
Is L-Glutamate Toxic to Neurons and Thereby Contributes to Neuronal Loss and Neurodegeneration? A Systematic Review


Abstract
: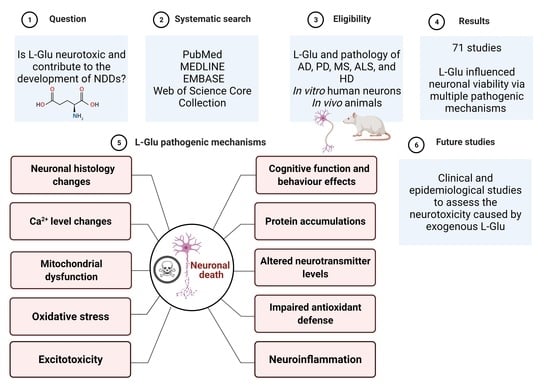
1. Introduction
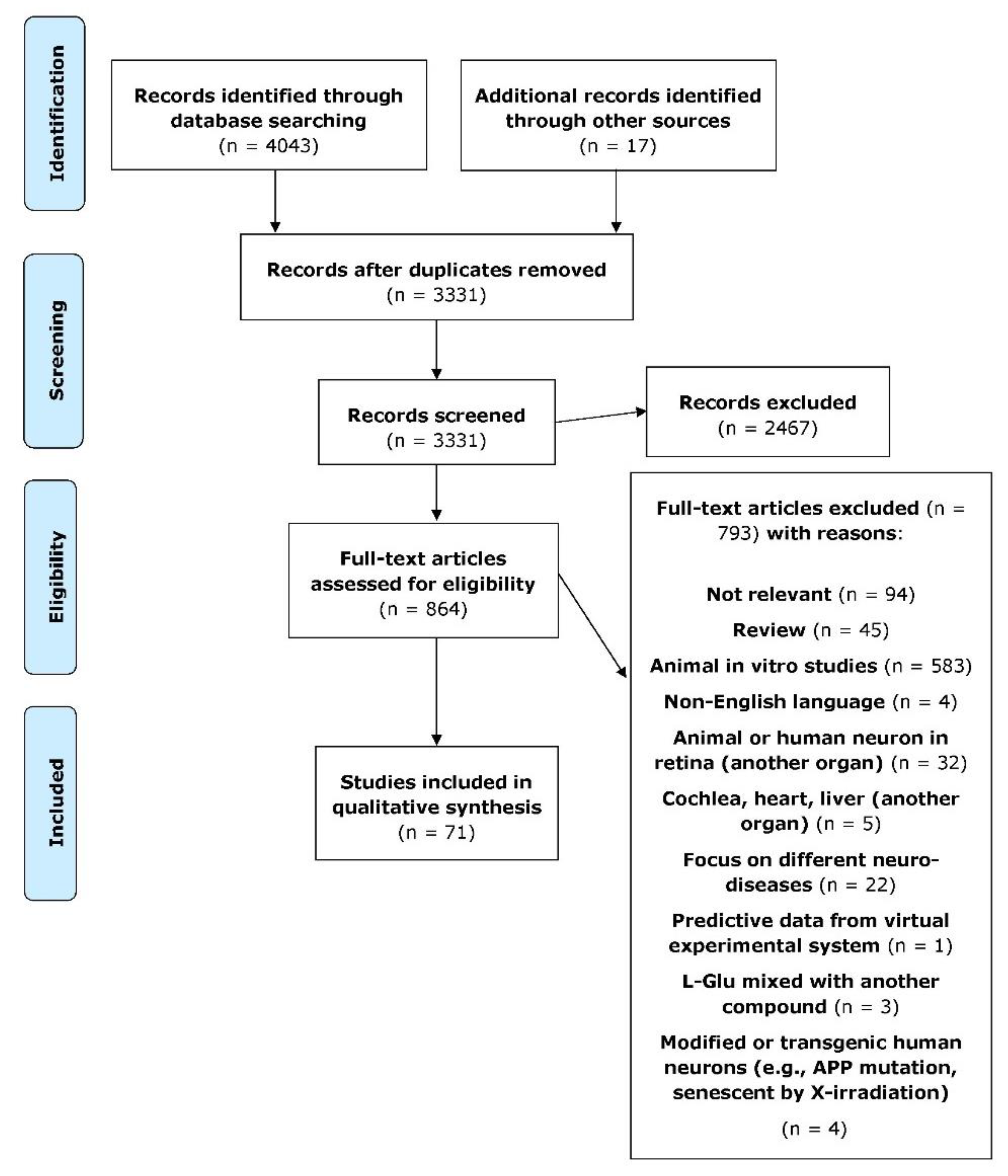
2. Materials and Methods
2.1. Search Scheme
2.2. Data Extraction and Collection
2.3. Eligibility Criteria
2.3.1. Inclusion Criteria
2.3.2. Exclusion Criteria
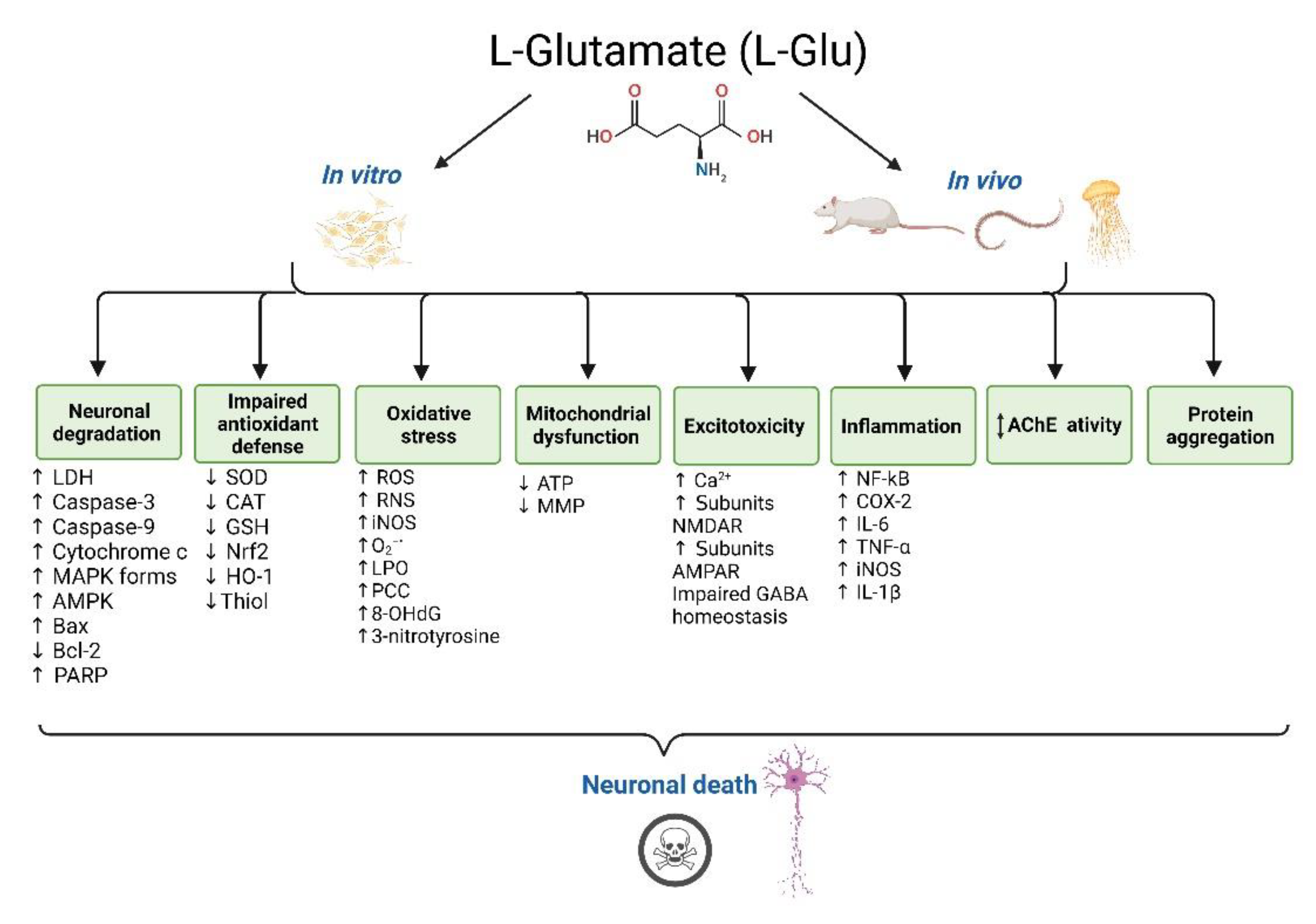
3. Results
3.1. In Vitro Studies Evaluating L-Glu Toxicity in Human Neurons
3.1.1. L-Glu Exposure Reduces Neuronal Viability
3.1.2. L-Glu Exposure Impairs Cellular Oxidant Defence and Stimulates Oxidative Stress
3.1.3. L-Glu Enhances Acetylcholinesterase (AChE) Activity
3.1.4. L-Glu Exposure Triggers Mitochondria Dysfunction and Neuronal Apoptosis
3.1.5. L-Glu Exposure Stimulates Excitotoxicity and Alters Neuronal Calcium Levels
3.1.6. L-Glu Exposure Triggers Neuroinflammation
3.1.7. L-Glu Exposure Affects the Morphological Characteristics of Neurons
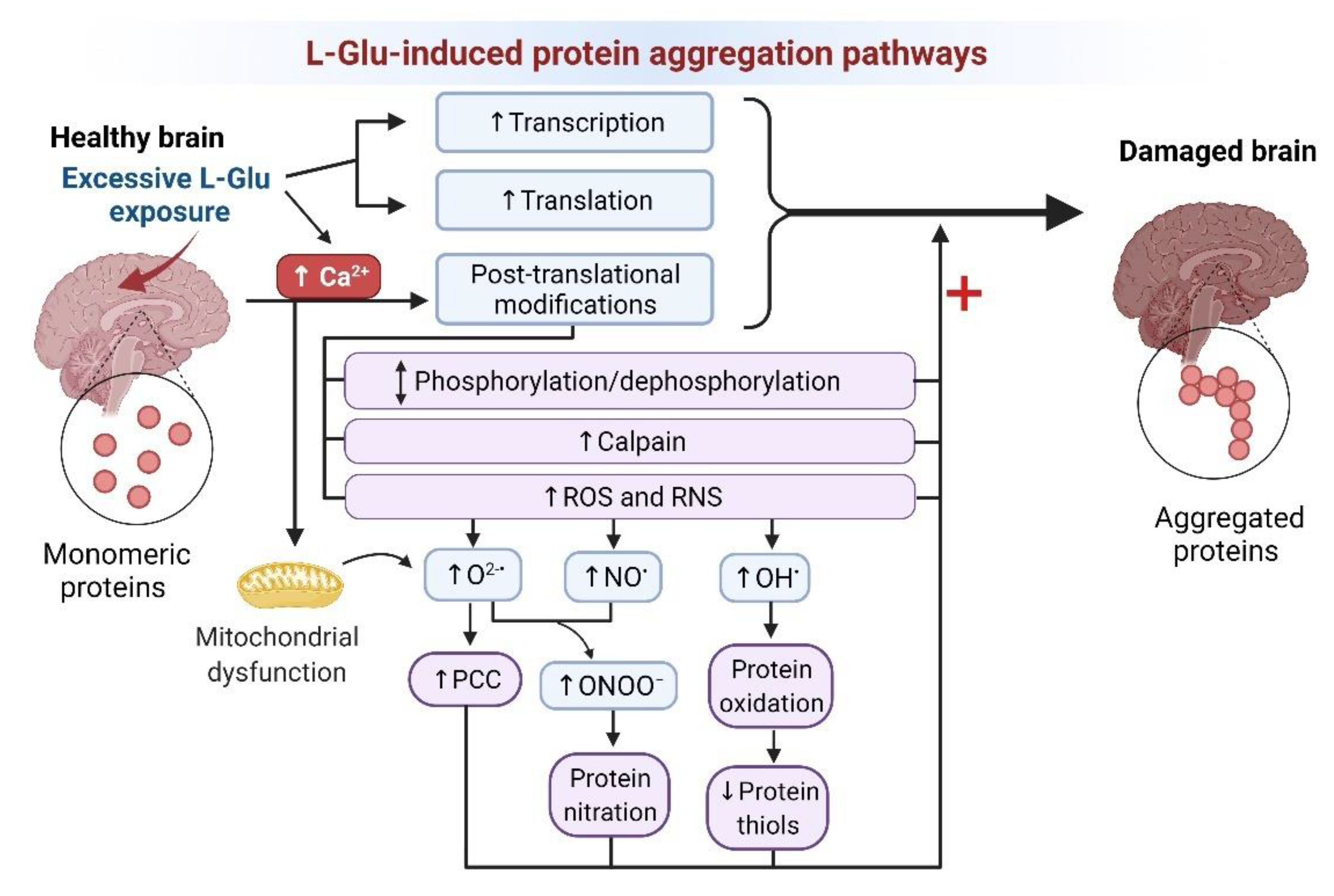
3.1.8. Protein Aggregation
3.2. In Vivo Studies Evaluating L-Glu Toxicity
3.2.1. Administration of L-Glu Directly to Animals
L-Glu Administration Reduces Neuronal Viability
L-Glu Administration Impairs Cellular Oxidant Defence and Stimulates Oxidative Stress
L-Glu Administration Influences Acetylcholinesterase (AChE) Activity
L-Glu Administration Influences Neurotransmitter Levels
L-Glu Administration Triggers Neuronal Apoptosis
Excitotoxicity, Calcium Level, and Other Ions in the Brain
Neuroinflammation
Histology Alteration
Behaviour and Cognitive Function
Protein Aggregation
Brain Weight
3.2.2. L-Glu Administration Directly to Animal Brains
Antioxidant and Oxidative Stress Markers
Neurotransmitter Levels
Mitochondrial Dysfunction and Apoptosis
Calcium Level
Neuroinflammation
Histological Abnormalities
Behaviour and Cognitive Function
Electroencephalogram (EEG)
L-Glu Administration Directly to an Animal’s Living Media
4. Discussion
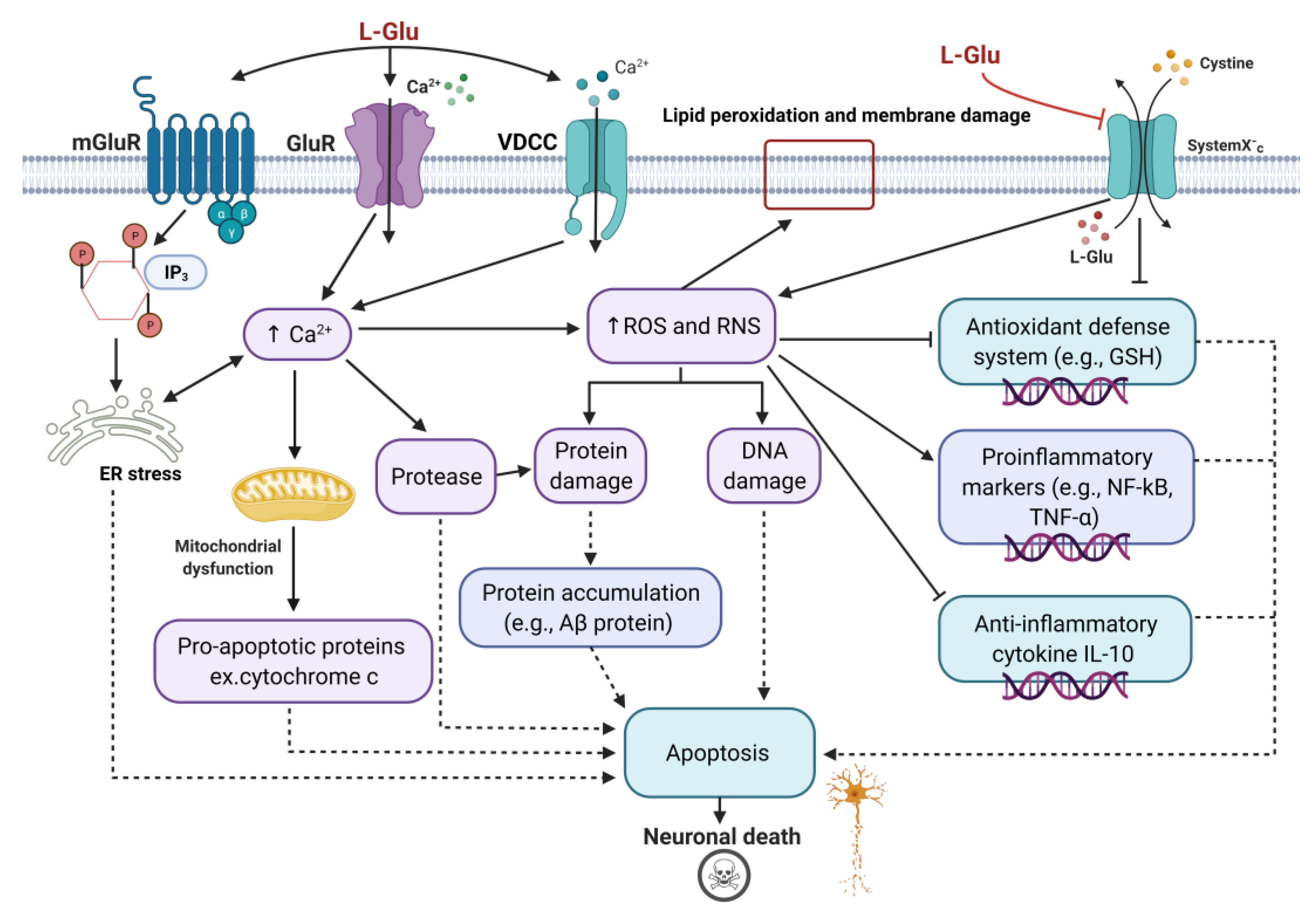
4.1. Common L-Glu Neurotoxic Pathways In Vitro and In Vivo
4.1.1. Cellular and Molecular Changes
4.1.2. Neural Structure Changes
4.2. Other Neuropathology Observed after L-Glu Administration In Vivo
4.2.1. Brain Structural Changes
4.2.2. Changes in Behaviour and Cognition
4.2.3. Brain Weight and Homeostasis Changes
4.3. Study Limitations
5. Summary and Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Zhou, Y.; Danbolt, N. Glutamate as a neurotransmitter in the healthy brain. J. Neural. Transm. 2014, 121, 799–817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willard, S.; Koochekpour, S. Glutamate, glutamate receptors, and downstream signaling pathways. Int. J. Biol. Sci. 2013, 9, 948–959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moldovan, O.-L.; Rusu, A.; Tanase, C.; Vari, C.-E. Glutamate—A multifaceted molecule: Endogenous neurotransmitter, controversial food additive, design compound for anti-cancer drugs. A critical appraisal. Food Chem. Toxicol. 2021, 153, 112290. [Google Scholar] [CrossRef] [PubMed]
- Egbenya, D.; Aidoo, E.; Kyei, G. Glutamate receptors in brain development. Childs Nerv. Syst. 2021, 37, 2753–2758. [Google Scholar] [CrossRef]
- Andersen, J.; Markussen, K.; Jakobsen, E.; Schousboe, A.; Waagepetersen, H.; Rosenberg, P.; Aldana, B. Glutamate metabolism and recycling at the excitatory synapse in health and neurodegeneration. Neuropharmacology 2021, 196, 108719. [Google Scholar] [CrossRef]
- Mahmoud, S.; Gharagozloo, M.; Simard, C.; Gris, D. Astrocytes maintain glutamate homeostasis in the CNS by controlling the balance between glutamate uptake and release. Cells 2019, 8, 184. [Google Scholar] [CrossRef] [Green Version]
- Iovino, L.; Tremblay, M.; Civiero, L. Glutamate-induced excitotoxicity in Parkinson’s disease: The role of glial cells. J. Pharmacol. Sci. 2020, 144, 151–164. [Google Scholar] [CrossRef]
- Moussawi, K.; Riegel, A.; Nair, S.; Kalivas, P. Extracellular glutamate: Functional compartments operate in different concentration ranges. Front. Syst. Neurosci. 2011, 5, 94. [Google Scholar] [CrossRef] [Green Version]
- Leibowitz, A.; Boyko, M.; Shapira, Y.; Zlotnik, A. Blood glutamate scavenging: Insight into neuroprotection. Int. J. Mol. Sci. 2012, 13, 10041–10066. [Google Scholar] [CrossRef] [Green Version]
- Hynd, M.; Scott, H.; Dodd, P. Glutamate-mediated excitotoxicity and neurodegeneration in Alzheimer’s disease. Neurochem. Int. 2004, 45, 583–595. [Google Scholar] [CrossRef]
- Zumkehr, J.; Rodriguez-Ortiz, C.; Cheng, D.; Kieu, Z.; Wai, T.; Hawkins, C.; Kilian, J.; Lim, S.; Medeiros, R.; Kitazawa, M. Ceftriaxone ameliorates tau pathology and cognitive decline via restoration of glial glutamate transporter in a mouse model of Alzheimer’s disease. Neurobiol. Aging 2015, 36, 2260–2271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Tan, F.; Xu, P.; Qu, S. Recent advance in the relationship between excitatory amino acid transporters and Parkinson’s disease. Neural. Plast. 2016, 2016, 8941327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Wang, F.; Mai, D.; Qu, S. Molecular mechanisms of glutamate toxicity in Parkinson’s disease. Front. Neurosci. 2020, 14, 585584. [Google Scholar] [CrossRef] [PubMed]
