
Synaptic Dysfunction by Mutations in GRIN2B: Influence of Triheteromeric NMDA Receptors on Gain-of-Function and Loss-of-Function Mutant Classification

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Molecular Biology
2.2. Animals
2.3. Slice Culture and Transfection
2.4. Slice Electrophysiology
2.5. HEK293T Transfection and Whole-Cell Patch-Clamp Electrophysiology
2.6. Statistics
3. Results
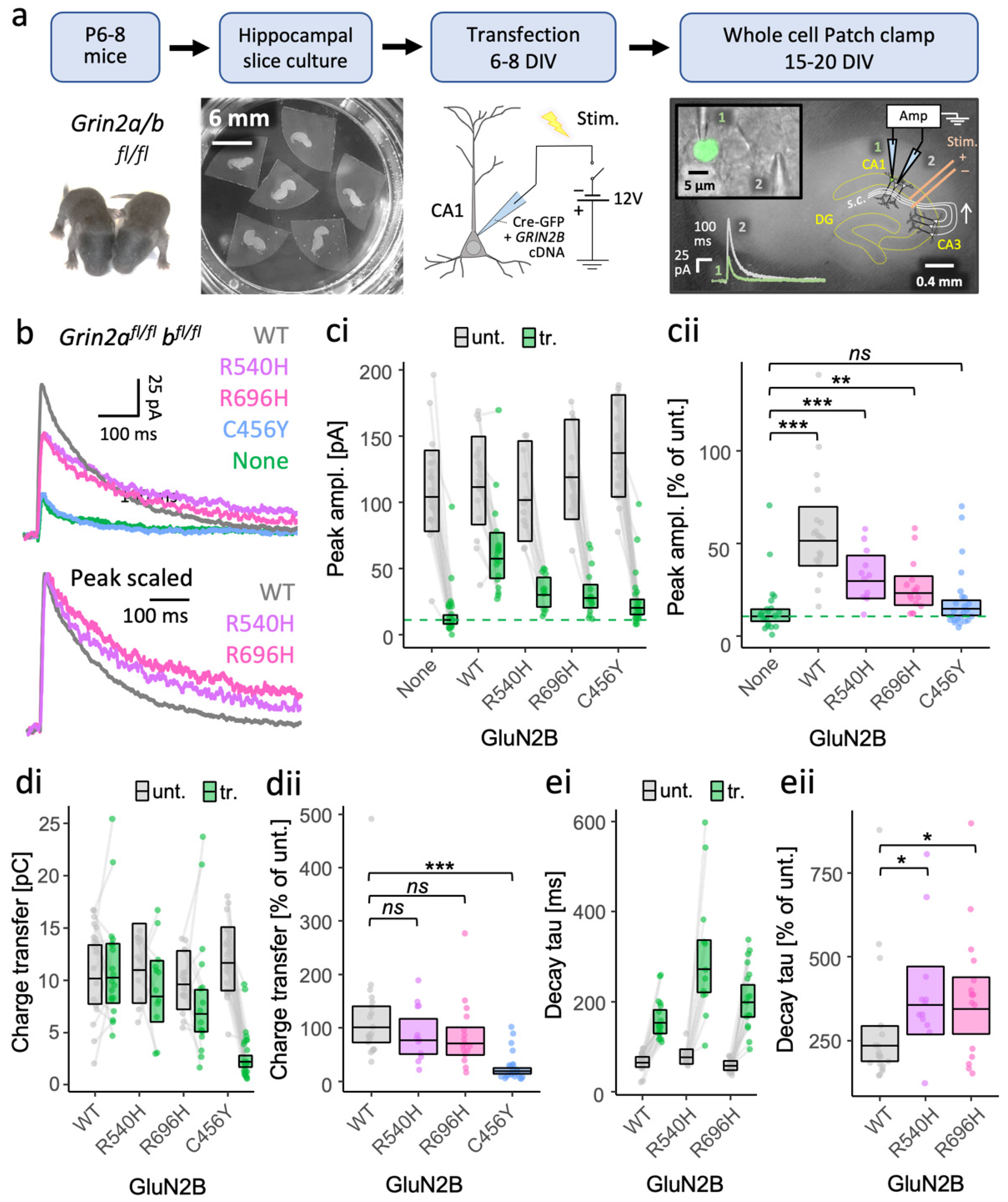
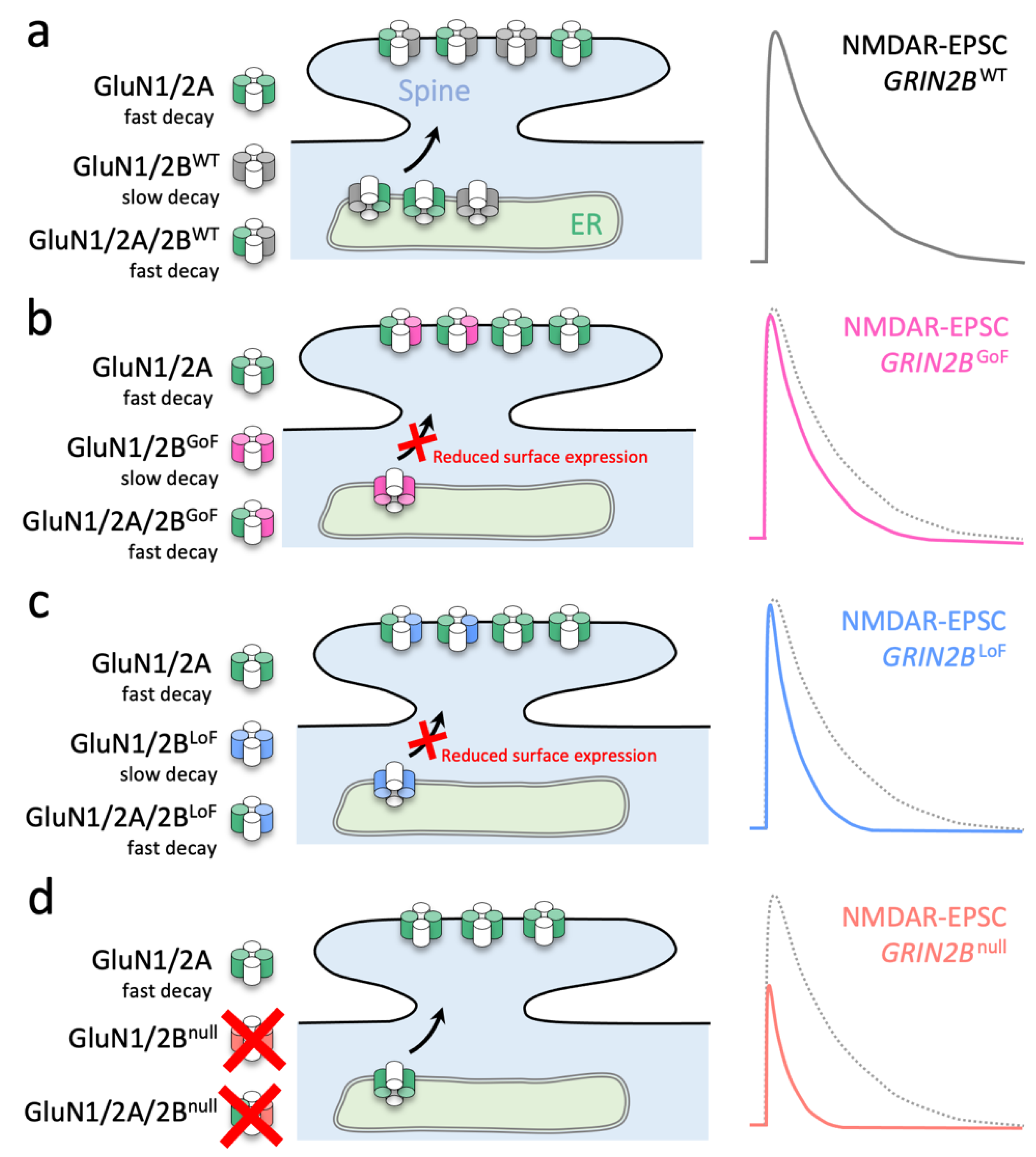
3.1. GoF GRIN2B Mutants Can Traffic to Synapses of Grin2a−/−b−/− Neurons and Produce NMDA-EPSCs with Prolonged Time Course
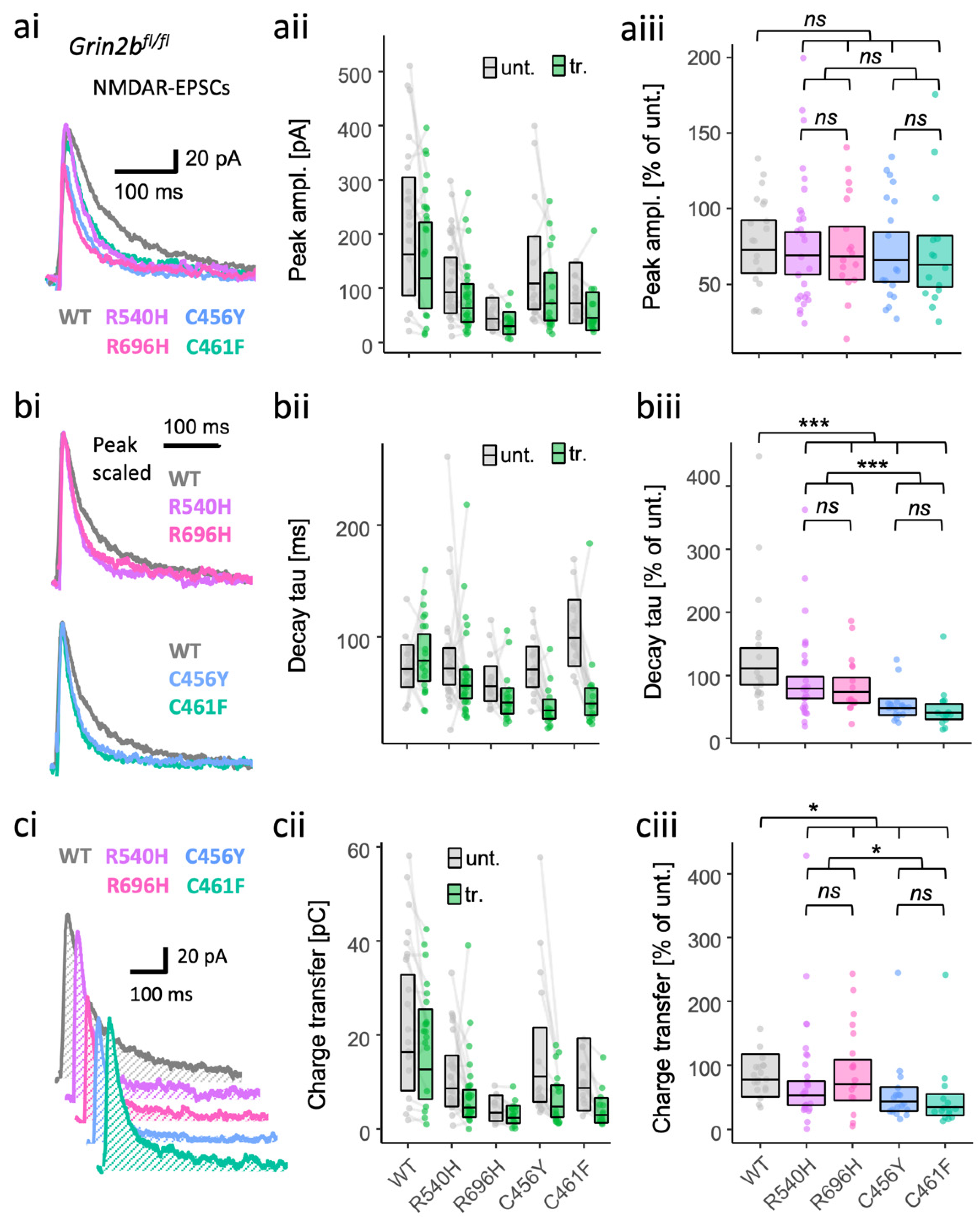
3.2. LoF and GoF GluN2B Mutant Rescue in Grin2b−/− Neurons Can Produce Similar Effects on NMDA-EPSCs


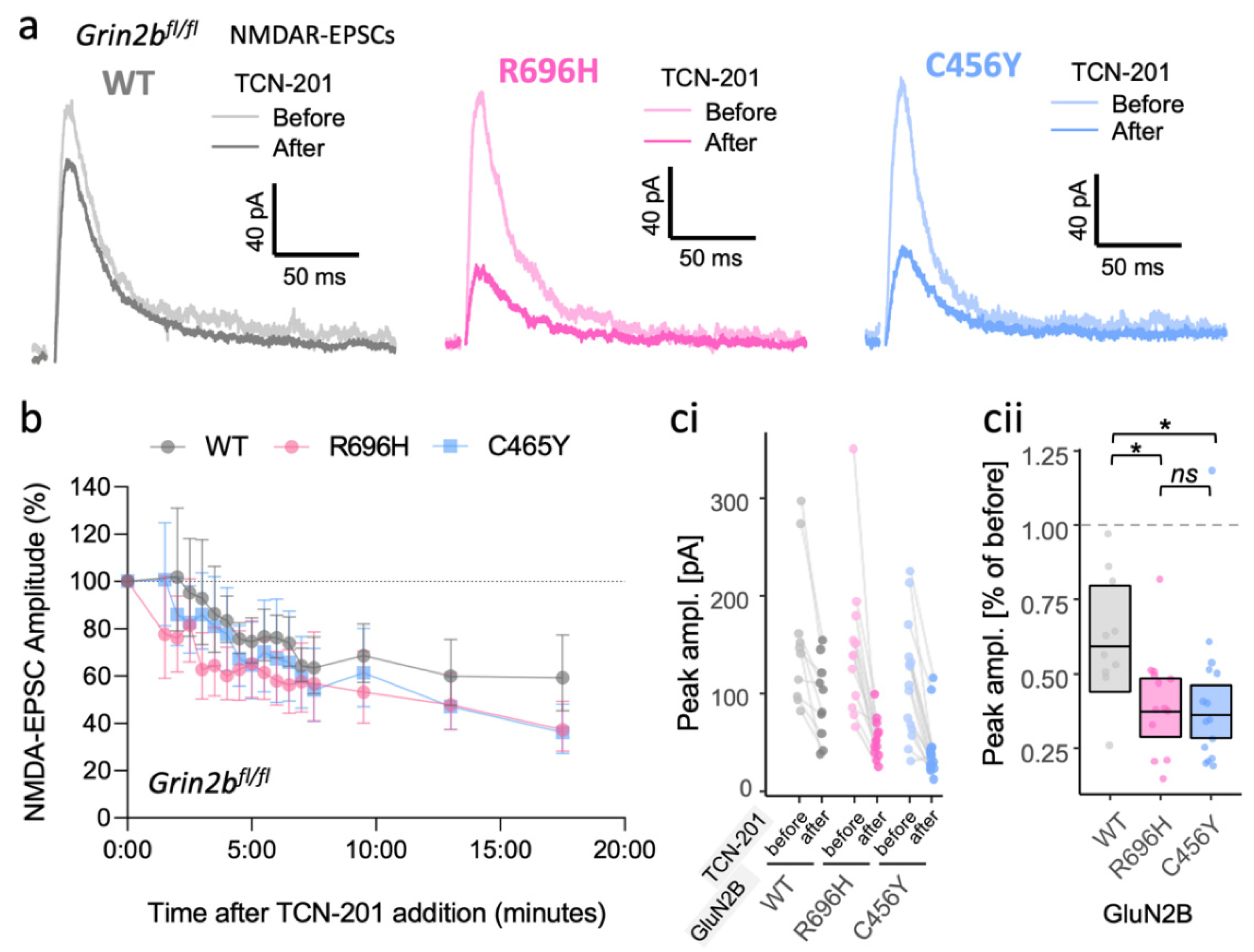
3.3. Inadequate Functional Synaptic Incorporation of GluN1/2B and Co-Assembly with GluN2A in GluN1/2A/2B Triheteromers Result in Similar Phenotypes for GluN2B GoF and LoF Mutants
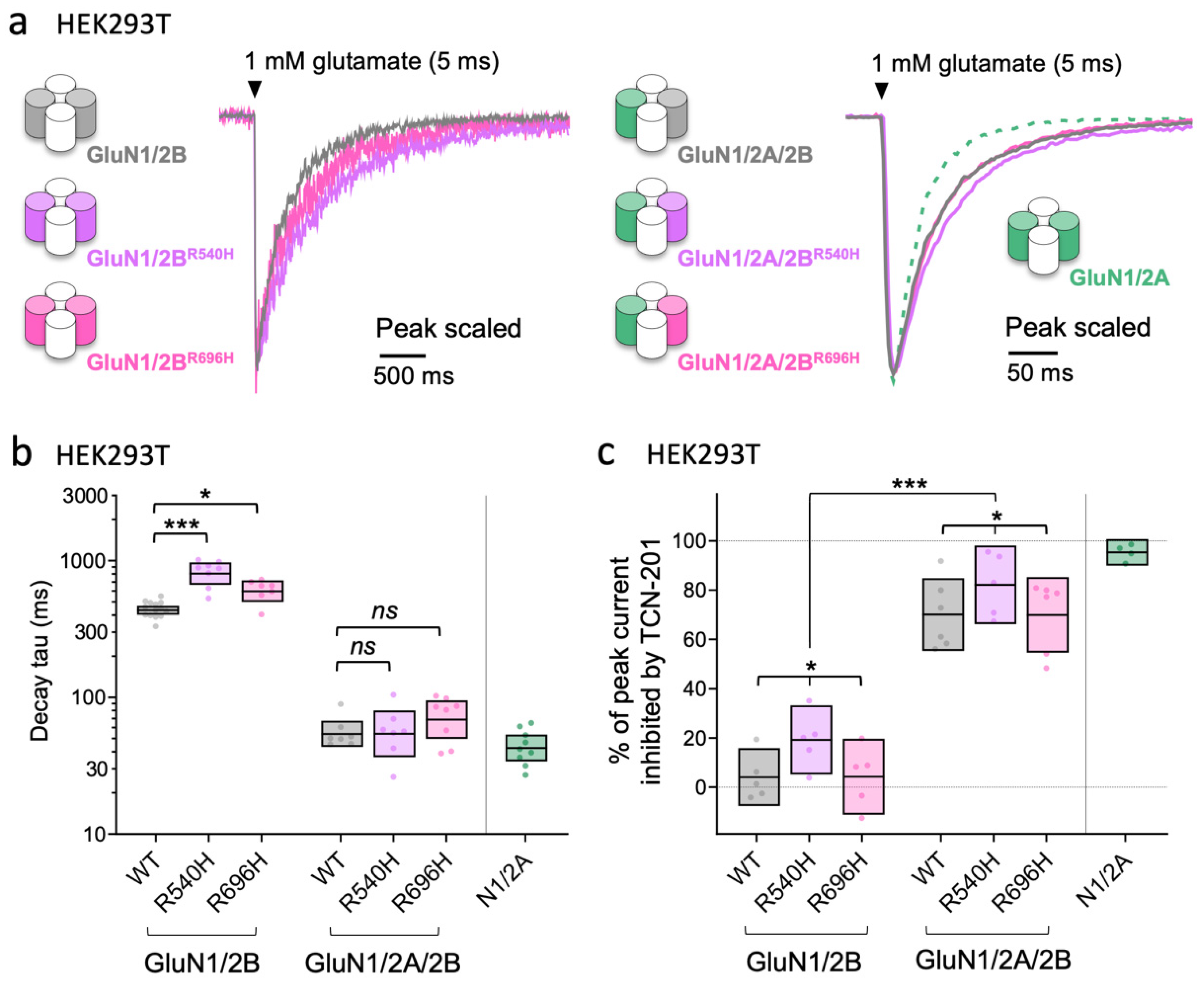
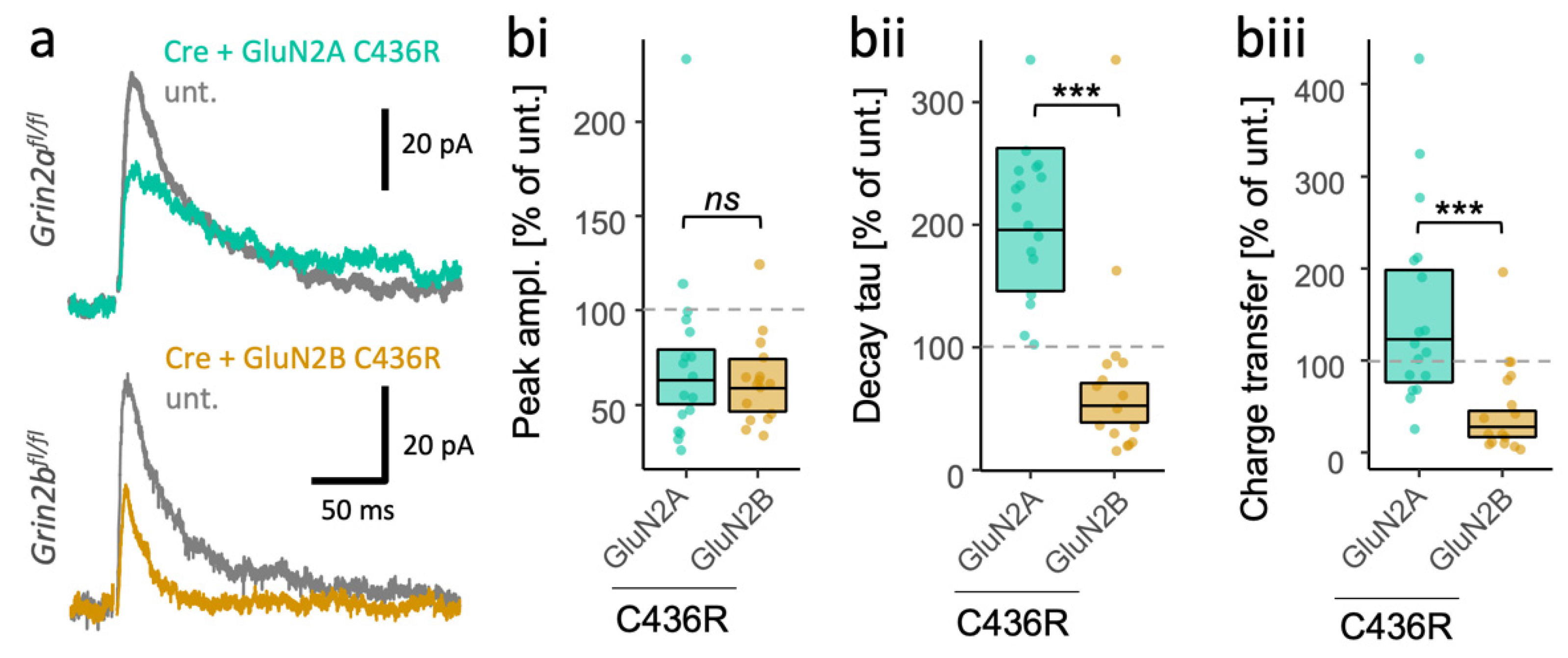
3.4. Different Consequences of the Same Missense LoF Mutation in GluN2A and GluN2B on the Time Course of NMDA-EPSCs
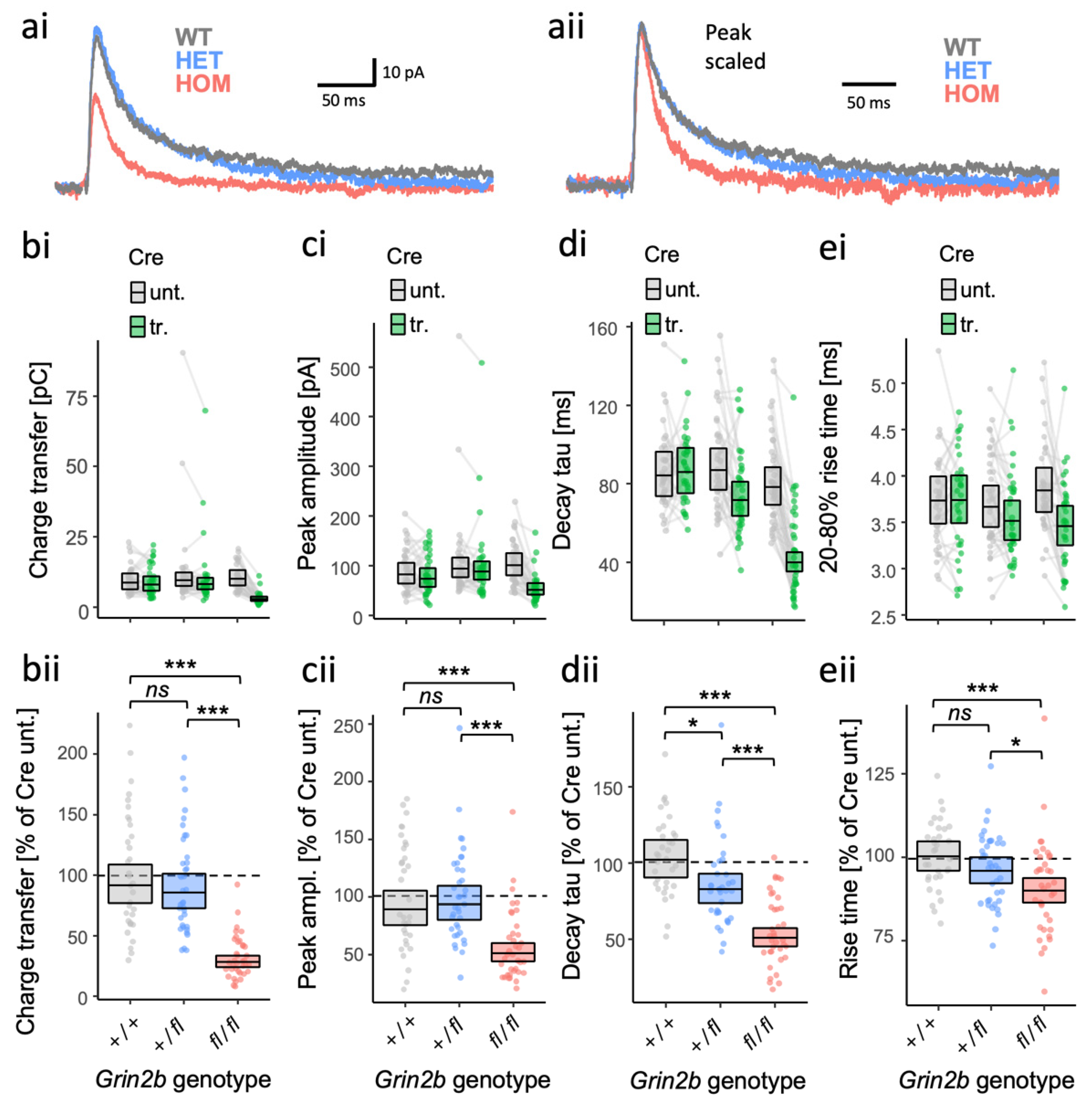
3.5. Partial Haploinsufficiency for a Grin2b Loss-of-Function Allele
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Paoletti, P.; Bellone, C.; Zhou, Q. NMDA Receptor Subunit Diversity: Impact on Receptor Properties, Synaptic Plasticity and Disease. Nat. Rev. Neurosci. 2013, 14, 383–400. [Google Scholar] [CrossRef] [PubMed]
- Hansen, K.B.; Wollmuth, L.P.; Bowie, D.; Furukawa, H.; Menniti, F.S.; Sobolevsky, A.I.; Swanson, G.T.; Swanger, S.A.; Greger, I.H.; Nakagawa, T.; et al. Structure, Function, and Pharmacology of Glutamate Receptor Ion Channels. Pharmacol. Rev. 2021, 73, 298–487. [Google Scholar] [CrossRef] [PubMed]
- Kew, J.N.; Koester, A.; Moreau, J.L.; Jenck, F.; Ouagazzal, A.M.; Mutel, V.; Richards, J.G.; Trube, G.; Fischer, G.; Montkowski, A.; et al. Functional Consequences of Reduction in NMDA Receptor Glycine Affinity in Mice Carrying Targeted Point Mutations in the Glycine Binding Site. J. Neurosci. 2000, 20, 4037–4049. [Google Scholar] [CrossRef] [Green Version]
- Kleckner, N.W.; Dingledine, R. Requirement for Glycine in Activation of NMDA-Receptors Expressed in Xenopus Oocytes. Science 1988, 241, 835–837. [Google Scholar] [CrossRef] [PubMed]
- Mothet, J.-P.; Parent, A.T.; Wolosker, H.; Brady, R.O.; Linden, D.J.; Ferris, C.D.; Rogawski, M.A.; Snyder, S.H. D-Serine Is an Endogenous Ligand for the Glycine Site of the N-Methyl-d-Aspartate Receptor. Proc. Natl. Acad. Sci. USA 2000, 97, 4926–4931. [Google Scholar] [CrossRef] [Green Version]
- Mayer, M.L.; Westbrook, G.L.; Guthrie, P.B. Voltage-Dependent Block by Mg2+ of NMDA Responses in Spinal Cord Neurones. Nature 1984, 309, 261–263. [Google Scholar] [CrossRef]
- Nowak, L.; Bregestovski, P.; Ascher, P.; Herbet, A.; Prochiantz, A. Magnesium Gates Glutamate-Activated Channels in Mouse Central Neurones. Nature 1984, 307, 462–465. [Google Scholar] [CrossRef]
- Mayer, M.L.; Westbrook, G.L. Permeation and Block of N-Methyl-D-Aspartic Acid Receptor Channels by Divalent Cations in Mouse Cultured Central Neurones. J. Physiol. 1987, 394, 501–527. [Google Scholar] [CrossRef] [Green Version]
