
Highly Potent, Selective, and Competitive Indole-Based MAO-B Inhibitors Protect PC12 Cells against 6-Hydroxydopamine- and Rotenone-Induced Oxidative Stress

 , , ,
, , ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemical Reagents, Purification, and Instrumentation
2.2. Synthesis of (3-fluorophenyl)(5-nitro-1H-indol-1-yl)methanone (2)
2.3. Synthesis of (5-amino-1H-indol-1-yl)(3-fluorophenyl)methanone (3)
2.4. General Procedure of Urea Derivatives (4a–n)
2.4.1. 1-Ethyl-3-(1-(3-fluorobenzoyl)-1H-indol-5-yl)urea (4a)
2.4.2. Ethyl ((1-(3-fluorobenzoyl)-1H-indol-5-yl)carbamoyl)glycinate (4b)
2.4.3. 1-(2-Chloroethyl)-3-(1-(3-fluorobenzoyl)-1H-indol-5-yl)urea (4c)
2.4.4. 1-Cyclohexyl-3-(1-(3-fluorobenzoyl)-1H-indol-5-yl)urea (4d)
2.4.5. 1-(1-(3-Fluorobenzoyl)-1H-indol-5-yl)-3-(4-nitrophenyl)urea (4e)
2.4.6. 1-(1-(3-Fluorobenzoyl)-1H-indol-5-yl)-3-(4-methoxyphenyl)urea (4f)
2.4.7. 1-(3,5-Dimethoxyphenyl)-3-(1-(3-fluorobenzoyl)-1H-indol-5-yl)urea (4g)
2.4.8. 1-(3,5-Dichlorophenyl)-3-(1-(3-fluorobenzoyl)-1H-indol-5-yl)urea (4h)
2.4.9. 1-(1-(3-Fluorobenzoyl)-1H-indol-5-yl)-3-(4-fluorophenyl)urea (4i)
2.4.10. 1-(3,4-Dichlorophenyl)-3-(1-(3-fluorobenzoyl)-1H-indol-5-yl)urea (4j)
2.4.11. 1-(4-Chloro-3-(trifluoromethyl)phenyl)-3-(1-(3-fluorobenzoyl)-1H-indol-5-yl)urea (4k)
2.4.12. 1-(1-(3-Fluorobenzoyl)-1H-indol-5-yl)-3-(4-fluorophenethyl)urea (4l)
2.4.13. 1-(1-(3-Fluorobenzoyl)-1H-indol-5-yl)-3-(4-phenoxyphenyl)urea (4m)
2.4.14. 1-(1-(3-Fluorobenzoyl)-1H-indol-5-yl)-3-(4-morpholinophenyl)urea (4n)
2.5. General Procedure of Thiourea Derivatives (5a–j)
2.5.1. 1-Ethoxycarbonyl-3-(1-(3-fluorobenzoyl)-1H-indol-5-yl)thiourea (5a)
2.5.2. 1-(2-Chloroethyl)-3-(1-(3-fluorobenzoyl)-1H-indol-5-yl)thiourea (5b)
2.5.3. 1-(1-(3-Fluorobenzoyl)-1H-indol-5-yl)-3-(4-morpholinophenyl)thiourea (5c)
2.5.4. 1-(1-(3-Fluorobenzoyl)-1H-indol-5-yl)-3-(2-morpholinoethyl)thiourea (5d)
2.5.5. 1-(1-(3-Fluorobenzoyl)-1H-indol-5-yl)-3-(2-(piperidin-1-yl)ethyl)thiourea (5e)
2.5.6. 1-(1-(3-Fluorobenzoyl)-1H-indol-5-yl)-3-(furan-2-ylmethyl)thiourea (5f)
2.5.7. 1-(1-(3-Fluorobenzoyl)-1H-indol-5-yl)-3-(4-nitrophenyl)thiourea (5g)