- Bolton, C.; Paul, C. Glutamate receptors in neuroinflammatory demyelinating disease. Mediators Inflamm. 2006, 2006, 93684. [Google Scholar] [CrossRef]
- Rajda, C.; Pukoli, D.; Bende, Z.; Majláth, Z.; Vécsei, L. Excitotoxins, mitochondrial and redox disturbances in multiple sclerosis. Int. J. Mol. Sci. 2017, 18, 353. [Google Scholar] [CrossRef] [Green Version]
- Stojanovic, I.; Kostic, M.; Ljubisavljevic, S. The role of glutamate and its receptors in multiple sclerosis. J. Neural. Transm. 2014, 121, 945–955. [Google Scholar] [CrossRef]
- Fomin, V.; Richard, P.; Hoque, M.; Li, C.; Gu, Z.; Fissore, O.; Leary, M.; Tian, B.; Prives, C.; Manley, J. The C9ORF72 Gene, implicated in amyotrophic lateral sclerosis and frontotemporal dementia, encodes a protein that functions in control of endothelin and glutamate signaling. Mol. Cell. Biol. 2018, 38, e00155-18. [Google Scholar] [CrossRef] [Green Version]
- Plaitakis, A.; Constantakakis, E. Altered metabolism of excitatory amino acids, N-acetyl-aspartate and N-acetyl-aspartylglutamate in amyotrophic lateral sclerosis. Brain Res. Bull. 1993, 30, 381–386. [Google Scholar] [CrossRef]
- Rothstein, J.; Martin, L.; Kuncl, R. Decreased glutamate transport by the brain and spinal cord in amyotrophic lateral sclerosis. N. Engl. J. Med. 1992, 326, 1464–1468. [Google Scholar] [CrossRef]
- Estrada Sánchez, A.; Mejía-Toiber, J.; Massieu, L. Excitotoxic neuronal death and the pathogenesis of Huntington’s disease. Arch. Med. Res. 2008, 39, 265–276. [Google Scholar] [CrossRef]
- Zeron, M.; Chen, N.; Moshaver, A.; Lee, A.; Wellington, C.; Hayden, M.; Raymond, L. Mutant huntingtin enhances excitotoxic cell death. Mol. Cell. Neurosci. 2001, 17, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Fukui, M.; Song, J.-H.; Choi, J.; Choi, H.; Zhu, B. Mechanism of glutamate-induced neurotoxicity in HT22 mouse hippocampal cells. Eur. J. Pharmacol. 2009, 617, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Pereira, C.; Oliveira, C. Glutamate toxicity on a PC12 cell line involves glutathione (GSH) depletion and oxidative stress. Free Radic. Biol. Med. 1997, 23, 637–647. [Google Scholar] [CrossRef]
- Kang, T.; Bae, K.-H.; Yu, M.-j.; Kim, W.-K.; Hwang, H.-R.; Jung, H.; Lee, P.; Kang, S.; Yoon, T.-S.; Park, S.; et al. Phosphoproteomic analysis of neuronal cell death by glutamate-induced oxidative stress. Proteomics 2007, 7, 2624–2635. [Google Scholar] [CrossRef]
- Murphy, T.; Miyamoto, M.; Sastre, A.; Schnaar, R.; Coyle, J. Glutamate toxicity in a neuronal cell line involves inhibition of cystine transport leading to oxidative stress. Neuron 1989, 2, 1547–1558. [Google Scholar] [CrossRef]
- Tobaben, S.; Grohm, J.; Seiler, A.; Conrad, M.; Plesnila, N.; Culmsee, C. Bid-mediated mitochondrial damage is a key mechanism in glutamate-induced oxidative stress and AIF-dependent cell death in immortalized HT-22 hippocampal neurons. Cell Death Differ. 2011, 18, 282–292. [Google Scholar] [CrossRef] [Green Version]
- Shivasharan, B.; Nagakannan, P.; Thippeswamy, B.; Veerapur, V. Protective effect of calendula officinalis L. flowers against monosodium glutamate induced oxidative stress and excitotoxic brain damage in rats. Indian J. Clin. Biochem. 2013, 28, 292–298. [Google Scholar] [CrossRef] [Green Version]
- Onaolapo, O.; Onaolapo, A.; Akanmu, M.; Gbola, O. Evidence of alterations in brain structure and antioxidant status following ‘low-dose’ monosodium glutamate ingestion. Pathophysiology 2016, 23, 147–156. [Google Scholar] [CrossRef]
- Lau, A.; Tymianski, M. Glutamate receptors, neurotoxicity and neurodegeneration. Pflugers Arch. 2010, 460, 525–542. [Google Scholar] [CrossRef]
- Dong, X.-x.; Wang, Y.; Qin, Z.-h. Molecular mechanisms of excitotoxicity and their relevance to pathogenesis of neurodegenerative diseases. Acta Pharmacol. Sin. 2009, 30, 379–387. [Google Scholar] [CrossRef] [Green Version]
- Hsa, E.; Ya, K. Glutamate excitotoxicity and neurodegeneration. J. Mol. Genet. Med. 2014, 8, 1747-0862. [Google Scholar] [CrossRef]
- Vincent, P.; Mulle, C. Kainate receptors in epilepsy and excitotoxicity. Neuroscience 2009, 158, 309–323. [Google Scholar] [CrossRef] [PubMed]
- Dief, A.; Kamha, E.; Baraka, A.; Elshorbagy, A. Monosodium glutamate neurotoxicity increases beta amyloid in the rat hippocampus: A potential role for cyclic AMP protein kinase. Neurotoxicology 2014, 42, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Hassaan, P.; Dief, A.; Zeitoun, T.; Baraka, A.; Deacon, R.; Elshorbagy, A. Cortical tau burden and behavioural dysfunctions in mice exposed to monosodium glutamate in early life. PLoS ONE 2019, 14, e0220720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahanger, I.; Bashir, S.; Parray, Z.; Alajmi, M.; Hussain, A.; Ahmad, F.; Hassan, M.; Islam, A.; Sharma, A. Rationalizing the role of monosodium glutamate in the protein aggregation through biophysical approaches: Potential impact on neurodegeneration. Front. Neurosci. 2021, 15, 636454. [Google Scholar] [CrossRef]
- Chaparro-Huerta, V.; Flores-Soto, M.; Gudino-Cabrera, G.; Rivera-Cervantes, M.; Bitzer-Quintero, O.; Beas-Zarate, C. Role of p38 MAPK and pro-inflammatory cytokines expression in glutamate-induced neuronal death of neonatal rats. Int. J. Dev. Neurosci. 2008, 26, 487–495. [Google Scholar] [CrossRef]