- Ascher, P.; Nowak, L. The Role of Divalent Cations in the N-Methyl-D-Aspartate Responses of Mouse Central Neurones in Culture. J. Physiol. 1988, 399, 247–266. [Google Scholar] [CrossRef]
- Malenka, R.C.; Kauer, J.A.; Zucker, R.S.; Nicoll, R.A. Postsynaptic Calcium Is Sufficient for Potentiation of Hippocampal Synaptic Transmission. Science 1988, 242, 81–84. [Google Scholar] [CrossRef]
- Collingridge, G.L.; Kehl, S.J.; McLennan, H. Excitatory Amino Acids in Synaptic Transmission in the Schaffer Collateral-Commissural Pathway of the Rat Hippocampus. J. Physiol. 1983, 334, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Kutsuwada, T.; Sakimura, K.; Manabe, T.; Takayama, C.; Katakura, N.; Kushiya, E.; Natsume, R.; Watanabe, M.; Inoue, Y.; Yagi, T.; et al. Impairment of Suckling Response, Trigeminal Neuronal Pattern Formation, and Hippocampal LTD in NMDA Receptor Epsilon 2 Subunit Mutant Mice. Neuron 1996, 16, 333–344. [Google Scholar] [CrossRef] [Green Version]
- Sakimura, K.; Kutsuwada, T.; Ito, I.; Manabe, T.; Takayama, C.; Kushiya, E.; Yagi, T.; Aizawa, S.; Inoue, Y.; Sugiyama, H. Reduced Hippocampal LTP and Spatial Learning in Mice Lacking NMDA Receptor Epsilon 1 Subunit. Nature 1995, 373, 151–155. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Erzurumlu, R.S.; Chen, C.; Jhaveri, S.; Tonegawa, S. Whisker-Related Neuronal Patterns Fail to Develop in the Trigeminal Brainstem Nuclei of NMDAR1 Knockout Mice. Cell 1994, 76, 427–437. [Google Scholar] [CrossRef]
- Mohn, A.R.; Gainetdinov, R.R.; Caron, M.G.; Koller, B.H. Mice with Reduced NMDA Receptor Expression Display Behaviors Related to Schizophrenia. Cell 1999, 98, 427–436. [Google Scholar] [CrossRef] [Green Version]
- Tsien, J.Z.; Huerta, P.T.; Tonegawa, S. The Essential Role of Hippocampal CA1 NMDA Receptor-Dependent Synaptic Plasticity in Spatial Memory. Cell 1996, 87, 1327–1338. [Google Scholar] [CrossRef] [Green Version]
- Single, F.N.; Rozov, A.; Burnashev, N.; Zimmermann, F.; Hanley, D.F.; Forrest, D.; Curran, T.; Jensen, V.; Hvalby, O.; Sprengel, R.; et al. Dysfunctions in Mice by NMDA Receptor Point Mutations NR1(N598Q) and NR1(N598R). J. Neurosci. 2000, 20, 2558–2566. [Google Scholar] [CrossRef] [Green Version]
- Rudhard, Y.; Kneussel, M.; Nassar, M.A.; Rast, G.F.; Annala, A.J.; Chen, P.E.; Tigaret, C.M.; Dean, I.; Roes, J.; Gibb, A.J.; et al. Absence of Whisker-Related Pattern Formation in Mice with NMDA Receptors Lacking Coincidence Detection Properties and Calcium Signaling. J. Neurosci. 2003, 23, 2323–2332. [Google Scholar] [CrossRef] [Green Version]
- Bertocchi, I.; Eltokhi, A.; Rozov, A.; Chi, V.N.; Jensen, V.; Bus, T.; Pawlak, V.; Serafino, M.; Sonntag, H.; Yang, B.; et al. Voltage-Independent GluN2A-Type NMDA Receptor Ca2+ Signaling Promotes Audiogenic Seizures, Attentional and Cognitive Deficits in Mice. Commun. Biol. 2021, 4, 59. [Google Scholar] [CrossRef]
- Duan, Y.; Wang, Q.; Zeng, Q.; Wang, J.; Chen, Z.; Xu, M.; Duan, Y.; Zhao, Z.; Xue, Q.; Cao, X. Striatal GluN2B Involved in Motor Skill Learning and Stimulus-Response Learning. Neuropharmacology 2018, 135, 73–85. [Google Scholar] [CrossRef]