2.5.8. 1-(3,5-Dimethoxyphenyl)-3-(1-(3-fluorobenzoyl)-1H-indol-5-yl)thiourea (5h)
2.5.9. 1-(3,4-Dichlorophenyl)-3-(1-(3-fluorobenzoyl)-1H-indol-5-yl)thiourea (5i)
2.5.10. 1-(1-(3-Fluorobenzoyl)-1H-indol-5-yl)-3-(pyridin-3-yl)thiourea (5j)
2.6. General Procedure of Amide Derivatives (6a–e)
2.6.1. N-(1-(3-Fluorobenzoyl)-1H-indol-5-yl)-2-nitroisonicotinamide (6a)
2.6.2. Ethyl (1-(3-fluorobenzoyl)-1H-indol-5-yl)carbamate (6b)
2.6.3. Ethyl 2-((1-(3-fluorobenzoyl)-1H-indol-5-yl)amino)-2-oxoacetate (6c)
2.6.4. 3,4-Dichloro-N-(1-(3-fluorobenzoyl)-1H-indol-5-yl)benzamide (6d)
2.6.5. N-(1-(3-fluorobenzoyl)-1H-indol-5-yl)-4-oxo-4H-chromene-3-carboxamide (6e)
2.7. Synthesis of Methyl 1H-indole-5-carboxylate (10)
2.8. General Procedure of Methyl Ester Derivatives (7a and 7b)
2.8.1. Methyl 1-(3-fluorobenzoyl)-1H-indole-5-carboxylate (7a)
2.8.2. Methyl 1-(3,4-dichlorobenzoyl)-1H-indole-5-carboxylate (7b)
2.9. General Procedure of the Free NH Indole Derivatives 8a–e
2.9.1. N-(4-iodophenyl)-1H-indole-5-carboxamide (8a)
2.9.2. N-(4-bromophenyl)-1H-indole-5-carboxamide (8b)
2.9.3. N-(3-chloro-4-fluorophenyl)-1H-indole-5-carboxamide (8c)
2.9.4. N-(benzo[d][1,3]dioxol-5-ylmethyl)-1H-indole-5-carboxamide (8d)
2.9.5. N-(benzo[d][1,3]dioxol-5-yl)-1H-indole-5-carboxamide (8e)
2.10. MAO Assays
2.11. Kinetic Study of MAO-B Inhibition Mode
2.12. Molecular Modeling Study
2.13. In Vitro Cellular and Cell-Free Bio-Assay
2.13.1. Materials
2.13.2. PC12 Cell Culture
2.13.3. Drug Treatment
2.13.4. Measurements of Cell Viability
2.13.5. Measurement of ROS Production Using H2-DCFDA
2.13.6. Assessment of DPPH Radical Scavenging Activity
2.13.7. Statistical Analysis
3. Results and Discussion
3.1. Chemical Synthesis
3.2. Primary Screening at Single Dose of 10 µM over MAO-B
3.3. Dose-Dependent Assay over MAO-B
3.4. Selectivity Assay of Compounds 7b, 8a, 8b, and 8e over Both Isoforms of MAO
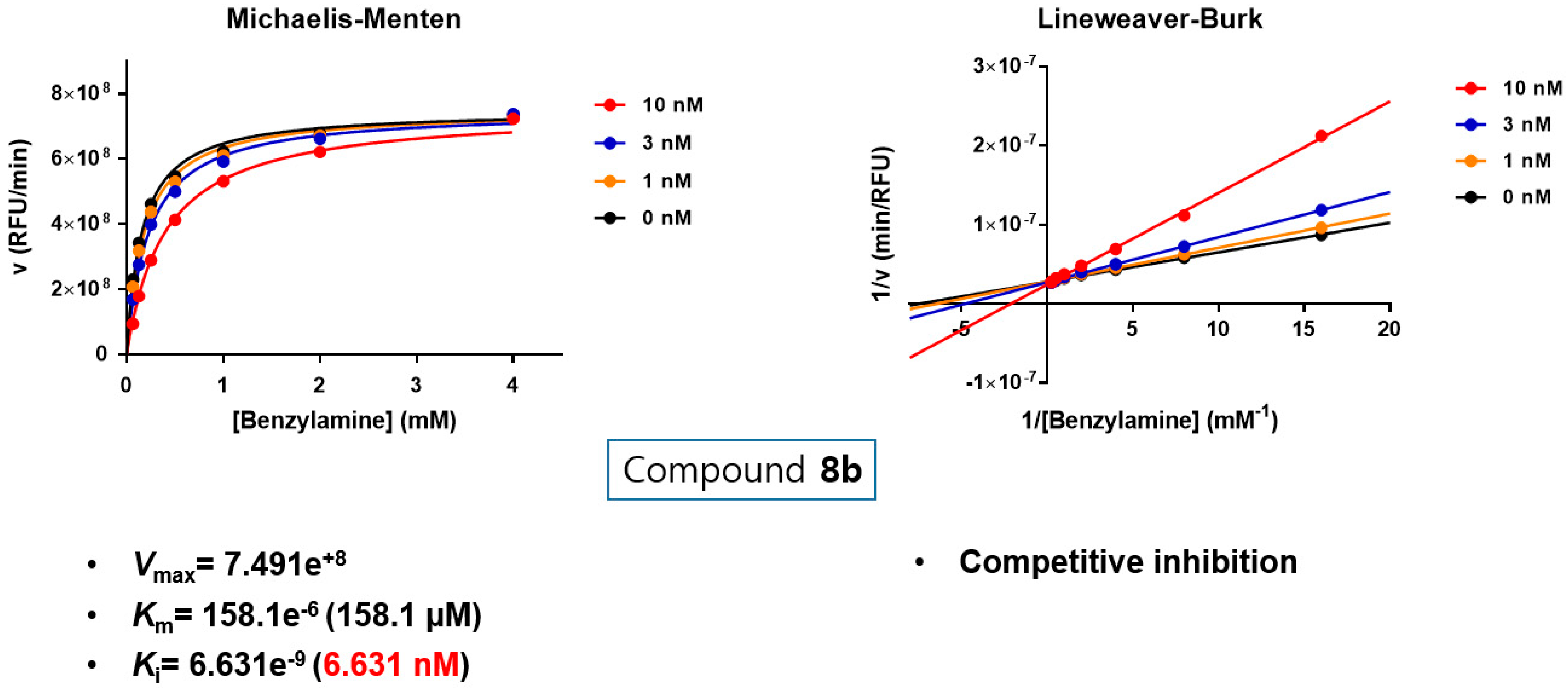
3.5. Kinetic Study to Define the Interaction Mode of Compounds 8a and 8b with MAO-B
3.6. Molecular Modeling Study
- The active N-substituted indole compound 7b has similar activity to the free (NH) indole derivatives.
