]
- Thayyil, S.; Pant, S.; Montaldo, P.; Shukla, D.; Oliveira, V.; Ivain, P.; Bassett, P.; Swamy, R.; Mendoza, J.; Moreno-Morales, M.; et al. Hypothermia for moderate or severe neonatal encephalopathy in low-income and middle-income countries (HELIX): A randomised controlled trial in India, Sri Lanka, and Bangladesh. Lancet Glob. Health 2021, 9, e1273–e1285. [Google Scholar] [CrossRef]
- Vittori, D.C.; Chamorro, M.E.; Hernández, Y.V.; Maltaneri, R.E.; Nesse, A.B. Erythropoietin and derivatives: Potential beneficial effects on the brain. J. Neurochem. 2021, 158, 1032–1057. [Google Scholar] [CrossRef]
- Pathipati, P.; Ferriero, D.M. The Differential Effects of Erythropoietin Exposure to Oxidative Stress on Microglia and Astrocytes in vitro. Dev. Neurosci. 2017, 39, 310–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genc, S.; Akhisaroglu, M.; Kuralay, F.; Genc, K. Erythropoietin restores glutathione peroxidase activity in 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine-induced neurotoxicity in C57BL mice and stimulates murine astroglial glutathione peroxidase production in vitro. Neurosci. Lett. 2002, 321, 73–76. [Google Scholar] [CrossRef]
- Solaroglu, I.; Solaroglu, A.; Kaptanoglu, E.; Dede, S.; Haberal, A.; Beskonakli, E.; Kilinc, K. Erythropoietin prevents ischemia-reperfusion from inducing oxidative damage in fetal rat brain. Childs. Nerv. Syst. 2003, 19, 19–22. [Google Scholar] [CrossRef]
- Kumral, A.; Baskin, H.; Gokmen, N.; Yilmaz, O.; Genc, K.; Genc, S.; Tatli, M.M.; Duman, N.; Ozer, E.; Ozkan, H. Selective inhibition of nitric oxide in hypoxic-ischemic brain model in newborn rats: Is it an explanation for the protective role of erythropoietin? Biol. Neonate 2004, 85, 51–54. [Google Scholar] [CrossRef]
- McAdams, R.M.; Juul, S.E. Neonatal Encephalopathy: Update on Therapeutic Hypothermia and Other Novel Therapeutics. Clin. Perinatol. 2016, 43, 485–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, C.; Kang, W.; Xu, F.; Cheng, X.; Zhang, Z.; Jia, L.; Ji, L.; Guo, X.; Xiong, H.; Simbruner, G.; et al. Erythropoietin Improved Neurologic Outcomes in Newborns with Hypoxic-Ischemic Encephalopathy. Pediatrics 2009, 124, e218–e226. [Google Scholar] [CrossRef]
- Malla, R.R.; Asimi, R.; Teli, M.A.; Shaheen, F.; Bhat, M.A. Erythropoietin monotherapy in perinatal asphyxia with moderate to severe encephalopathy: A randomized placebo-controlled trial. J. Perinatol. 2017, 37, 596–601. [Google Scholar] [CrossRef]
- Avasiloaiei, A.; Dimitriu, C.; Moscalu, M.; Paduraru, L.; Stamatin, M. High-dose phenobarbital or erythropoietin for the treatment of perinatal asphyxia in term newborns. Pediatr. Int. 2013, 55, 589–593. [Google Scholar] [CrossRef] [PubMed]
- El Shimi, M.S.; Awad, H.A.; Hassanein, S.M.A.; Gad, G.I.; Imam, S.S.; Shaaban, H.A.; El Maraghy, M.O. Single dose recombinant erythropoietin versus moderate hypothermia for neonatal hypoxic ischemic encephalopathy in low resource settings. J. Matern. Neonatal Med. 2014, 27, 1295–1300. [Google Scholar] [CrossRef]
- Elmahdy, H.; El-Mashad, A.-R.; El-Bahrawy, H.; El-Gohary, T.; El-Barbary, A.; Aly, H. Human Recombinant Erythropoietin in Asphyxia Neonatorum: Pilot Trial. Pediatrics 2010, 125, e1135–e1142. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.-J.; Pan, K.-L.; Zhao, X.-L.; Qiang, H.; Cheng, S.-Q. Therapeutic effects of erythropoietin on hypoxic-ischemic encephalopathy in neonates. Zhongguo Dang Dai Er Ke Za Zhi 2011, 13, 855–858. [Google Scholar]