- Von Engelhardt, J.; Doganci, B.; Jensen, V.; Hvalby, Ø.; Göngrich, C.; Taylor, A.; Barkus, C.; Sanderson, D.J.; Rawlins, J.N.P.; Seeburg, P.H.; et al. Contribution of Hippocampal and Extra-Hippocampal NR2B-Containing NMDA Receptors to Performance on Spatial Learning Tasks. Neuron 2008, 60, 846–860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.-C.; Held, R.G.; Chang, S.-C.; Yang, L.; Delpire, E.; Ghosh, A.; Hall, B.J. A Critical Role for GluN2B-Containing NMDA Receptors in Cortical Development and Function. Neuron 2011, 72, 789–805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vicini, S.; Wang, J.F.; Li, J.H.; Zhu, W.J.; Wang, Y.H.; Luo, J.H.; Wolfe, B.B.; Grayson, D.R. Functional and Pharmacological Differences between Recombinant N-Methyl-D-Aspartate Receptors. J. Neurophysiol. 1998, 79, 555–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansen, K.B.; Ogden, K.K.; Yuan, H.; Traynelis, S.F. Distinct Functional and Pharmacological Properties of Triheteromeric GluN1/GluN2A/GluN2B NMDA Receptors. Neuron 2014, 81, 1084–1096. [Google Scholar] [CrossRef] [Green Version]
- Yi, F.; Bhattacharya, S.; Thompson, C.M.; Traynelis, S.F.; Hansen, K.B. Functional and Pharmacological Properties of Triheteromeric GluN1/2B/2D NMDA Receptors. J. Physiol. 2019, 597, 5495–5514. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Khatri, A.; Swanger, S.A.; DiRaddo, J.O.; Yi, F.; Hansen, K.B.; Yuan, H.; Traynelis, S.F. Triheteromeric GluN1/GluN2A/GluN2C NMDARs with Unique Single-Channel Properties Are the Dominant Receptor Population in Cerebellar Granule Cells. Neuron 2018, 99, 315–328.e5. [Google Scholar] [CrossRef] [Green Version]
- Rumbaugh, G.; Prybylowski, K.; Wang, J.F.; Vicini, S. Exon 5 and Spermine Regulate Deactivation of NMDA Receptor Subtypes. J. Neurophysiol. 2000, 83, 1300–1306. [Google Scholar] [CrossRef]
- Yi, F.; Zachariassen, L.G.; Dorsett, K.N.; Hansen, K.B. Properties of Triheteromeric N-Methyl-d-Aspartate Receptors Containing Two Distinct GluN1 Isoforms. Mol. Pharmacol. 2018, 93, 453–467. [Google Scholar] [CrossRef] [Green Version]
- Traynelis, S.F.; Hartley, M.; Heinemann, S.F. Control of Proton Sensitivity of the NMDA Receptor by RNA Splicing and Polyamines. Science 1995, 268, 873–876. [Google Scholar] [CrossRef]
- Maki, B.A.; Cole, R.; Popescu, G.K. Two Serine Residues on GluN2A C-Terminal Tails Control NMDA Receptor Current Decay Times. Channels 2013, 7, 126–132. [Google Scholar] [CrossRef] [Green Version]
- Amico-Ruvio, S.A.; Murthy, S.E.; Smith, T.P.; Popescu, G.K. Zinc Effects on NMDA Receptor Gating Kinetics. Biophys. J. 2011, 100, 1910–1918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrovski, S.; Wang, Q.; Heinzen, E.L.; Allen, A.S.; Goldstein, D.B. Genic Intolerance to Functional Variation and the Interpretation of Personal Genomes. PLoS Genet. 2013, 9, e1003709. [Google Scholar] [CrossRef]
- Pierson, T.M.; Yuan, H.; Marsh, E.D.; Fuentes-Fajardo, K.; Adams, D.R.; Markello, T.; Golas, G.; Simeonov, D.R.; Holloman, C.; Tankovic, A.; et al. GRIN2A Mutation and Early-Onset Epileptic Encephalopathy: Personalized Therapy with Memantine. Ann. Clin. Transl. Neurol. 2014, 1, 190–198. [Google Scholar] [CrossRef] [PubMed]
- Platzer, K.; Yuan, H.; Schütz, H.; Winschel, A.; Chen, W.; Hu, C.; Kusumoto, H.; Heyne, H.O.; Helbig, K.L.; Tang, S.; et al. GRIN2B Encephalopathy: Novel Findings on Phenotype, Variant Clustering, Functional Consequences and Treatment Aspects. J. Med. Genet. 2017, 54, 460–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- XiangWei, W.; Kannan, V.; Xu, Y.; Kosobucki, G.J.; Schulien, A.J.; Kusumoto, H.; Moufawad El Achkar, C.; Bhattacharya, S.; Lesca, G.; Nguyen, S.; et al. Heterogeneous Clinical and Functional Features of GRIN2D-Related Developmental and Epileptic Encephalopathy. Brain 2019, 142, 3009–3027. [Google Scholar] [CrossRef]