- The 3,4-dichlorophenyl substituent is superior to the alkyl substitutions.
- Among the 3,4-dichlorophenyl compounds, the thiourea linker is the most active, whereas in the alkyl substituent series it is the least active.
- The active N-substituted indole and free (NH) indole compounds are highly selective towards MAO-B.
3.7. Biological Evaluation Using PC12 Cells and Cell-Free Bio-Assay
3.7.1. Assessment of the Cytotoxicity over PC12 Cells
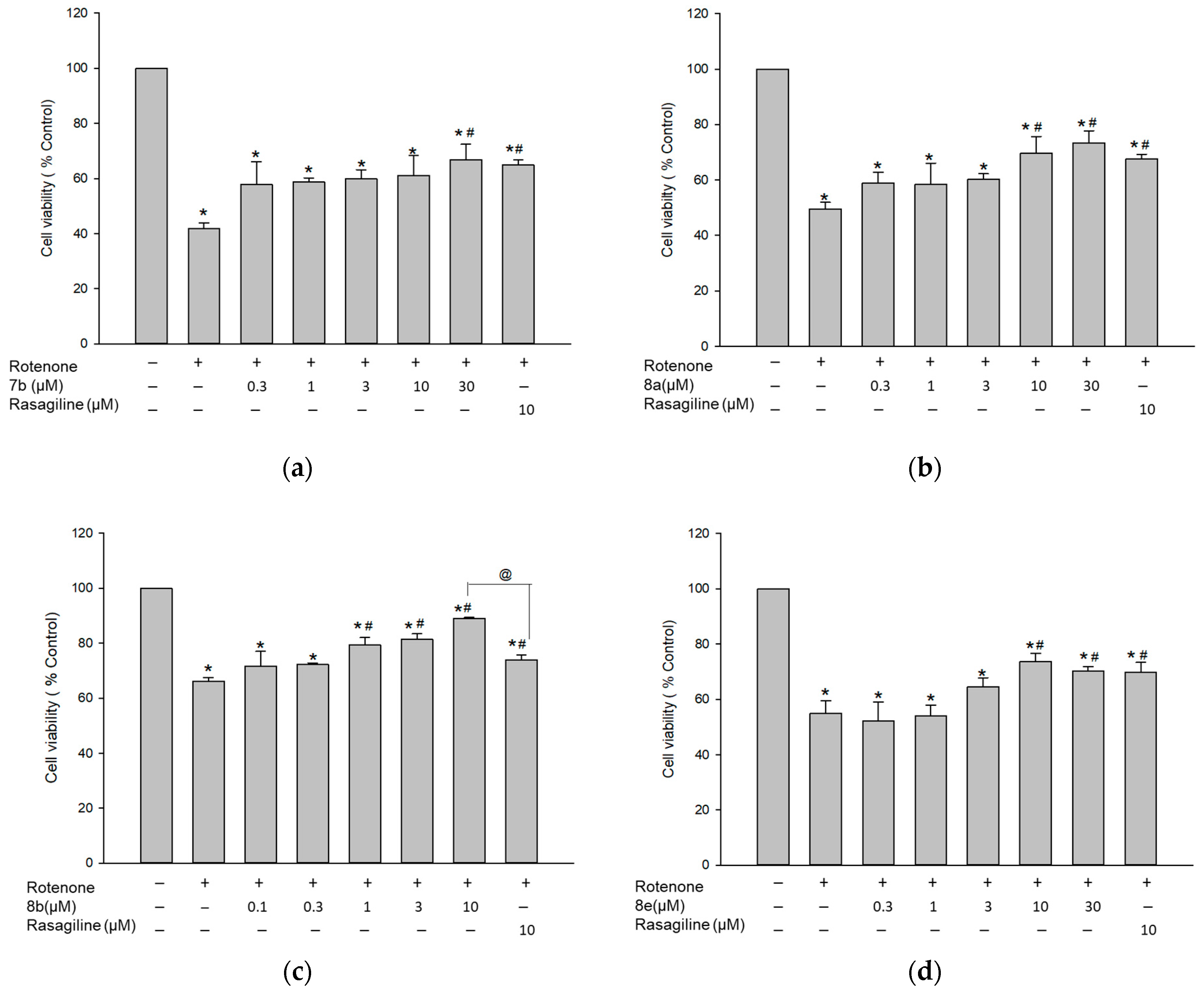
3.7.2. Protective Effect against 6-OHDA-Induced Cytotoxicity in PC12 Cells
3.7.3. Protective Effect against Rotenone-Induced Cytotoxicity in PC12 Cells
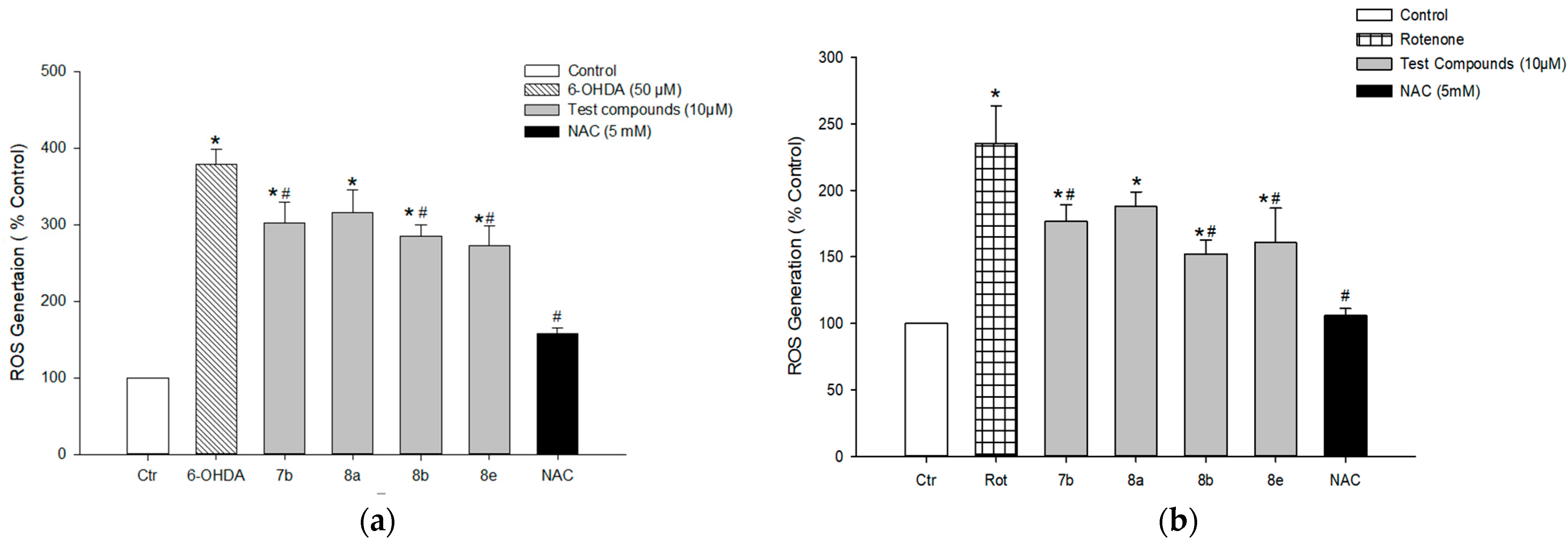
3.7.4. Effect on 6-OHDA- or Rotenone-Induced ROS Generation in PC12 Cells
3.7.5. DPPH Radical Scavenging Activity
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mhyre, T.R.; Boyd, J.T.; Hamill, R.W.; Maguire-Zeiss, K.A. Parkinson’s Disease. In Protein Aggregation and Fibrillogenesis in Cerebral and Systemic Amyloid Disease; Harris, J.R., Ed.; Springer: Dordrecht, The Netherlands, 2012; pp. 389–455. [Google Scholar] [CrossRef] [Green Version]
- Van Den Eeden, S.K.; Tanner, C.M.; Bernstein, A.L.; Fross, R.D.; Leimpeter, A.; Bloch, D.A.; Nelson, L.M. Incidence of Parkinson’s Disease: Variation by Age, Gender, and Race/Ethnicity. Am. J. Epidemiol. 2003, 157, 1015–1022. [Google Scholar] [CrossRef]
- Miller, I.N.; Cronin-Golomb, A. Gender differences in Parkinson’s disease: Clinical characteristics and cognition. Mov. Disord. 2010, 25, 2695–2703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rocca, W.A. The burden of Parkinson’s disease: A worldwide perspective. Lancet Neurol. 2018, 17, 928–929. [Google Scholar] [CrossRef] [Green Version]
- Pringsheim, T.; Jette, N.; Frolkis, A.; Steeves, T.D.L. The prevalence of Parkinson’s disease: A systematic review and meta-analysis. Mov. Disord. 2014, 29, 1583–1590. [Google Scholar] [CrossRef] [PubMed]
- Grosch, J.; Winkler, J.; Kohl, Z. Early Degeneration of Both Dopaminergic and Serotonergic Axons—A Common Mechanism in Parkinson’s Disease. Front. Cell. Neurosci. 2016, 10, 293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jankovic, J.; Aguilar, L.G. Current approaches to the treatment of Parkinson’s disease. Neuropsychiatr. Dis. Treat. 2008, 4, 743–757. [Google Scholar] [CrossRef] [Green Version]