- Chen, X.; Guo, C.; Kong, J. Oxidative stress in neurodegenerative diseases. Neural. Regen. Res. 2012, 7, 376. [Google Scholar] [CrossRef]
- Denzer, I.; Muench, G.; Friedland, K. Modulation of mitochondrial dysfunction in neurodegenerative diseases via activation of nuclear factor erythroid-2-related factor 2 by food-derived compounds. Pharmacol. Res. 2016, 103, 80–94. [Google Scholar] [CrossRef]
- Kovacs, G. Molecular pathological classification of neurodegenerative diseases: Turning towards precision medicine. Int. J. Mol. Sci. 2016, 17, 189. [Google Scholar] [CrossRef]
- Lewerenz, J.; Maher, P. Chronic glutamate toxicity in neurodegenerative diseases—what is the evidence? Front. Neurosci. 2015, 9, 469. [Google Scholar] [CrossRef]
- Cynober, L. Metabolism of dietary glutamate in adults. Ann. Nutr. Metab. 2018, 73, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Populin, T.; Moret, S.; Truant, S.; Conte, L. A survey on the presence of free glutamic acid in foodstuffs, with and without added monosodium glutamate. Food Chem. 2007, 104, 1712–1717. [Google Scholar] [CrossRef]
- Nepalia, A.; Singh, A.; Mathur, N.; Pareek, S. Baby foods can also have toxic side effects: A review. Asian J. Sci. Technol. 2017, 8, 4386–4393. [Google Scholar]
- Mortensen, A.; Aguilar, F.; Crebelli, R.; Di Domenico, A.; Dusemund, B.; Frutos, M.; Galtier, P.; Gott, D.; Gundert-Remy, U. Re-evaluation of glutamic acid (E 620), sodium glutamate (E 621), potassium glutamate (E 622), calcium glutamate (E 623), ammonium glutamate (E 624) and magnesium glutamate (E 625) as food additives. EFSA J. 2017, 15, e04910. [Google Scholar] [CrossRef]
- CDC. Ingredients of vaccines—Fact sheet. Available online: https://www.cdc.gov/vaccines/vac-gen/additives.htm (accessed on 20 March 2019).
- Niaz, K.; Zaplatic, E.; Spoor, J. Extensive use of monosodium glutamate: A threat to public health? EXCLI J. 2018, 17, 273–278. [Google Scholar] [CrossRef]
- Zanfirescu, A.; Ungurianu, A.; Tsatsakis, A.; Nițulescu, G.; Kouretas, D.; Veskoukis, A.; Tsoukalas, D.; Engin, A.; Aschner, M.; Margină, D. A review of the alleged health hazards of monosodium glutamate. Compr. Rev. Food Sci. Food Saf. 2019, 18, 1111–1134. [Google Scholar] [CrossRef] [Green Version]
- Page, M.; Moher, D.; Bossuyt, P.; Boutron, I.; Hoffmann, T.; Mulrow, C.; Shamseer, L.; Tetzlaff, J.; Akl, E.; Brennan, S.; et al. PRISMA 2020 explanation and elaboration: Updated guidance and exemplars for reporting systematic reviews. BMJ 2021, 372, n160. [Google Scholar] [CrossRef]
- Page, M.; McKenzie, J.; Bossuyt, P.; Boutron, I.; Hoffmann, T.; Mulrow, C.; Shamseer, L.; Tetzlaff, J.; Akl, E.; Brennan, S.; et al. The PRISMA 2020 statement: An updated guideline for reporting systematic reviews. BMJ 2021, 10, 89. [Google Scholar] [CrossRef]
- Moher, D.; Liberati, A.; Tetzlaff, J.; Altman, D.; Group, P. Preferred reporting items for systematic reviews and meta-analyses: The PRISMA statement. J. Clin. Epidemiol. 2009, 6, e1000097. [Google Scholar] [CrossRef]
- Hu, Y.; Li, J.; Liu, P.; Chen, X.; Guo, D.; Li, Q.S.; Rahman, K. Protection of SH-SY5Y neuronal cells from glutamate-induced apoptosis by 3,6′-disinapoyl sucrose, a bioactive compound isolated from Radix Polygala. J. Biomed. Biotechnol. 2012, 2012, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Petroni, D.; Tsai, J.; Mondal, D.; George, W. Attenuation of low dose methylmercury and glutamate induced-cytotoxicity and Tau phosphorylation by an N-methyl-D-aspartate antagonist in human neuroblastoma (SHSY5Y) cells. Environ. Toxicol. 2013, 28, 700–706. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.; Kang, S.; Kim, J.; Yu, K.; Lee, I.; Lee, Y.; Lee, J.; Lee, N.; Jeong, Y.; Kim, D.; et al. Protective effects of poly(lactic-co-glycolic acid) nanoparticles loaded with erythropoietin stabilized by sodium cholate against glutamate-induced neurotoxicity. J. Nanosci. Nanotechnol. 2014, 14, 8365–8371. [Google Scholar] [CrossRef] [PubMed]
- Nampoothiri, M.; Reddy, N.; John, J.; Kumar, N.; Kutty Nampurath, G.; Rao Chamallamudi, M. Insulin blocks glutamate-induced neurotoxicity in differentiated SH-SY5Y neuronal cells. Behav. Neurol. 2014, 2014, 674164–674168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brizi, C.; Santulli, C.; Micucci, M.; Budriesi, R.; Chiarini, A.; Aldinucci, C.; Frosini, M. Neuroprotective effects of Castanea sativa Mill. bark extract in human neuroblastoma cells subjected to oxidative stress. J. Cell. Biochem. 2016, 117, 510–520. [Google Scholar] [CrossRef]
- Shah, S.; Amin, F.; Khan, M.; Abid, M.; Rehman, S.; Kim, T.; Kim, M.; Kim, M. Anthocyanins abrogate glutamate-induced AMPK activation, oxidative stress, neuroinflammation, and neurodegeneration in postnatal rat brain. J. Neuroinflamm. 2016, 13, 16. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Shi, X.; Lu, L.; Jiang, Y.; Liu, B. Stimulus-dependent neuronal cell responses in SH-SY5Y neuroblastoma cells. Mol. Med. Rep. 2016, 13, 2215–2220. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Wang, K.; Zhang, K.; Lin, X.; Zhu, L.; Zhou, F. Puerarin protects human neuroblastoma SH-SY5Y cells against glutamate-induced oxidative stress and mitochondrial dysfunction. J. Biochem. Mol. Toxicol. 2016, 30, 22–28. [Google Scholar] [CrossRef]