- Amador, A.; Bostick, C.D.; Olson, H.; Peters, J.; Camp, C.R.; Krizay, D.; Chen, W.; Han, W.; Tang, W.; Kanber, A.; et al. Modelling and Treating GRIN2A Developmental and Epileptic Encephalopathy in Mice. Brain 2020, 143, 2039–2057. [Google Scholar] [CrossRef]
- Xu, Y.; Song, R.; Chen, W.; Strong, K.; Shrey, D.; Gedela, S.; Traynelis, S.F.; Zhang, G.; Yuan, H. Recurrent Seizure-Related GRIN1 Variant: Molecular Mechanism and Targeted Therapy. Ann. Clin. Transl. Neurol. 2021, 8, 1480–1494. [Google Scholar] [CrossRef]
- Soto, D.; Olivella, M.; Grau, C.; Armstrong, J.; Alcon, C.; Gasull, X.; Santos-Gómez, A.; Locubiche, S.; de Salazar, M.G.; García-Díaz, R.; et al. L-Serine Dietary Supplementation Is Associated with Clinical Improvement of Loss-of-Function GRIN2B-Related Pediatric Encephalopathy. Sci. Signal. 2019, 12, eaaw0936. [Google Scholar] [CrossRef]
- Li, D.; Yuan, H.; Ortiz-Gonzalez, X.R.; Marsh, E.D.; Tian, L.; McCormick, E.M.; Kosobucki, G.J.; Chen, W.; Schulien, A.J.; Chiavacci, R.; et al. GRIN2D Recurrent De Novo Dominant Mutation Causes a Severe Epileptic Encephalopathy Treatable with NMDA Receptor Channel Blockers. Am. J. Hum. Genet. 2016, 99, 802–816. [Google Scholar] [CrossRef] [Green Version]
- Elmasri, M.; Hunter, D.W.; Winchester, G.; Bates, E.E.; Aziz, W.; Van Der Does, D.M.; Karachaliou, E.; Sakimura, K.; Penn, A.C. Common Synaptic Phenotypes Arising from Diverse Mutations in the Human NMDA Receptor Subunit GluN2A. Commun. Biol. 2022, 5, 174. [Google Scholar] [CrossRef]
- XiangWei, W.; Jiang, Y.; Yuan, H. De Novo Mutations and Rare Variants Occurring in NMDA Receptors. Curr. Opin. Physiol. 2018, 2, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Swanger, S.A.; Chen, W.; Wells, G.; Burger, P.B.; Tankovic, A.; Bhattacharya, S.; Strong, K.L.; Hu, C.; Kusumoto, H.; Zhang, J.; et al. Mechanistic Insight into NMDA Receptor Dysregulation by Rare Variants in the GluN2A and GluN2B Agonist Binding Domains. Am. J. Hum. Genet. 2016, 99, 1261–1280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemke, J.R.; Hendrickx, R.; Geider, K.; Laube, B.; Schwake, M.; Harvey, R.J.; James, V.M.; Pepler, A.; Steiner, I.; Hörtnagel, K.; et al. GRIN2B Mutations in West Syndrome and Intellectual Disability with Focal Epilepsy. Ann. Neurol. 2014, 75, 147–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Roak, B.J.; Vives, L.; Fu, W.; Egertson, J.D.; Stanaway, I.B.; Phelps, I.G.; Carvill, G.; Kumar, A.; Lee, C.; Ankenman, K.; et al. Multiplex Targeted Sequencing Identifies Recurrently Mutated Genes in Autism Spectrum Disorders. Science 2012, 338, 1619–1622. [Google Scholar] [CrossRef] [Green Version]
- CFERV Center for Functional Evaluation of Rare Variants. Available online: http://functionalvariants.emory.edu/index.html (accessed on 21 August 2019).
- Lemke, J.R.; Lal, D.; Reinthaler, E.M.; Steiner, I.; Nothnagel, M.; Alber, M.; Geider, K.; Laube, B.; Schwake, M.; Finsterwalder, K.; et al. Mutations in GRIN2A Cause Idiopathic Focal Epilepsy with Rolandic Spikes. Nat. Genet. 2013, 45, 1067–1072. [Google Scholar] [CrossRef]