- Bortolato, M.; Chen, K.; Shih, J.C. Monoamine oxidase inactivation: From pathophysiology to therapeutics. Adv. Drug Deliv. Rev. 2008, 60, 1527–1533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
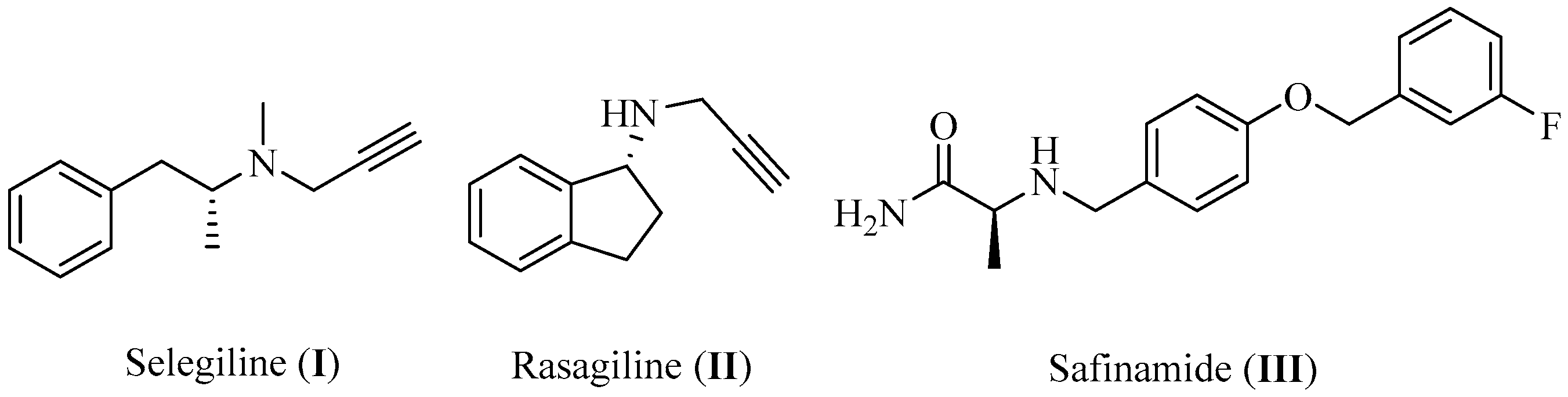
- Youdim, M.B.; Gross, A.; Finberg, J.P. Rasagiline [N-propargyl-1R(+)-aminoindan], a selective and potent inhibitor of mitochondrial monoamine oxidase B. Br. J. Pharm. 2001, 132, 500–506. [Google Scholar] [CrossRef] [PubMed]
- Caccia, C.; Maj, R.; Calabresi, M.; Maestroni, S.; Faravelli, L.; Curatolo, L.; Salvati, P.; Fariello, R.G. Safinamide. Neurology 2006, 67, S18. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-H.; Ju, Y.H.; Choi, J.W.; Song, H.J.; Jang, B.K.; Woo, J.; Chun, H.; Kim, H.J.; Shin, S.J.; Yarishkin, O.; et al. Newly developed reversible MAO-B inhibitor circumvents the shortcomings of irreversible inhibitors in Alzheimer’s disease. Sci. Adv. 2019, 5, eaav0316. [Google Scholar] [CrossRef] [Green Version]
- Guglielmi, P.; Carradori, S.; Ammazzalorso, A.; Secci, D. Novel approaches to the discovery of selective human monoamine oxidase-B inhibitors: Is there room for improvement? Expert Opin. Drug Discov. 2019, 14, 995–1035. [Google Scholar] [CrossRef]
- Fernando Rodrigues de Sa, A.; Eliezer, J.B.; Carlos Alberto Manssour, F. From Nature to Drug Discovery: The Indole Scaffold as a ‘Privileged Structure’. Mini-Rev. Med. Chem. 2009, 9, 782–793. [Google Scholar] [CrossRef]
- Sravanthi, T.V.; Manju, S.L. Indoles—A promising scaffold for drug development. Eur. J. Pharm. Sci. 2016, 91, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.; Li, Y.; Yan, C.; Yan, M.; Tang, Z. Indole: A privileged scaffold for the design of anti-cancer agents. Eur. J. Med. Chem. 2019, 183, 111691. [Google Scholar] [CrossRef] [PubMed]
- Elkamhawy, A.; Park, J.E.; Hassan, A.H.E.; Pae, A.N.; Lee, J.; Park, B.G.; Roh, E.J. Synthesis and evaluation of 2-(3-arylureido)pyridines and 2-(3-arylureido)pyrazines as potential modulators of Aβ-induced mitochondrial dysfunction in Alzheimer’s disease. Eur. J. Med. Chem. 2018, 144, 529–543. [Google Scholar] [CrossRef] [PubMed]
- Elkamhawy, A.; Park, J.E.; Hassan, A.H.E.; Ra, H.; Pae, A.N.; Lee, J.; Park, B.G.; Moon, B.; Park, H.M.; Roh, E.J. Discovery of 1-(3-(benzyloxy)pyridin-2-yl)-3-(2-(piperazin-1-yl)ethyl)urea: A new modulator for amyloid beta-induced mitochondrial dysfunction. Eur. J. Med. Chem. 2017, 128, 56–69. [Google Scholar] [CrossRef]