- Li, H.; Han, W.; Wang, H.; Ding, F.; Xiao, L.; Shi, R.; Ai, L.; Huang, Z. Tanshinone IIA inhibits glutamate-induced oxidative toxicity through prevention of mitochondrial dysfunction and suppression of MAPK activation in SH-SY5Y human neuroblastoma cells. Oxid. Med. Cell. Longev. 2017, 2017, 13. [Google Scholar] [CrossRef]
- Bharate, S.; Kumar, V.; Singh, G.; Singh, A.; Gupta, M.; Singh, D.; Kumar, A.; Vishwakarma, R.; Bharate, S. Preclinical development of Crocus sativus-based botanical lead IIIM-141 for Alzheimer’s Disease: Chemical standardization, efficacy, formulation development, pharmacokinetics, and safety pharmacology. ACS Omega 2018, 3, 9572–9585. [Google Scholar] [CrossRef] [Green Version]
- de Oliveira, M.; Duarte, A.; Chenet, A.; de Almeida, F.; Andrade, C. Carnosic acid pretreatment attenuates mitochondrial dysfunction in SH-SY5Y Cells in an experimental model of glutamate-induced excitotoxicity. Neurotox. Res. 2019, 36, 551–562. [Google Scholar] [CrossRef]
- Lee, H.; Spandidos, D.; Tsatsakis, A.; Margina, D.; Izotov, B.; Yang, S. Neuroprotective effects of Scrophularia buergeriana extract against glutamate-induced toxicity in SH-SY5Y cells. Int. J. Mol. Med. 2019, 43, 2144–2152. [Google Scholar] [CrossRef] [PubMed]
- Xin, H.; Cui, Y.; An, Z.; Yang, Q.; Zou, X.; Yu, N. Attenuated glutamate induced ROS production by antioxidative compounds in neural cell lines. RSC Adv. 2019, 9, 34735–34743. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Han, A.; Kim, E.; Yang, J.; Ahn, J.; Na, J.; Cho, S. KHG21834 attenuates glutamate-induced mitochondrial damage, apoptosis, and NLRP3 inflammasome activation in SH-SY5Y human neuroblastoma cells. Eur. J. Pharmacol. 2019, 856, 10. [Google Scholar] [CrossRef] [PubMed]
- Yuksel, T.; Yayla, M.; Halici, Z.; Cadirci, E.; Polat, B.; Kose, D. Protective effect of 5-HT7 receptor activation against glutamate-induced neurotoxicity in human neuroblastoma SH-SY5Y cells via antioxidative and antiapoptotic pathways. Neurotoxicol. Teratol. 2019, 72, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Bebitoglu, B.; Oguz, E.; Gokce, A. Effect of valproic acid on oxidative stress parameters of glutamate-induced excitotoxicity in SH-SY5Y cells. Exp. Ther. Med. 2020, 20, 1321–1328. [Google Scholar] [CrossRef] [PubMed]
- Fallarini, S.; Miglio, G.; Paoletti, T.; Minassi, A.; Amoruso, A.; Bardelli, C.; Brunelleschi, S.; Lombardi, G. Clovamide and rosmarinic acid induce neuroprotective effects in in vitro models of neuronal death. Br. J. Pharmacol. 2009, 157, 1072–1084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kataria, H.; Wadhwa, R.; Kaul, S.; Kaur, G. Water extract from the leaves of Withania somnifera protect RA differentiated C6 and IMR-32 cells against glutamate-induced excitotoxicity. PLoS ONE 2012, 7, e37080. [Google Scholar] [CrossRef] [Green Version]
- Occhiuto, F.; Zangla, G.; Samperi, S.; Palumbo, D.; Pino, A.; De Pasquale, R.; Circosta, C. The phytoestrogenic isoflavones from Trifolium pratense L. (Red clover) protects human cortical neurons from glutamate toxicity. Phytomedicine 2008, 15, 676–682. [Google Scholar] [CrossRef]
- Palumbo, D.; Occhiuto, F.; Spadaro, F.; Circosta, C. Rhodiola rosea extract protects human cortical neurons against glutamate and hydrogen peroxide-induced cell death through reduction in the accumulation of intracellular calcium. Phytother. Res. 2012, 26, 878–883. [Google Scholar] [CrossRef]
- Gupta, K.; Hardingham, G.; Chandran, S. NMDA receptor-dependent glutamate excitotoxicity in human embryonic stem cell-derived neurons. Neurosci. Lett. 2013, 543, 95–100. [Google Scholar] [CrossRef] [Green Version]
- Yon, J.; Kim, Y.; Park, D. The ethanol fraction of white rose petal extract abrogates excitotoxicity-induced neuronal damage in vivo and in vitro through inhibition of oxidative stress and proinflammation. Nutrients 2018, 10, 1375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chao, C.; Hu, S. Tumor necrosis factor-alpha potentiates glutamate neurotoxicity in human fetal brain cell cultures. Dev. Neurosci. 1994, 16, 172–179. [Google Scholar] [CrossRef] [PubMed]
- de Vera, N.; Martínez, E.; Sanfeliu, C. Spermine induces cell death in cultured human embryonic cerebral cortical neurons through N-methyl-D-aspartate receptor activation. J. Neurosci. Res. 2008, 86, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Babu, G.; Bawari, M.; Ali, M. Lipid peroxidation potential and antioxidant status of circumventricular organs of rat brain following neonatal monosodium glutamate. Neurotoxicology 1994, 15, 773–777. [Google Scholar]
- Ferger, B.; van Amsterdam, C.; Seyfried, C.; Kuschinsky, K. Effects of alpha-phenyl-tert-butylnitrone and selegiline on hydroxyl free radicals in rat striatum produced by local application of glutamate. J. Neurochem. 1998, 70, 276–280. [Google Scholar] [CrossRef]
- Singh, P.; Mann, K.; Mangat, H.; Kaur, G. Prolonged glutamate excitotoxicity: Effects on mitochondrial antioxidants and antioxidant enzymes. Mol. Cell. Biochem. 2003, 243, 139–145. [Google Scholar] [CrossRef]
- Rivera-Cervantes, M.; Torres, J.; Feria-Velasco, A.; Armendariz-Borunda, J.; Beas-Zárate, C. NMDA and AMPA receptor expression and cortical neuronal death are associated with p38 in glutamate-induced excitotoxicity in vivo. J. Neurosci. Res. 2004, 76, 678–687. [Google Scholar] [CrossRef]