- Yi, F.; Traynelis, S.F.; Hansen, K.B. Selective Cell-Surface Expression of Triheteromeric NMDA Receptors. Methods Mol. Biol. 2017, 1677, 145–162. [Google Scholar] [CrossRef]
- Akashi, K.; Kakizaki, T.; Kamiya, H.; Fukaya, M.; Yamasaki, M.; Abe, M.; Natsume, R.; Watanabe, M.; Sakimura, K. NMDA Receptor GluN2B (GluR Epsilon 2/NR2B) Subunit Is Crucial for Channel Function, Postsynaptic Macromolecular Organization, and Actin Cytoskeleton at Hippocampal CA3 Synapses. J. Neurosci. 2009, 29, 10869–10882. [Google Scholar] [CrossRef]
- Gray, J.A.; Shi, Y.; Usui, H.; During, M.J.; Sakimura, K.; Nicoll, R.A. Distinct Modes of AMPA Receptor Suppression at Developing Synapses by GluN2A and GluN2B: Single-Cell NMDA Receptor Subunit Deletion In Vivo. Neuron 2011, 71, 1085–1101. [Google Scholar] [CrossRef] [Green Version]
- Rathenberg, J.; Nevian, T.; Witzemann, V. High-Efficiency Transfection of Individual Neurons Using Modified Electrophysiology Techniques. J. Neurosci. Methods 2003, 126, 91–98. [Google Scholar] [CrossRef]
- Campagnola, L.; Kratz, M.B.; Manis, P.B. ACQ4: An Open-Source Software Platform for Data Acquisition and Analysis in Neurophysiology Research. Front. Neuroinform. 2014, 8, 3. [Google Scholar] [CrossRef]
- Traynelis, S.F. Software-Based Correction of Single Compartment Series Resistance Errors. J. Neurosci. Methods 1998, 86, 25–34. [Google Scholar] [CrossRef]
- Malachowski, G.C.; Clegg, R.M.; Redford, G.I. Analytic Solutions to Modelling Exponential and Harmonic Functions Using Chebyshev Polynomials: Fitting Frequency-Domain Lifetime Images with Photobleaching. J. Microsc. 2007, 228, 282–295. [Google Scholar] [CrossRef] [PubMed]
- Hansen, K.B.; Tajima, N.; Risgaard, R.; Perszyk, R.E.; Jørgensen, L.; Vance, K.M.; Ogden, K.K.; Clausen, R.P.; Furukawa, H.; Traynelis, S.F. Structural Determinants of Agonist Efficacy at the Glutamate Binding Site of N-Methyl-D-Aspartate Receptors. Mol. Pharmacol. 2013, 84, 114–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verboven, S.; Hubert, M. LIBRA: A MATLAB Library for Robust Analysis. Chemom. Intell. Lab. Syst. 2005, 75, 127–136. [Google Scholar] [CrossRef]
- Hubert, M.; Rousseeuw, P.J.; Branden, K.V. ROBPCA: A New Approach to Robust Principal Component Analysis. Technometrics 2005, 47, 64–79. [Google Scholar] [CrossRef]
- Bates, D.; Mächler, M.; Bolker, B.; Walker, S. Fitting Linear Mixed-Effects Models Using Lme4. J. Stat. Softw. 2015, 67, 1–48. [Google Scholar] [CrossRef]
- Sun, W.; Hansen, K.B.; Jahr, C.E. Allosteric Interactions between NMDA Receptor Subunits Shape the Developmental Shift in Channel Properties. Neuron 2017, 94, 58–64.e3. [Google Scholar] [CrossRef] [Green Version]
- Lü, W.; Du, J.; Goehring, A.; Gouaux, E. Cryo-EM Structures of the Triheteromeric NMDA Receptor and Its Allosteric Modulation. Science 2017, 355, eaal3729. [Google Scholar] [CrossRef] [Green Version]
- Edman, S.; McKay, S.; MacDonald, L.J.; Samadi, M.; Livesey, M.R.; Hardingham, G.E.; Wyllie, D.J.A. TCN 201 Selectively Blocks GluN2A-Containing NMDARs in a GluN1 Co-Agonist Dependent but Non-Competitive Manner. Neuropharmacology 2012, 63, 441–449. [Google Scholar] [CrossRef] [Green Version]