- Elkamhawy, A.; Paik, S.; Kim, H.J.; Park, J.-H.; Londhe, A.M.; Lee, K.; Pae, A.N.; Park, K.D.; Roh, E.J. Discovery of N-(1-(3-fluorobenzoyl)-1H-indol-5-yl)pyrazine-2-carboxamide: A novel, selective, and competitive indole-based lead inhibitor for human monoamine oxidase B. J. Enzym. Inhib. Med. Chem. 2020, 35, 1568–1580. [Google Scholar] [CrossRef]
- Tzvetkov, N.T.; Hinz, S.; Küppers, P.; Gastreich, M.; Müller, C.E. Indazole- and indole-5-carboxamides: Selective and reversible monoamine oxidase B inhibitors with subnanomolar potency. J. Med. Chem. 2014, 57, 6679–6703. [Google Scholar] [CrossRef]
- Ye, Y.; Suo, Y.; Yang, F.; Yang, Y.; Han, L. Microwave-Assisted Synthesis and Molecular Recognition Properties of Novel Indole Acylhydrazone Receptors. J. Chem. Res. 2015, 39, 296–299. [Google Scholar] [CrossRef]
- Choi, J.W.; Jang, B.K.; Cho, N.C.; Park, J.H.; Yeon, S.K.; Ju, E.J.; Lee, Y.S.; Han, G.; Pae, A.N.; Kim, D.J.; et al. Synthesis of a series of unsaturated ketone derivatives as selective and reversible monoamine oxidase inhibitors. Bioorg. Med. Chem. 2015, 23, 6486–6496. [Google Scholar] [CrossRef]
- Binda, C.; Wang, J.; Pisani, L.; Caccia, C.; Carotti, A.; Salvati, P.; Edmondson, D.E.; Mattevi, A. Structures of human monoamine oxidase B complexes with selective noncovalent inhibitors: Safinamide and coumarin analogs. J. Med. Chem. 2007, 50, 5848–5852. [Google Scholar] [CrossRef]
- Wagmann, L.; Brandt, S.D.; Kavanagh, P.V.; Maurer, H.H.; Meyer, M.R. In vitro monoamine oxidase inhibition potential of alpha-methyltryptamine analog new psychoactive substances for assessing possible toxic risks. Toxicol. Lett. 2017, 272, 84–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Son, S.Y.; Ma, J.; Kondou, Y.; Yoshimura, M.; Yamashita, E.; Tsukihara, T. Structure of human monoamine oxidase A at 2.2-A resolution: The control of opening the entry for substrates/inhibitors. Proc. Natl. Acad. Sci. USA 2008, 105, 5739–5744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Wu, J.; Yang, X.; Cai, P.; Liu, Q.; Wang, K.D.G.; Kong, L.; Wang, X. Neuroprotective effects of benzyloxy substituted small molecule monoamine oxidase B inhibitors in Parkinson’s disease. Bioorg. Med. Chem. 2016, 24, 5929–5940. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Li, E.; Park, S. Insulin-like growth factor-1 inhibits 6-hydroxydopamine-mediated endoplasmic reticulum stress-induced apoptosis via regulation of heme oxygenase-1 and Nrf2 expression in PC12 cells. Int. J. Neurosci. 2012, 122, 641–649. [Google Scholar] [CrossRef]