- Chaparro-Huerta, V.; Rivera-Cervantes, M.; Flores-Soto, M.; Gomez-Pinedo, U.; Beas-Zarate, C. Proinflammatory cytokines and apoptosis following glutamate-induced excitotoxicity mediated by p38 MAPK in the hippocampus of neonatal rats. J. Neuroimmunol. 2005, 165, 53–62. [Google Scholar] [CrossRef]
- Mejía-Toiber, J.; Montiel, T.; Massieu, L. D-beta-hydroxybutyrate prevents glutamate-mediated lipoperoxidation and neuronal damage elicited during glycolysis inhibition in vivo. Neurochem. Res. 2006, 31, 1399–1408. [Google Scholar] [CrossRef]
- Segura Torres, J.; Chaparro-Huerta, V.; Rivera Cervantres, M.; Montes-González, R.; Flores Soto, M.; Beas-Zárate, C. Neuronal cell death due to glutamate excitotocity is mediated by p38 activation in the rat cerebral cortex. Neurosci. Lett. 2006, 403, 233–238. [Google Scholar] [CrossRef]
- Del Río, P.; Massieu, L. Mild mitochondrial inhibition in vivo enhances glutamate-induced neuronal damage through calpain but not caspase activation: Role of ionotropic glutamate receptors. Exp. Neurol. 2008, 212, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Hashem, H.; Safwat, M.; Algaidi, S. The effect of monosodium glutamate on the cerebellar cortex of male albino rats and the protective role of vitamin C (histological and immunohistochemical study). J. Mol. Histol. 2012, 43, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Swamy, A.; Patel, N.; Gadad, P.; Koti, B.; Patel, U.M.; Thippeswamy, A.; Manjula, D. Neuroprotective activity of Pongamia pinnata in monosodium glutamate-induced neurotoxicity in rats. Indian J. Pharm. Sci. 2013, 75, 657–663. [Google Scholar] [PubMed]
- Thonda, V.; Kumar, S.; Handral, M.; Sonowal, A. Neuroprotective evaluation of ethanolic leaf extract of Dalbergia sissoo in monosodium glutamate induced neurotoxicity in rats. Int. J. Pharm. Sci. Res. 2014, 5, 829–838. [Google Scholar] [CrossRef]
- Rivera-Cervantes, M.; Castañeda-Arellano, R.; Castro-Torres, R.; Gudiño-Cabrera, G.; Feria y Velasco, A.; Camins, A.; Beas-Zárate, C. P38 MAPK inhibition protects against glutamate neurotoxicity and modifies NMDA and AMPA receptor subunit expression. J. Mol. Neurosci. 2015, 55, 596–608. [Google Scholar] [CrossRef]
- Khalil, R.; Khedr, N. Curcumin protects against monosodium glutamate neurotoxicity and decreasing NMDA2B and mGluR5 expression in rat hippocampus. NeuroSignals 2016, 24, 81–87. [Google Scholar] [CrossRef]
- Sadek, K.; Abouzed, T.; Nasr, S. Lycopene modulates cholinergic dysfunction, Bcl-2/Bax balance, and antioxidant enzymes gene transcripts in monosodium glutamate (E621) induced neurotoxicity in a rat model. Can. J. Physiol. Pharmacol. 2016, 94, 394–401. [Google Scholar] [CrossRef]
- Hussein, U.; Hassan, N.; Elhalwagy, M.; Zaki, A.; Abubakr, H.; Nagulapalli Venkata, K.; Jang, K.; Bishayee, A. Ginger and propolis exert neuroprotective effects against monosodium glutamate-induced neurotoxicity in rats. Molecules 2017, 22, 1928. [Google Scholar] [CrossRef] [Green Version]
- Abdel Moneim, W.; Yassa, H.; Makboul, R.; Mohamed, N. Monosodium glutamate affects cognitive functions in male albino rats. Egypt J. Forensic. Sci. 2018, 8, 9. [Google Scholar] [CrossRef] [Green Version]
- Fouad, I.; Sharaf, N.; Abdelghany, R.; El Sayed, N. Neuromodulatory effect of thymoquinone in attenuating glutamate-mediated neurotoxicity targeting the amyloidogenic and apoptotic pathway. Front. Neurol. 2018, 9, 12. [Google Scholar] [CrossRef] [Green Version]
- Hazzaa, S.; Abdelaziz, S.; Abd Eldaim, M.; Abdel-Daim, M.; Elgarawany, G. Neuroprotective potential of Allium sativum against monosodium glutamate-induced excitotoxicity: Impact on short-term memory, gliosis, and oxidative stress. Nutrients 2020, 12, 1028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burde, R.; Schainker, B.; Kayes, J. Acute effect of oral and subcutaneous administration of monosodium glutamate on the arcuate nucleus of the hypothalamus in mice and rats. Nature 1971, 233, 58–60. [Google Scholar] [CrossRef] [PubMed]
- Greene, J.; Greenamyre, J. Exacerbation of NMDA, AMPA, and L-glutamate excitotoxicity by the succinate dehydrogenase inhibitor malonate. J. Neurochem. 1995, 64, 2332–2338. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Tsai, P.; Lin, N.; Kuo, J. Elevated extracellular glutamate concentrations increased malondialdehyde production in anesthetized rat brain cortex. Neurosci. Lett. 1998, 243, 33–36. [Google Scholar] [CrossRef]
- Bodnár, I.; Göõz, P.; Okamura, H.; Tóth, B.; Vecsernyé, M.; Halász, B.; Nagy, G. Effect of neonatal treatment with monosodium glutamate on dopaminergic and L-DOPA-ergic neurons of the medial basal hypothalamus and on prolactin and MSH secretion of rats. Brain Res. Bull. 2001, 55, 767–774. [Google Scholar] [CrossRef]
- Kumar, A.; Babu, G. In vivo neuroprotective effects of peripheral kynurenine on acute neurotoxicity induced by glutamate in rat cerebral cortex. Neurochem. Res. 2010, 35, 636–644. [Google Scholar] [CrossRef]
- Kumar, A.; Singh, R.; Babu, G. Cell death mechanisms in the early stages of acute glutamate neurotoxicity. Neurosci. Res. 2010, 66, 271–278. [Google Scholar] [CrossRef]
- Nagesh Babu, G.; Kumar, A.; Singh, R. Chronic pretreatment with acetyl-L-carnitine and +/-DL-alpha-lipoic acid protects against acute glutamate-induced neurotoxicity in rat brain by altering mitochondrial function. Neurotox. Res. 2011, 19, 319–329. [Google Scholar] [CrossRef]