- Wu, Y.; Zhu, Z.; Fang, X.; Yin, L.; Liu, Y.; Xu, S.; Li, A. The Association between NOS3 Gene Polymorphisms and Hypoxic-Ischemic Encephalopathy Susceptibility and Symptoms in Chinese Han Population. Biomed. Res. Int. 2016, 2016, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.W.; Mathur, A.M.; Chang, T.; McKinstry, R.C.; Mulkey, S.B.; Mayock, D.E.; Van Meurs, K.P.; Rogers, E.E.; Gonzalez, F.F.; Comstock, B.A.; et al. High-Dose Erythropoietin and Hypothermia for Hypoxic-Ischemic Encephalopathy: A Phase II Trial. Pediatrics 2016, 137, e20160191. [Google Scholar] [CrossRef] [Green Version]
- Ohlsson, A.; Aher, S.M. Early erythropoiesis-stimulating agents in preterm or low birth weight infants. Cochrane Database Syst. Rev. 2020, 2020, CD004863. [Google Scholar] [CrossRef]
- Leuchter, R.H.V.; Gui, L.; Poncet, A.; Hagmann, C.; Lodygensky, G.A.; Martin, E.; Koller, B.; Darqué, A.; Bucher, H.U.; Hüppi, P.S. Association between early administration of high-dose erythropoietin in preterm infants and brain MRI abnormality at term-equivalent age. JAMA J. Am. Med. Assoc. 2014, 312, 817–824. [Google Scholar] [CrossRef] [PubMed]
- O’Gorman, R.L.; Bucher, H.U.; Held, U.; Koller, B.M.; Hüppi, P.S.; Hagmann, C.F. Tract-based spatial statistics to assess the neuroprotective effect of early erythropoietin on white matter development in preterm infants. Brain 2015, 138, 388–397. [Google Scholar] [CrossRef] [Green Version]
- Song, J.; Wang, Y.; Xu, F.; Sun, H.; Zhang, X.; Xia, L.; Zhang, S.; Li, K.; Peng, X.; Li, B.; et al. Erythropoietin Improves Poor Outcomes in Preterm Infants with Intraventricular Hemorrhage. CNS Drugs 2021, 35, 681–690. [Google Scholar] [CrossRef] [PubMed]
- Rüegger, C.M.; Hagmann, C.F.; Bührer, C.; Held, L.; Bucher, H.U.; Wellmann, S. Erythropoietin for the repair of cerebral injury in very preterm infants (EpoRepair). Neonatology 2015, 108, 198–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oorschot, D.E.; Sizemore, R.J.; Amer, A.R. Treatment of neonatal hypoxic-ischemic encephalopathy with erythropoietin alone, and erythropoietin combined with hypothermia: History, current status, and future research. Int. J. Mol. Sci. 2020, 21, 1487. [Google Scholar] [CrossRef] [Green Version]
- Fauchère, J.C.; Koller, B.M.; Tschopp, A.; Dame, C.; Ruegger, C.; Bucher, H.U.; Zeilinger, G.; Pasquier, S.; Bührer, C. Safety of early high-dose recombinant erythropoietin for neuroprotection in very preterm infants. J Pediatrics 2015, 167, 52–57.e3. [Google Scholar] [CrossRef]
- Ohls, R.K.; Kamath-Rayne, B.D.; Christensen, R.D.; Wiedmeier, S.E.; Rosenberg, A.; Fuller, J.; Lacy, C.B.; Roohi, M.; Lambert, D.K.; Burnett, J.J.; et al. Cognitive outcomes of preterm infants randomized to darbepoetin, erythropoietin, or placebo. Pediatrics 2014, 133, 1023–1030. [Google Scholar] [CrossRef] [Green Version]
- Ohls, R.K.; Christensen, R.D.; Kamath-Rayne, B.D.; Rosenberg, A.; Wiedmeier, S.E.; Roohi, M.; Lacy, C.B.; Lambert, D.K.; Burnett, J.J.; Pruckler, B.; et al. A randomized, masked, placebo-controlled study of darbepoetin alfa in preterm infants. Pediatrics 2013, 132, e119–e127. [Google Scholar] [CrossRef] [Green Version]