- Stroebel, D.; Casado, M.; Paoletti, P. Triheteromeric NMDA Receptors: From Structure to Synaptic Physiology. Curr. Opin. Physiol. 2018, 2, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Zhang, J.; Tang, W.; Mizu, R.K.; Kusumoto, H.; Xiangwei, W.; Xu, Y.; Chen, W.; Amin, J.B.; Hu, C.; et al. De Novo GRIN Variants in NMDA Receptor M2 Channel Pore-Forming Loop Are Associated with Neurological Diseases. Hum. Mutat. 2019, 40, 2393–2413. [Google Scholar] [CrossRef] [PubMed]
- She, K.; Ferreira, J.S.; Carvalho, A.L.; Craig, A.M. Glutamate Binding to the GluN2B Subunit Controls Surface Trafficking of N-Methyl-d-Aspartate (NMDA) Receptors. J. Biol. Chem. 2012, 287, 27432–27445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mah, S.J.; Cornell, E.; Mitchell, N.A.; Fleck, M.W. Glutamate Receptor Trafficking: Endoplasmic Reticulum Quality Control Involves Ligand Binding and Receptor Function. J. Neurosci. 2005, 25, 2215–2225. [Google Scholar] [CrossRef] [PubMed]
- Gill, M.B.; Vivithanaporn, P.; Swanson, G.T. Glutamate Binding and Conformational Flexibility of Ligand-Binding Domains Are Critical Early Determinants of Efficient Kainate Receptor Biogenesis. J. Biol. Chem. 2009, 284, 14503–14512. [Google Scholar] [CrossRef] [Green Version]
- Penn, A.C.; Williams, S.R.; Greger, I.H. Gating Motions Underlie AMPA Receptor Secretion from the Endoplasmic Reticulum. Embo J. 2008, 27, 3056–3068. [Google Scholar] [CrossRef]
- Kenny, A.V.; Cousins, S.L.; Pinho, L.; Stephenson, F.A. The Integrity of the Glycine Co-Agonist Binding Site of N-Methyl-D-Aspartate Receptors Is a Functional Quality Control Checkpoint for Cell Surface Delivery. J. Biol. Chem. 2009, 284, 324–333. [Google Scholar] [CrossRef] [Green Version]
- Skrenkova, K.; Hemelikova, K.; Kolcheva, M.; Kortus, S.; Kaniakova, M.; Krausova, B.; Horak, M. Structural Features in the Glycine-Binding Sites of the GluN1 and GluN3A Subunits Regulate the Surface Delivery of NMDA Receptors. Sci. Rep. 2019, 9, 12303. [Google Scholar] [CrossRef] [Green Version]
- Coleman, S.K.; Möykkynen, T.; Hinkkuri, S.; Vaahtera, L.; Korpi, E.R.; Pentikäinen, O.T.; Keinänen, K. Ligand-Binding Domain Determines Endoplasmic Reticulum Exit of AMPA Receptors. J. Biol. Chem. 2010, 285, 36032–36039. [Google Scholar] [CrossRef] [Green Version]
- Coleman, S.K.; Möykkynen, T.; Jouppila, A.; Koskelainen, S.; Rivera, C.; Korpi, E.R.; Keinänen, K. Agonist Occupancy Is Essential for Forward Trafficking of AMPA Receptors. J. Neurosci. 2009, 29, 303–312. [Google Scholar] [CrossRef]
- Vyklicky, V.; Krausova, B.; Cerny, J.; Ladislav, M.; Smejkalova, T.; Kysilov, B.; Korinek, M.; Danacikova, S.; Horak, M.; Chodounska, H.; et al. Surface Expression, Function, and Pharmacology of Disease-Associated Mutations in the Membrane Domain of the Human GluN2B Subunit. Front. Mol. Neurosci. 2018, 11, 110. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Keramidas, A.; Harvey, R.J.; Lynch, J.W. Effects of GluN2A and GluN2B Gain-of-Function Epilepsy Mutations on Synaptic Currents Mediated by Diheteromeric and Triheteromeric NMDA Receptors. Neurobiol. Dis. 2020, 140, 104850. [Google Scholar] [CrossRef] [PubMed]
- Fedele, L.; Newcombe, J.; Topf, M.; Gibb, A.; Harvey, R.J.; Smart, T.G. Disease-Associated Missense Mutations in GluN2B Subunit Alter NMDA Receptor Ligand Binding and Ion Channel Properties. Nat. Commun. 2018, 9, 957. [Google Scholar] [CrossRef] [PubMed]