- Morales, I.; Rodriguez, M. Self-induced accumulation of glutamate in striatal astrocytes and basal ganglia excitotoxicity. Glia 2012, 60, 1481–1494. [Google Scholar] [CrossRef]
- Kim, E.; Choi, J.; Han, A.; Choi, S.; Hahn, H.; Cho, S. Anti-oxidative and anti-inflammatory effects of 2-cyclopropylimino-3-methyl-1,3-thiazoline hydrochloride on glutamate-induced neurotoxicity in rat brain. Neurotoxicology 2013, 38, 106–114. [Google Scholar] [CrossRef]
- Shah, S.; Yoon, G.; Kim, H.; Kim, M. Vitamin C neuroprotection against dose-dependent glutamate-induced neurodegeneration in the postnatal brain. Neurochem. Res. 2015, 40, 875–884. [Google Scholar] [CrossRef] [PubMed]
- Calis, I.; Cosan, D.; Saydam, F.; Kolac, U.; Soyocak, A.; Kurt, H.; Gunes, H.; Sahinturk, V.; Mutlu, F.; Koroglu, Z.; et al. The effects of monosodium glutamate and tannic acid on adult rats. Iran Red Crescent Med. J. 2016, 18, e37912. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Kim, E.; Chang, M.; Kim, J.; Na, J.; Choi, S.; Cho, S. N-Adamantyl-4-Methylthiazol-2-Amine attenuates glutamate-induced oxidative stress and inflammation in the brain. Neurotox. Res. 2017, 32, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Firgany, A.; Sarhan, N. Quercetin mitigates monosodium glutamate-induced excitotoxicity of the spinal cord motoneurons in aged rats via p38 MAPK inhibition. Acta Histochem. 2020, 122, 10. [Google Scholar] [CrossRef] [PubMed]
- Hamza, R.; Al-Salmi, F.; El-Shenawy, N. Evaluation of the effects of the green nanoparticles zinc oxide on monosodium glutamate-induced toxicity in the brain of rats. PeerJ 2019, 7, e7460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohan, M.; Gangurde, S.; Kadam, V. Protective effect of Solanum torvum on monosodium glutamate-induced neurotoxicity in mice. Indian J. Nat. Prod. Resour. 2017, 8, 351–359. [Google Scholar]
- Estrada-Sánchez, A.; Montiel, T.; Segovia, J.; Massieu, L. Glutamate toxicity in the striatum of the R6/2 Huntington’s disease transgenic mice is age-dependent and correlates with decreased levels of glutamate transporters. Neurobiol. Dis. 2009, 34, 78–86. [Google Scholar] [CrossRef]
- Estrada-Sánchez, A.; Montiel, T.; Massieu, L. Glycolysis inhibition decreases the levels of glutamate transporters and enhances glutamate neurotoxicity in the R6/2 Huntington’s disease mice. Neurochem. Res. 2010, 35, 1156–1163. [Google Scholar] [CrossRef]
- Guemez-Gamboa, A.; Estrada-Sánchez, A.; Montiel, T.; Páramo, B.; Massieu, L.; Morán, J. Activation of NOX2 by the stimulation of ionotropic and metabotropic glutamate receptors contributes to glutamate neurotoxicity in vivo through the production of reactive oxygen species and calpain activation. J. Neuropathol. Exp. Neurol. 2011, 70, 1020–1035. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Zhang, F.; Li, L.; Tang, F.; Siedlak, S.; Fujioka, H.; Liu, Y.; Su, B.; Pi, Y.; Wang, X. MFN2 couples glutamate excitotoxicity and mitochondrial dysfunction in motor neurons. J. Biol. Chem. 2015, 290, 168–182. [Google Scholar] [CrossRef] [Green Version]
- Zou, J.; Wang, M.; Uchiumi, O.; Shui, Y.; Ishigaki, Y.; Liu, X.; Tajima, N.; Akai, T.; Iizuka, H.; Kato, N. Learning impairment by minimal cortical injury in a mouse model of Alzheimer’s disease. Brain Res. 2016, 1637, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Zhang, Y.; Ma, R.; Bao, L.; Fang, J.; Yu, T. Potent protection of ferulic acid against excitotoxic effects of maternal intragastric administration of monosodium glutamate at a late stage of pregnancy on developing mouse fetal brain. Eur. Neuropsychopharmacol. 2006, 16, 170–177. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Yu, L.; Ma, R.; Zhang, Y.; Fang, J.; Zhang, X.; Yu, T. Repair of glutamate-induced excitotoxic neuronal damage mediated by intracerebroventricular transplantation of neural stem cells in adult mice. Neurosci. Bull. 2007, 23, 209–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penugonda, S.; Ercal, N. Comparative evaluation of N-acetylcysteine (NAC) and N-acetylcysteine amide (NACA) on glutamate and lead-induced toxicity in CD-1 mice. Toxicol. Lett. 2011, 201, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, Q.; Li, Y.; Hua, Z.; Wang, Y.; Yu, X.; Jia, R.; Chen, W.; Zheng, X. Tetrastigma hemsleyanum vine flavone ameliorates glutamic acid-induced neurotoxicity via MAPK pathways. Oxid. Med. Cell. Longev. 2020, 2020, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spangenberg, D.; Lattanzio, F.; Navarro, G. D-Methionine and gold chloride alleviate adverse effects of glutamate on motility of ephyrae of Aurelia aurita (Linnaeus, 1758) (Scyphozoa: Semaeostomeae). Hydrobiologia 2004, 530, 355–363. [Google Scholar] [CrossRef]
- Roberts, A.; Lynch, B.; Rietjens, I. Risk assessment paradigm for glutamate. Ann. Nutr. Metab. 2018, 73, 53–64. [Google Scholar] [CrossRef]
- Beyreuther, K.; Biesalski, H.; Fernstrom, J.; Grimm, P.; Hammes, W.; Heinemann, U.; Kempski, O.; Stehle, P.; Steinhart, H.; Walker, R. Consensus meeting: Monosodium glutamate—An update. Eur. J. Clin. Nutr. 2007, 61, 304–313. [Google Scholar] [CrossRef]