- Warwood, T.L.; Ohls, R.K.; Wiedmeier, S.E.; Lambert, D.K.; Jones, C.; Scoffield, S.H.; Neeraj, G.; Veng-Pedersen, P.; Christensen, R.D. Single-dose darbepoetin administration to anemic preterm neonates. J. Perinatol. 2005, 25, 725–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, L.; Li, N.; Bo, L.; Xu, Z. Melatonin and Hypothalamic-Pituitary-Gonadal Axis. Curr. Med. Chem. 2013, 20, 2017–2031. [Google Scholar] [CrossRef]
- Reiter, R.J.; Tan, D.X.; Osuna, C.; Gitto, E. Actions of melatonin in the reduction of oxidative stress. A review. J. Biomed. Sci. 2000, 7, 444–458. [Google Scholar] [CrossRef]
- Blanco, S.; Hernández, R.; Franchelli, G.; Ramos-Álvarez, M.M.; Peinado, M.Á. Melatonin influences NO/NOS pathway and reduces oxidative and nitrosative stress in a model of hypoxic-ischemic brain damage. Nitric Oxide 2017, 62, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Fulia, F.; Gitto, E.; Cuzzocrea, S.; Reiter, R.J.; Dugo, L.; Gitto, P.; Barberi, S.; Cordaro, S.; Barberi, I. Increased levels of malondialdehyde and nitrite/nitrate in the blood of asphyxiated newborns: Reduction by melatonin. J. Pineal Res. 2001, 31, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Aly, H.; Elmahdy, H.; El-Dib, M.; Rowisha, M.; Awny, M.; El-Gohary, T.; Elbatch, M.; Hamisa, M.; El-Mashad, A.R. Melatonin use for neuroprotection in perinatal asphyxia: A randomized controlled pilot study. J. Perinatol. 2015, 35, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Balduini, W.; Weiss, M.D.; Carloni, S.; Rocchi, M.; Sura, L.; Rossignol, C.; Longini, M.; Bazzini, F.; Perrone, S.; Ott, D.; et al. Melatonin pharmacokinetics and dose extrapolation after enteral infusion in neonates subjected to hypothermia. J. Pineal Res. 2019, 66, e12565. [Google Scholar] [CrossRef]
- Van Bel, F.; Shadid, M.; Moison, R.M.; Dorrepaal, C.A.; Fontijn, J.; Monteiro, L.; Van De Bor, M.; Berger, H.M. Effect of allopurinol on postasphyxial free radical formation, cerebral hemodynamics, and electrical brain activity. Pediatrics 1998, 101, 185–193. [Google Scholar] [CrossRef]
- Benders, M.J.N.L.; Bos, A.F.; Rademaker, C.M.A.; Rijken, M.; Torrance, H.L.; Groenendaal, F.; van Bel, F. Early postnatal allopurinol does not improve short term outcome after severe birth asphyxia. Arch. Dis. Child. Fetal Neonatal Ed. 2006, 91, F163–F165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaandorp, J.J.; van Bel, F.; Veen, S.; Derks, J.B.; Groenendaal, F.; Rijken, M.; Roze, E.; Venema, M.M.U.; Rademaker, C.M.; Bos, A.F.; et al. Long-term neuroprotective effects of allopurinol after moderate perinatal asphyxia: Follow-up of two randomised controlled trials. Arch. Dis. Child.-Fetal Neonatal Ed. 2012, 97, F162–F166. [Google Scholar] [CrossRef] [PubMed]
- Torrance, H.L.; Benders, M.J.; Derks, J.B.; Rademaker, C.M.A.; Bos, A.F.; Van Den Berg, P.; Longini, M.; Buonocore, G.; Venegas, M.; Baquero, H.; et al. Maternal allopurinol during fetal hypoxia lowers cord blood levels of the brain injury marker S-100B. Pediatrics 2009, 124, 350–357. [Google Scholar] [CrossRef] [Green Version]