- Shin, W.; Kim, K.; Serraz, B.; Cho, Y.S.; Kim, D.; Kang, M.; Lee, E.-J.; Lee, H.; Bae, Y.C.; Paoletti, P.; et al. Early Correction of Synaptic Long-Term Depression Improves Abnormal Anxiety-like Behavior in Adult GluN2B-C456Y-Mutant Mice. PLoS Biol. 2020, 18, e3000717. [Google Scholar] [CrossRef] [PubMed]
- Bell, S.; Maussion, G.; Jefri, M.; Peng, H.; Theroux, J.-F.; Silveira, H.; Soubannier, V.; Wu, H.; Hu, P.; Galat, E.; et al. Disruption of GRIN2B Impairs Differentiation in Human Neurons. Stem Cell Rep. 2018, 11, 183–196. [Google Scholar] [CrossRef] [PubMed]
- Sceniak, M.P.; Fedder, K.N.; Wang, Q.; Droubi, S.; Babcock, K.; Patwardhan, S.; Wright-Zornes, J.; Pham, L.; Sabo, S.L. A GluN2B Mutation Identified in Autism Prevents NMDA Receptor Trafficking and Interferes with Dendrite Growth. J. Cell. Sci. 2019, 132, jcs232892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bahry, J.A.; Fedder-Semmes, K.N.; Sceniak, M.P.; Sabo, S.L. An Autism-Associated de Novo Mutation in GluN2B Destabilizes Growing Dendrites by Promoting Retraction and Pruning. Front. Cell. Neurosci. 2021, 15, 290. [Google Scholar] [CrossRef]
- Sheng, M.; Cummings, J.; Roldan, L.A.; Jan, Y.N.; Jan, L.Y. Changing Subunit Composition of Heteromeric NMDA Receptors during Development of Rat Cortex. Nature 1994, 368, 144–147. [Google Scholar] [CrossRef]
- Monyer, H.; Burnashev, N.; Laurie, D.J.; Sakmann, B.; Seeburg, P.H. Developmental and Regional Expression in the Rat Brain and Functional Properties of Four NMDA Receptors. Neuron 1994, 12, 529–540. [Google Scholar] [CrossRef]
- Rauner, C.; Köhr, G. Triheteromeric NR1/NR2A/NR2B Receptors Constitute the Major N-Methyl-D-Aspartate Receptor Population in Adult Hippocampal Synapses. J. Biol. Chem. 2011, 286, 7558–7566. [Google Scholar] [CrossRef] [Green Version]
- Von Engelhardt, J.; Bocklisch, C.; Tönges, L.; Herb, A.; Mishina, M.; Monyer, H. GluN2D-Containing NMDA Receptors-Mediate Synaptic Currents in Hippocampal Interneurons and Pyramidal Cells in Juvenile Mice. Front. Cell. Neurosci. 2015, 9, 95. [Google Scholar] [CrossRef] [Green Version]
- Perszyk, R.E.; DiRaddo, J.O.; Strong, K.L.; Low, C.-M.; Ogden, K.K.; Khatri, A.; Vargish, G.A.; Pelkey, K.A.; Tricoire, L.; Liotta, D.C.; et al. GluN2D-Containing N-Methyl-d-Aspartate Receptors Mediate Synaptic Transmission in Hippocampal Interneurons and Regulate Interneuron Activity. Mol. Pharmacol. 2016, 90, 689–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swanger, S.A.; Vance, K.M.; Acker, T.M.; Zimmerman, S.S.; DiRaddo, J.O.; Myers, S.J.; Bundgaard, C.; Mosley, C.A.; Summer, S.L.; Menaldino, D.S.; et al. A Novel Negative Allosteric Modulator Selective for GluN2C/2D-Containing NMDA Receptors Inhibits Synaptic Transmission in Hippocampal Interneurons. ACS Chem. Neurosci. 2018, 9, 306–319. [Google Scholar] [CrossRef] [PubMed]
- Piña-Crespo, J.C.; Gibb, A.J. Subtypes of NMDA Receptors in New-Born Rat Hippocampal Granule Cells. J. Physiol. 2002, 541, 41–64. [Google Scholar] [CrossRef]
- Logan, S.M.; Partridge, J.G.; Matta, J.A.; Buonanno, A.; Vicini, S. Long-Lasting NMDA Receptor-Mediated EPSCs in Mouse Striatal Medium Spiny Neurons. J. Neurophysiol. 2007, 98, 2693–2704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brickley, S.G.; Misra, C.; Mok, M.H.S.; Mishina, M.; Cull-Candy, S.G. NR2B and NR2D Subunits Coassemble in Cerebellar Golgi Cells to Form a Distinct NMDA Receptor Subtype Restricted to Extrasynaptic Sites. J. Neurosci. 2003, 23, 4958–4966. [Google Scholar] [CrossRef] [PubMed]