- Maluly, H.; Arisseto-Bragotto, A.; Reyes, F. Monosodium glutamate as a tool to reduce sodium in foodstuffs: Technological and safety aspects. Food Sci. Nutr. 2017, 5, 1039–1048. [Google Scholar] [CrossRef]
- Mattson, M. Excitotoxic and excitoprotective mechanisms. Neuromolecular Med. 2003, 3, 65–94. [Google Scholar] [CrossRef] [Green Version]
- Lewerenz, J.; Hewett, S.J.; Huang, Y.; Lambros, M.; Gout, P.W.; Kalivas, P.W.; Massie, A.; Smolders, I.; Methner, A.; Pergande, M.; et al. The cystine/glutamate antiporter system x(c)(-) in health and disease: From molecular mechanisms to novel therapeutic opportunities. Antioxid. Redox Signal. 2013, 18, 522–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattson, M. Calcium and neurodegeneration. Aging cell 2007, 6, 337–350. [Google Scholar] [CrossRef] [PubMed]
- Bahar, E.; Kim, H.; Yoon, H. ER Stress-Mediated Signaling: Action Potential and Ca(2+) as Key Players. Int. J. Mol. Sci. 2016, 17, 1558. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S. Does the Interdependence between Oxidative Stress and Inflammation Explain the Antioxidant Paradox? Oxid. Med. Cell. Longev. 2016, 2016, 5698931. [Google Scholar] [CrossRef] [Green Version]
- Schaffert, L.; Carter, W. Do Post-Translational Modifications Influence Protein Aggregation in Neurodegenerative Diseases: A Systematic Review. Brain Sci. 2020, 10, 232. [Google Scholar] [CrossRef]
- Jin, L.; Li, Y.-P.; Feng, Q.; Ren, L.; Wang, F.; Bo, G.-J.; Wang, L. Cognitive deficits and Alzheimer-like neuropathological impairments during adolescence in a rat model of type 2 diabetes mellitus. Neural. Regen. Res. 2018, 13, 1995–2004. [Google Scholar] [CrossRef]
- Jin, L.; Lin, L.; Li, G.; Liu, S.; Luo, D.; Feng, Q.; Sun, D.; Wang, W.; Liu, J.; Wang, Q.; et al. Monosodium glutamate exposure during the neonatal period leads to cognitive deficits in adult Sprague-Dawley rats. Neurosci. Lett. 2018, 682, 39–44. [Google Scholar] [CrossRef]
- Špolcová, A.; Mikulášková, B.; Holubová, M.; Nagelová, V.; Pirnik, Z.; Zemenová, J.; Haluzík, M.; Železná, B.; Galas, M.; Maletínská, L. Anorexigenic lipopeptides ameliorate central insulin signaling and attenuate tau phosphorylation in haippocampi of mice with monosodium glutamate-induced obesity. J. Alzheimers Dis. 2015, 45, 823–835. [Google Scholar] [CrossRef]
- Kobayashi, S.; Tanaka, T.; Soeda, Y.; Takashima, A. Enhanced Tau Protein Translation by Hyper-Excitation. Front. Aging Neurosci. 2019, 11, 322. [Google Scholar] [CrossRef] [Green Version]
- Levy, E.; El Banna, N.; Baille, D.; Heneman-Masurel, A.; Truchet, S.; Rezaei, H.; Huang, M.; Beringue, V.; Martin, D.; Vernis, L. Causative Links between Protein Aggregation and Oxidative Stress: A Review. Int. J. Mol. Sci. 2019, 20, 3896. [Google Scholar] [CrossRef] [Green Version]
- Kritis, A.; Stamoula, E.; Paniskaki, K.; Vavilis, T. Researching glutamate—Induced cytotoxicity in different cell lines: A comparative/collective analysis/study. Front. Cell. Neurosci. 2015, 9, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheldon, A.; Robinson, M. The role of glutamate transporters in neurodegenerative diseases and potential opportunities for intervention. Neurochem. Int. 2007, 51, 333–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbosa, D.; Capela, J.; de Lourdes Bastos, M.; Carvalho, F. In vitro models for neurotoxicology research. Toxicol. Res. 2015, 4, 801–842. [Google Scholar] [CrossRef]
- Ganesan, K.; Sukalingam, K.; Balamurali, K.; Ponnusamy, K.; Ariffin, I.; Gani, S. A studies on monosodium L- glutamate toxicity in animal models- A review. Int. J. Pharm. Chem. Biol. Sci. 2013, 3, 1257–1268. [Google Scholar]
- Samuels, A. Dose dependent toxicity of glutamic acid: A review. Int. J. Food Prop. 2020, 23, 412–419. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study Reference | In Vitro Model | L-Glu Treatment and Duration | Study Outcomes | Level of Significance |
|---|---|---|---|---|
| Hu et al. (2012) [51] | SH-SY5Y undifferentiated | 8 mM; 0.5 h or 12 h | ↓ cell viability ↑ LDH levels Morphological alterations ↑ apoptosis ↑ Bax expression ↓ Bcl-2 expression | (p < 0.01) (p < 0.01) ND ND (p < 0.01) (p < 0.01) |
| Petroni et al. (2013) [52] | SH-SY5Y undifferentiated | 1 mM; 6 h or 24 h | ↓ cell viability ↑ tau protein phosphorylation | (p < 0.05) NS |
| Jeong et al. (2014) [53] | SH-SY5Y undifferentiated | 0.01–6 mM (MSG); 24 h | ↓ cell viability (5 and 6 mM) Morphological changes: pyknosis, nuclear condensation, and cytoplasmic shrinkage | (p < 0.01–0.001) ND |
| Nampoothiri et al. (2014) [54] | SH-SY5Y undifferentiated and differentiated | 5–80 mM; 48 h | ↓ cell viability | Undifferentiated (p < 0.001) Differentiated (p < 0.05–0.001) |
| SH-SY5Y differentiated | 20 mM; 48 h | ↓ cell viability ↑ apoptosis ↓ neurite length ↑ ROS | (p < 0.05) (p < 0.01) (p < 0.001) (p < 0.001) | |
| Brizi et al. (2016) [55] | SH-SY5Y undifferentiated | 1–100 mM; 24 h | ↓ cell viability ↑ apoptosis (50 mM) ↓ growth (50 and 80 mM) ↑ ROS (50 mM) Morphological alteration in neurons and nuclear material | ND (p < 0.01) (p < 0.01), (p < 0.001) ND ND |
| Shah et al. (2016) [56] | SH-SY5Y undifferentiated | 10–30 mM; 3 h | ↓ cell viability | (p < 0.05–0.001) |