- Buratta, S.; Chiaradia, E.; Tognoloni, A.; Gambelunghe, A.; Meschini, C.; Palmieri, L.; Muzi, G.; Urbanelli, L.; Emiliani, C.; Tancini, B. Effect of Curcumin on Protein Damage Induced by Rotenone in Dopaminergic PC12 Cells. Int. J. Mol. Sci. 2020, 21, 2761. [Google Scholar] [CrossRef]
- Do, H.T.T.; Bui, B.P.; Sim, S.; Jung, J.-K.; Lee, H.; Cho, J. Anti-Inflammatory and Anti-Migratory Activities of Isoquinoline-1-Carboxamide Derivatives in LPS-Treated BV2 Microglial Cells via Inhibition of MAPKs/NF-κB Pathway. Int. J. Mol. Sci. 2020, 21, 2319. [Google Scholar] [CrossRef] [Green Version]
- Heusinkveld, H.J.; Molendijk, J.; van den Berg, M.; Westerink, R.H. Azole fungicides disturb intracellular Ca2+ in an additive manner in dopaminergic PC12 cells. Toxicol. Sci. 2013, 134, 374–381. [Google Scholar] [CrossRef] [Green Version]
- Oh, Y.; Do, H.T.T.; Kim, S.; Kim, Y.M.; Chin, Y.W.; Cho, J. Memory-Enhancing Effects of Mangosteen Pericarp Water Extract through Antioxidative Neuroprotection and Anti-Apoptotic Action. Antioxidants 2020, 10, 34. [Google Scholar] [CrossRef]
- Ferino, G.; Vilar, S.; Matos, M.J.; Uriarte, E.; Cadoni, E. Monoamine oxidase inhibitors: Ten years of docking studies. Curr. Top. Med. Chem. 2012, 12, 2145–2162. [Google Scholar] [CrossRef]
- Prins, L.H.; Petzer, J.P.; Malan, S.F. Inhibition of monoamine oxidase by indole and benzofuran derivatives. Eur. J. Med. Chem. 2010, 45, 4458–4466. [Google Scholar] [CrossRef] [PubMed]
- Walkinshaw, G.; Waters, C.M. Neurotoxin-induced cell death in neuronal PC12 cells is mediated by induction of apoptosis. Neuroscience 1994, 63, 975–987. [Google Scholar] [CrossRef]
- Zhang, G.; Buchler, I.P.; DePasquale, M.; Wormald, M.; Liao, G.; Wei, H.; Barrow, J.C.; Carr, G.V. Development of a PC12 Cell Based Assay for Screening Catechol-O-methyltransferase Inhibitors. ACS Chem. Neurosci. 2019, 10, 4221–4226. [Google Scholar] [CrossRef] [PubMed]
- Rai, S.N.; Singh, P. Advancement in the modelling and therapeutics of Parkinson’s disease. J. Chem. Neuroanat. 2020, 104, 101752. [Google Scholar] [CrossRef]