- Kaandorp, J.J.; Benders, M.J.; Rademaker, C.M.; Torrance, H.L.; Oudijk, M.A.; de Haan, T.R.; Bloemenkamp, K.W.; Rijken, M.; van Pampus, M.G.; Bos, A.F.; et al. Antenatal allopurinol for reduction of birth asphyxia induced brain damage (ALLO-Trial); a randomized double blind placebo controlled multicenter study. BMC Pregnancy Childbirth 2010, 10, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaandorp, J.J.; van den Broek, M.P.H.; Benders, M.J.N.L.; Oudijk, M.A.; Porath, M.M.; Bambang Oetomo, S.; Wouters, M.G.A.J.; van Elburg, R.; Franssen, M.T.M.; Bos, A.F.; et al. Rapid target allopurinol concentrations in the hypoxic fetus after maternal administration during labour. Arch. Dis. Child.-Fetal Neonatal Ed. 2014, 99, F144–F148. [Google Scholar] [CrossRef]
- Kaandorp, J.J.; Benders, M.J.N.L.; Schuit, E.; Rademaker, C.M.A.; Oudijk, M.A.; Porath, M.M.; Oetomo, S.B.; Wouters, M.G.A.J.; van Elburg, R.M.; Franssen, M.T.M.; et al. Maternal allopurinol administration during suspected fetal hypoxia: A novel neuroprotective intervention? A multicentre randomised placebo controlled trial. Arch. Dis. Child.-Fetal Neonatal Ed. 2015, 100, F216–F223. [Google Scholar] [CrossRef]
- Chaudhari, T.; McGuire, W. Allopurinol for preventing mortality and morbidity in newborn infants with hypoxic-ischaemic encephalopathy. Cochrane Database Syst. Rev. 2012, CD006817. [Google Scholar] [CrossRef] [PubMed]
- Russell, G.A.B.; Cooke, R.W.I. Randomised controlled trial of allopurinol prophylaxis in very preterm infants. Arch. Dis. Child. 1995, 73, F27–F31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rushworth, G.F.; Megson, I.L. Existing and potential therapeutic uses for N-acetylcysteine: The need for conversion to intracellular glutathione for antioxidant benefits. Pharmacol. Ther. 2014, 141, 150–159. [Google Scholar] [CrossRef]
- Khan, M.; Sekhon, B.; Jatana, M.; Giri, S.; Gilg, A.G.; Sekhon, C.; Singh, I.; Singh, A.K. Administration of N-acetylcysteine after focal cerebral ischemia protects brain and reduces inflammation in a rat model of experimental stroke. J. Neurosci. Res. 2004, 76, 519–527. [Google Scholar] [CrossRef]
- Moss, H.G.; Brown, T.R.; Wiest, D.B.; Jenkins, D.D. N-Acetylcysteine rapidly replenishes central nervous system glutathione measured via magnetic resonance spectroscopy in human neonates with hypoxic-ischemic encephalopathy. J. Cereb. Blood Flow Metab. 2018, 38, 950–958. [Google Scholar] [CrossRef]
- Jenkins, D.D.; Wiest, D.B.; Mulvihill, D.M.; Hlavacek, A.M.; Majstoravich, S.J.; Brown, T.R.; Taylor, J.J.; Buckley, J.R.; Turner, R.P.; Rollins, L.G.; et al. Fetal and Neonatal Effects of N-Acetylcysteine When Used for Neuroprotection in Maternal Chorioamnionitis. J. Pediatr. 2016, 168, 67–76.e6. [Google Scholar] [CrossRef] [Green Version]
- Soghier, L.M.; Brion, L.P. Cysteine, cystine or N-acetylcysteine supplementation in parenterally fed neonates. Cochrane Database Syst. Rev. 2006, CD004869. [Google Scholar] [CrossRef]
- Regan, R.F.; Jasper, E.; Guo, Y.; Panter, S.S. The effect of magnesium on oxidative neuronal injury in vitro. J. Neurochem. 1998, 70, 77–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohammadi, H.; Shamshirian, A.; Eslami, S.; Shamshirian, D.; Ebrahimzadeh, M.A. Magnesium Sulfate Attenuates Lethality and Oxidative Damage Induced by Different Models of Hypoxia in Mice. Biomed. Res. Int. 2020, 2020, 2624734. [Google Scholar] [CrossRef] [PubMed]