- Swanger, S.A.; Vance, K.M.; Pare, J.-F.; Sotty, F.; Fog, K.; Smith, Y.; Traynelis, S.F. NMDA Receptors Containing the GluN2D Subunit Control Neuronal Function in the Subthalamic Nucleus. J. Neurosci. 2015, 35, 15971–15983. [Google Scholar] [CrossRef] [Green Version]
- Jones, S.; Gibb, A.J. Functional NR2B- and NR2D-Containing NMDA Receptor Channels in Rat Substantia Nigra Dopaminergic Neurones. J. Physiol. 2005, 569, 209–221. [Google Scholar] [CrossRef]
- Brothwell, S.L.C.; Barber, J.L.; Monaghan, D.T.; Jane, D.E.; Gibb, A.J.; Jones, S. NR2B- and NR2D-Containing Synaptic NMDA Receptors in Developing Rat Substantia Nigra Pars Compacta Dopaminergic Neurones. J. Physiol. 2008, 586, 739–750. [Google Scholar] [CrossRef]
- Huang, Z.; Gibb, A.J. Mg2+ Block Properties of Triheteromeric GluN1–GluN2B–GluN2D NMDA Receptors on Neonatal Rat Substantia Nigra Pars Compacta Dopaminergic Neurones. J. Physiol. 2014, 592, 2059–2078. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Ma, Y.; Dunn, A.R.; Bradner, J.M.; Scimemi, A.; Miller, G.W.; Wichmann, T.; Traynelis, S.F. NMDA Receptor Blockade Ameliorates Abnormalities of Spike Firing of Subthalamic Nucleus Neurons in a Parkinsonian Non-Human Primate. J. Neurosci. Res. 2018, 96, 1324–1335. [Google Scholar] [CrossRef]
- Hildebrand, M.E.; Pitcher, G.M.; Harding, E.K.; Li, H.; Beggs, S.; Salter, M.W. GluN2B and GluN2D NMDARs Dominate Synaptic Responses in the Adult Spinal Cord. Sci. Rep. 2014, 4, 4094. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Subunit | Mutation | Phenotype * | Surface Expression $ | Current Density $ | Deactivation $ | Glu Potency $ | Gly Potency $ | Charge Transfer $ | Class |
|---|---|---|---|---|---|---|---|---|---|
| GRIN2B | R540H | ID, fSz (ASD, gSz) | −2.4 | −1.2 | +2.0 | 2.3 | +1.5 | +1.4 | GoF |
| R696H | ID, ASD (gSz) | −1.3 | −4.1 | +3.6 | 4.5 | −1.2 | +1.5 | GoF | |
| C456Y | ID, ASD | −11.8 | −1367 | n.d. | 3.8 | −2.6 | n.d. | LoF | |
| C461F | ID, ASD, gSz | −6.1 | −9.8 | −20.4 | −113 | 2.5 | −171 | LoF | |
| C436R | ID, ASD, fSz, gSz | −8.7 | −820 | n.d. | n.d. | n.d. | n.d. | LoF | |
| GRIN2A | C436R | gSz | −66.7 | −3400 | n.d. | −1.5 | +1.5 | n.d. | LoF |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elmasri, M.; Lotti, J.S.; Aziz, W.; Steele, O.G.; Karachaliou, E.; Sakimura, K.; Hansen, K.B.; Penn, A.C. Synaptic Dysfunction by Mutations in GRIN2B: Influence of Triheteromeric NMDA Receptors on Gain-of-Function and Loss-of-Function Mutant Classification. Brain Sci. 2022, 12, 789. https://doi.org/10.3390/brainsci12060789
Elmasri M, Lotti JS, Aziz W, Steele OG, Karachaliou E, Sakimura K, Hansen KB, Penn AC. Synaptic Dysfunction by Mutations in GRIN2B: Influence of Triheteromeric NMDA Receptors on Gain-of-Function and Loss-of-Function Mutant Classification. Brain Sciences. 2022; 12(6):789. https://doi.org/10.3390/brainsci12060789
Chicago/Turabian StyleElmasri, Marwa, James S. Lotti, Wajeeha Aziz, Oliver G. Steele, Eirini Karachaliou, Kenji Sakimura, Kasper B. Hansen, and Andrew C. Penn. 2022. "Synaptic Dysfunction by Mutations in GRIN2B: Influence of Triheteromeric NMDA Receptors on Gain-of-Function and Loss-of-Function Mutant Classification" Brain Sciences 12, no. 6: 789. https://doi.org/10.3390/brainsci12060789