- Bankiewicz, K.S.; Sanchez-Pernaute, R.; Oiwa, Y.; Kohutnicka, M.; Cummins, A.; Eberling, J. Preclinical models of Parkinson’s disease. Curr. Protoc. Neurosci. 2001, 9, Unit9.4. [Google Scholar] [CrossRef]
- Magalingam, K.B.; Radhakrishnan, A.; Haleagrahara, N. Protective effects of flavonol isoquercitrin, against 6-hydroxy dopamine (6-OHDA)-induced toxicity in PC12 cells. BMC Res. Notes 2014, 7, 49. [Google Scholar] [CrossRef] [Green Version]
- Msibi, Z.N.P.; Mabandla, M.V. Oleanolic Acid Mitigates 6-Hydroxydopamine Neurotoxicity by Attenuating Intracellular ROS in PC12 Cells and Striatal Microglial Activation in Rat Brains. Front. Physiol. 2019, 10, 1059. [Google Scholar] [CrossRef] [Green Version]
- Cui, G.; Shan, L.; Chen, Y.; Zhou, H.; Wang, Y.; Lee, S.M.-Y. A New Danshensu Derivative Protects Against 6-Hydroxydopamine-Induced Neurotoxicity In Vitro and In Vivo. Am. J. Chin. Med. 2016, 44, 1349–1361. [Google Scholar] [CrossRef]
- Uversky, V.N. Neurotoxicant-induced animal models of Parkinson’s disease: Understanding the role of rotenone, maneb and paraquat in neurodegeneration. Cell Tissue Res. 2004, 318, 225–241. [Google Scholar] [CrossRef] [PubMed]
- Jing, X.; Shi, Q.; Bi, W.; Zeng, Z.; Liang, Y.; Wu, X.; Xiao, S.; Liu, J.; Yang, L.; Tao, E. Rifampicin Protects PC12 Cells from Rotenone-Induced Cytotoxicity by Activating GRP78 via PERK-eIF2α-ATF4 Pathway. PLoS ONE 2014, 9, e92110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, Y.-J.; Song, Y.-Z.; Zhu, Y.; Zuo, W.-Q.; Zhao, Y.-F.; Shen, X.; Wang, W.-J.; Liu, Y.-I.; Wu, J.-C.; Liang, Z.-Q. Neuroprotective effects of olanzapine against rotenone-induced toxicity in PC12 cells. Acta Pharmacol. Sin. 2020, 41, 508–515. [Google Scholar] [CrossRef] [PubMed]
- Betarbet, R.; Sherer, T.B.; MacKenzie, G.; Garcia-Osuna, M.; Panov, A.V.; Greenamyre, J.T. Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat. Neurosci. 2000, 3, 1301–1306. [Google Scholar] [CrossRef] [PubMed]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comp. | C5 Linker | R (C5 Substitution) | N1 Indole Substitution | % Inhibition of MAO-B at 10 µM |
|---|---|---|---|---|
| 4a |  |  | 3-Fluorobenzoyl | 46.2 ± 1.1 |
| 4b |  | 9.9 ± 1.5 | ||
| 4c |  | 26.6 ± 0.6 | ||
| 4d |  | 44.0 ± 0.8 | ||
| 4e |  | 47.9 ± 1.6 | ||
| 4f |  | 32.5 ± 1.4 | ||
| 4g |  | 55.7 ± 0.2 | ||
| 4h |  | 38.6 ± 1.6 | ||
| 4i |  | 37.0 ± 0.9 | ||
| 4j |  | 53.5 ± 0.3 | ||
| 4k |  | 42.1 ± 1.7 | ||