- Abad, C.; Vargas, F.R.; Zoltan, T.; Proverbio, T.; Piñero, S.; Proverbio, F.; Marín, R. Magnesium sulfate affords protection against oxidative damage during severe preeclampsia. Placenta 2015, 36, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Doyle, L.W.; Crowther, C.A.; Middleton, P.; Marret, S.; Rouse, D. Magnesium sulphate for women at risk of preterm birth for neuroprotection of the fetus. Cochrane Database Syst. Rev. 2009, CD004661. [Google Scholar] [CrossRef]
- Conde-Agudelo, A.; Romero, R. Antenatal magnesium sulfate for the prevention of cerebral palsy in preterm infants less than 34 weeks’ gestation: A systematic review and metaanalysis. Am. J. Obstet. Gynecol. 2009, 200, 595–609. [Google Scholar] [CrossRef] [Green Version]
- Wolf, H.T.; Huusom, L.D.; Henriksen, T.B.; Hegaard, H.K.; Brok, J.; Pinborg, A. Magnesium sulphate for fetal neuroprotection at imminent risk for preterm delivery: A systematic review with meta-analysis and trial sequential analysis. BJOG An. Int. J. Obstet. Gynaecol. 2020, 127, 1180–1188. [Google Scholar] [CrossRef]
- Crowther, C.A.; Middleton, P.F.; Voysey, M.; Askie, L.; Duley, L.; Pryde, P.G.; Marret, S.; Doyle, L.W. Assessing the neuroprotective benefits for babies of antenatal magnesium sulphate: An individual participant data meta-analysis. PLoS Med. 2017, 14, e1002398. [Google Scholar] [CrossRef]
- Chollat, C.; Enser, M.; Houivet, E.; Provost, D.; Bénichou, J.; Marpeau, L.; Marret, S. School-age outcomes following a randomized controlled trial of magnesium sulfate for neuroprotection of preterm infants. J. Pediatr. 2014, 165, 398–400.e3. [Google Scholar] [CrossRef] [PubMed]
- Doyle, L.W.; Anderson, P.J.; Haslam, R.; Lee, K.J.; Crowther, C.; Darlow, B.; Austin, N.; French, N.; Campbell, C.; Carse, E.; et al. School-age outcomes of very preterm infants after antenatal treatment with magnesium sulfate vs placebo. JAMA J. Am. Med. Assoc. 2014, 312, 1105–1113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, T.M.N.; Crowther, C.A.; Wilkinson, D.; Bain, E. Magnesium sulphate for women at term for neuroprotection of the fetus. Cochrane Database Syst. Rev. 2013, 2013, CD009395. [Google Scholar] [CrossRef] [PubMed]
- Tagin, M.; Shah, P.S.; Lee, K.S. Magnesium for newborns with hypoxic-ischemic encephalopathy: A systematic review and meta-analysis. J. Perinatol. 2013, 33, 663–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iqbal, N.; Younus, J.; Malik, M.; Fatima, B.; Imran, A.; Maqbool, S.; Waheed, K.A.I.; Haque, K. The Neuroprotective Efficacy of Postnatal Magnesium Sulfate in Term or Near-Term Infants with Moderate-to-Severe Birth Asphyxia. Cureus 2021, 13, e16826. [Google Scholar] [CrossRef]
- Rahman, S.U.; Canpolat, F.E.; Oncel, M.Y.; Evli, A.; Dilmen, U.; Parappil, H.; Anabrees, J.; Hassan, K.; Khashaba, M.; Noor, I.A.; et al. Multicenter randomized controlled trial of therapeutic hypothermia plus magnesium sulfate versus therapeutic hypothermia plus placebo in the management of term and near-term infants with hypoxic ischemic encephalopathy (The Mag Cool study): A pilot study. J. Clin. Neonatol. 2015, 4, 158–163. [Google Scholar] [CrossRef]
- Siddiqui, M.A.S.; Butt, T.K. Role of intravenous magnesium sulphate in term neonates with hypoxic ischemic encephalopathy (hie) in a low-income country: A randomised clinical trial. J. Coll. Physicians Surg. Pakistan 2021, 31, 817–820. [Google Scholar]
- Galinsky, R.; Dean, J.M.; Lingam, I.; Robertson, N.J.; Mallard, C.; Bennet, L.; Gunn, A.J. A systematic review of magnesium sulfate for perinatal neuroprotection: What have we learnt from the past decade? Front. Neurol. 2020, 11, 449. [Google Scholar] [CrossRef]


{kind=link}
| Biomarkers | Plasma/Serum | Urine | Cerebro-Spinal Fluid | |
|---|---|---|---|---|
| Lipid peroxidation | Malondialdehyde |
| ||
| Isoprostanes | Not available | Not available | ||
| Protein oxidation | Protein carbonyls (PC) Advanced oxidation protein products (AOPP) | Not available | Not available | |
| Nucleic acid oxidation | 8-hydroxy-2-deoxyguanosine | Not available | Increased levels in HIE infants [92] | Increased levels in HIE infants (2-fold) [92] |
| Antioxidant enzymes | Superoxide dismutase (SOD)
Glutathione peroxidase (GP) Catalase (CAT) | Not available | ||
| Non-protein-bound iron (NPBI) | Not available | Increased levels in HIE infants [69] | ||
| Uric acid (UA) | Not available | |||
| Nitrosative biomarkers | Nitric oxide (NO)
Nitrate/nitrite ratio | Not available | Increased NO at 0–24 h in severe HIE [109] | |
| Biomarkers | Plasma/Serum | Urine | Cerebro-Spinal Fluid | |
|---|---|---|---|---|
| Lipid peroxidation | Malondialdehyde (MDA) | Increased in preterm infants who developed IVH [76] | Conflicting results in relation to IVH/PVL development [77,78] | Not available |
| Isoprostanes | Not available | Trend toward higher 8-isoprostane levels (not significant) in infants developing white matter injury [84] | ||
| Protein oxidation | Advanced oxidation protein products (AOPP) | Increased IVH risk for cord blood AOPP > 90.70 μmol/L [88] | Not available | Positive correlation between AOPP levels and white matter injury severity at term MRI [84] |
| Nucleic acid oxidation | 8-hydroxy-2-deoxyguanosine | Not available | Not available | Not available |
| Antioxidant enzymes | Superoxide dismutase (SOD) Glutathione peroxidase (GP) Catalase (CAT) | No difference in cord blood SOD in preterm infants developing early PVL vs. controls [94] | Not available | Not available |
| Non-protein-bound iron (NPBI) | Not available | |||
| Uric acid (UA) | Conflicting data on the association with severe IVH/PVL [105,106,107] | Not available | Increased levels in preterm infants with IVH grade 2–4 (mean age: 8 days) [107] | |
| Melatonin | Not available | Not available | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martini, S.; Castellini, L.; Parladori, R.; Paoletti, V.; Aceti, A.; Corvaglia, L. Free Radicals and Neonatal Brain Injury: From Underlying Pathophysiology to Antioxidant Treatment Perspectives. Antioxidants 2021, 10, 2012. https://doi.org/10.3390/antiox10122012
Martini S, Castellini L, Parladori R, Paoletti V, Aceti A, Corvaglia L. Free Radicals and Neonatal Brain Injury: From Underlying Pathophysiology to Antioxidant Treatment Perspectives. Antioxidants. 2021; 10(12):2012. https://doi.org/10.3390/antiox10122012
Chicago/Turabian StyleMartini, Silvia, Laura Castellini, Roberta Parladori, Vittoria Paoletti, Arianna Aceti, and Luigi Corvaglia. 2021. "Free Radicals and Neonatal Brain Injury: From Underlying Pathophysiology to Antioxidant Treatment Perspectives" Antioxidants 10, no. 12: 2012. https://doi.org/10.3390/antiox10122012