| 4l |  | 42.0 ± 0.6 | ||
| 4m |  | 38.1 ± 0.5 | ||
| 4n |  | 32.9 ± 0.3 | ||
| 5a |  |  | 3-Fluorobenzoyl | 15.5 ± 1.1 |
| 5b |  | 45.6 ± 1.2 | ||
| 5c |  | 43.9 ± 1.1 | ||
| 5d |  | 46.8 ± 0.2 | ||
| 5e |  | 44.7 ± 0.5 | ||
| 5f |  | 34.1 ± 0.7 | ||
| 5g |  | 30.6 ± 0.9 | ||
| 5h |  | 32.5 ± 0.5 | ||
| 5i |  | 59.8 ± 0.4 | ||
| 5j |  | 51.1 ± 0.4 | ||
| 6a |  |  | 3-Fluorobenzoyl | 26.8 ± 1.3 |
| 6b |  | 33.8 ± 2.3 | ||
| 6c |  | 35.5 ± 1.5 | ||
| 6d |  | 53.0 ± 0.3 | ||
| 6e |  | 59.6 ± 0.3 | ||
| 7a |  |  | 3-Fluorobenzoyl | 49.8 ± 0.3 |
| 7b |  | 3,4-Dichlorobenzoyl | 84.1 ± 0.0 | |
| 8a |  |  | Free NH | 99.3 ± 0.0 |
| 8b |  | 99.4 ± 0.0 | ||
| 8c |  | 34.2 ± 0.2 | ||
| 8d |  | 15.3 ± 0.8 | ||
| 8e |  | 89.6 ± 0.4 |
| MAO-A | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Cpd | Inhibition% (100 µM) | S.E.M | Inhibition% (30 µM) | S.E.M | Inhibition% (10 µM) | S.E.M | MAO-A IC50 (µM) | MAO-B IC50 (µM) | Selectivity Index (SI) a |
| 7b | <10 | 2.789 | <10 | 1.449 | <10 | 0.334 | >100 | 0.3278 | >305 |
| 8a | 23.48 | 21.66 | 21.66 | 0.920 | 17.16 | 0.687 | >100 | 0.0274 | >3649 |
| 8b | 30.07 | 1.474 | 27.88 | 0.965 | 15.07 | 0.764 | >100 | 0.0305 | >3278 |
| 8e | <10 | 1.959 | <10 | 1.009 | <10 | 0.689 | >100 | 0.4532 | >220 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elsherbeny, M.H.; Kim, J.; Gouda, N.A.; Gotina, L.; Cho, J.; Pae, A.N.; Lee, K.; Park, K.D.; Elkamhawy, A.; Roh, E.J. Highly Potent, Selective, and Competitive Indole-Based MAO-B Inhibitors Protect PC12 Cells against 6-Hydroxydopamine- and Rotenone-Induced Oxidative Stress. Antioxidants 2021, 10, 1641. https://doi.org/10.3390/antiox10101641
Elsherbeny MH, Kim J, Gouda NA, Gotina L, Cho J, Pae AN, Lee K, Park KD, Elkamhawy A, Roh EJ. Highly Potent, Selective, and Competitive Indole-Based MAO-B Inhibitors Protect PC12 Cells against 6-Hydroxydopamine- and Rotenone-Induced Oxidative Stress. Antioxidants. 2021; 10(10):1641. https://doi.org/10.3390/antiox10101641
Chicago/Turabian StyleElsherbeny, Mohamed H., Jushin Kim, Noha A. Gouda, Lizaveta Gotina, Jungsook Cho, Ae Nim Pae, Kyeong Lee, Ki Duk Park, Ahmed Elkamhawy, and Eun Joo Roh. 2021. "Highly Potent, Selective, and Competitive Indole-Based MAO-B Inhibitors Protect PC12 Cells against 6-Hydroxydopamine- and Rotenone-Induced Oxidative Stress" Antioxidants 10, no. 10: 1641. https://doi.org/10.3390/antiox10101641