| 30 mM; 3 h | ↑ apoptosis ↑ p-AMPK protein ↓ Nrf2 protein ↓ HO-1 protein ↑ ROS ↑ p-NF-kB protein ↑ COX-2 protein ↑ caspase-3 protein | (p < 0.001) (p < 0.001) (p < 0.01) (p < 0.01) (p < 0.001) (p < 0.001) (p < 0.001) (p < 0.001) | ||
| Sun et al. (2016) [57] | SH-SY5Y undifferentiated | 10–50 mM; 1, 2, 4, 6 or 8 h | ↓ cell viability ↑ apoptosis and necrosis ↑ Ca2+ ↓ MMP (10 or 15 mM) ↑ MMP (25 or 50 mM) ↑ RIP kinase 1 protein | (p < 0.05) (p < 0.05) (p < 0.05) (p < 0.05) (p < 0.05) (p < 0.05) |
| Zhu et al. (2016) [58] | SH-SY5Y undifferentiated | 5–40 mM (MSG); 24 h | ↓ cell viability | (p < 0.05–0.01) |
| 20 mM (MSG); 24 h | ↑ apoptosis ↑ ROS ↑ Bax protein ↓ Bcl-2 protein ↓ MMP ↓ cytosolic cytochrome c protein ↑ mitochondrial cytochrome c protein ↑ cleaved caspase-9 protein ↑ cleaved caspase-3 protein | (p < 0.01) (p < 0.01) ND ND (p < 0.01) ND ND ND ND | ||
| Li et al. (2017) [59] | SH-SY5Y undifferentiated | 10 mM; 24 h | ↓ cell viability ↑ LDH ↑ ROS ↑ PCC ↑ LPO ↓ SOD ↓ CAT ↓ MMP ↓ ATP ↑ mitochondrial PCC ↑ apoptosis ↑ Bax protein ↑ cleaved caspase-3 protein ↓ Bcl-2 protein ↑ p-MAPKs protein | ND ND (p < 0.05) (p < 0.05) (p < 0.05) (p < 0.05) (p < 0.05) ND ND ND ND ND ND ND ND |
| Bharate et al. (2018) [60] | SH-SY5Y differentiated | 250 µM; 24 h | ↓ cell viability | ND |
| de Oliveira et al. (2019) [61] | SH-SY5Y undifferentiated | 10–80 mM; 24 h | ↓ cell viability (40–80 mM) | (p < 0.05) |
| 80 mM; 24 or 6 h | ↑ cleaved PARP level ↑ DNA fragmentation ↑ LPO ↑ protein nitration ↑ PCC ↓ protein thiol ↑ 8-oxo-dG level ↓ MMP ↓ ATP ↑ MC I and V activities ↓ mitochondrial enzyme activities ↑ Bax protein ↑ cytosolic cytochrome c content ↓ mitochondrial cytochrome c content ↑ caspase-9 activity ↑ caspase-3 activity ↑ O2−• ↑ NO• ↑ ROS | (p < 0.05) (p < 0.05) (p < 0.05) (p < 0.05) (p < 0.05) (p < 0.05) (p < 0.05) (p < 0.05) (p < 0.05) (p < 0.05) (p < 0.05) (p < 0.05) (p < 0.05) (p < 0.05) (p < 0.05) (p < 0.05) (p < 0.05) (p < 0.05) (p < 0.05) | ||
| Lee et al. (2019) [62] | SH-SY5Y undifferentiated | 12.5–100 mM; 3 h | ↓ cell viability Nuclear condensation ↑ DNA fragmentation | (p < 0.01) ND (p < 0.01) |
| SH-SY5Y differentiated | 100 mM; 3 h | ↑ AChE activity | (p < 0.01) | |
| SH-SY5Y undifferentiated | ↓ GSH level ↓ SOD protein ↓ GPx protein ↑ pp38 protein ↑ Bax protein ↑ cleaved caspase-3 protein ↑ cleaved PARP protein ↓ Bcl-2 protein | (p < 0.01) (p < 0.01) (p < 0.01) (p < 0.01) (p < 0.01) (p < 0.01) (p < 0.01) (p < 0.01) | ||
| Xin et al. (2019) [63] | SH-SY5Y undifferentiated | 0.1–100 mM; 12 h | ↓ cell viability ↑ ROS | ND ND |
| Yang et al. (2019) [64] | SH-SY5Y undifferentiated | 10 mM; 24 h | ↓ cell viability ↑ LDH ↑ ROS ↑ LPO ↓ SOD, GPx activities, and GSH level ↓ MMP and ATP ↑ Ca2+ ↑ CHOP, GRP78 proteins, and caspase-4 activity ↑ NLRP3 protein ↑ IL-1β and IL-6 ↑ Bax/Bcl-2 ratio ↑ cleaved caspase-1 and caspase-3 proteins ↑ p-MAPKs protein ↑ apoptosis | ND ND ND ND ND ND ND ND ND ND ND ND ND ND |
| Yuksel et al. (2019) [65] | SH-SY5Y undifferentiated | 80 mM (MSG); 24, 48 or 72 h | ↑ cell toxicity ↑ LPO ↓ SOD activity ↓ GSH level ↑ TNF-α ↑ caspase 3 and caspase 9 mRNA | ND (p < 0.001) (p < 0.05) (p < 0.001) (p < 0.001) (p < 0.001) |
| Bebitoglu et al. (2020) [66] | SH-SY5Y undifferentiated | 1–50 mM; 3 h or 24 h | ↓ cell viability (15-50 mM) ↓ CAT ↓ SOD ↑ H2O2 ↑ LPO Morphological alteration | (p < 0.05–0.0001) (p < 0.05) NS (p < 0.0001) (p < 0.0001) ND |
| Fallarini et al. (2009) [67] | Differentiated SK-N-BE(2) | 1 mM; 24 h | ↑ LDH ↑ Ca2+ ↑ c-fos and c-jun mRNA | (p < 0.01) (p < 0.01) (p < 0.01) |
| Kataria et al. (2012) [68] | Differentiated IMR-32 human neuroblastoma | 0.06–10 mM; 24 h | Morphological changes, cell shrinkage, and rounding ↓ cell viability ↑ LDH ↓ NF200 mRNA and protein (0.25 and 0.5 mM) ↑ HSP70 mRNA and protein (0.25 and 0.5 mM) ↑ PSA-NCAM expression (0.5 mM) ↑ PST mRNA (0.25 and 0.5 mM) | ND (p < 0.05) (p < 0.05) (p < 0.05) (p < 0.05) (p < 0.05) (p < 0.05) |
| Occhiuto et al. (2008) [69] | Differentiated HCN-1A cell line | 0.01–5 mM; 24 h | ↓ cell viability | ND |
| 0.1 mM; 24 h | ↓ cell viability ↑ LDH Neuron morphological alteration | (p < 0.01) (p < 0.05) ND | ||
| Palumbo et al. (2012) [70] | Differentiated HCN-1A cell line | 0.1 mM; 24 h | ↑ LDH ↓ cell viability Neuron morphological alteration ↑ Ca2+ | (p < 0.05) (p < 0.05) ND (p < 0.01) |
| Gupta et al. (2013) [71] | Human embryonic stem cell (HESC) line H9 | 20-200 µM; 24 h | ↑ neuronal death (20–80 µM) ↑ Ca2+ influx (200 µM) | ND (p < 0.001) |
| Yon et al. (2018) [72] | Human neural stem cell (NSC) culture HB1.F3 | 0.8–50 mM (MSG); 2 h | ↑ LDH | ND |
| 2.5 mM (MSG); 2 h | ↑ LDH ↑ NF-κB mRNA ↑ TNF-α mRNA ↑ IL-6 mRNA ↑ iNOS mRNA and protein ↑ COX-2 mRNA and protein ↑ TGF-β protein ↑ HMGB1 protein | (p < 0.05) (p < 0.05) (p < 0.05) (p < 0.05) (p < 0.05) (p < 0.05) (p < 0.05) (p < 0.05) | ||
| Chao and Hu (1994) [73] | Human fetal brain tissue | 1.6–5000 µM; 6 d | ↑ LDH ↓ GABA uptake (as marker of GABAergic neuron integrity) | ND ND |
| de Vera et al. (2008) [74] | Human fetal cortical brain tissue 14–42 DIV | 1–10 mM; 24 h | ↑ neuronal death (5 mM at 26, 32, and 42 DIV) ↑ swelling of astrocyte nuclei (5 mM at 32 DIV) | (p < 0.05) (p < 0.05) |
| Reference | Species and Strain, Size of L-Glu Treatment Group | L-Glu Treatment and Duration (Dose, Route of Drug Application) | Study Outcomes | Level of Significance |
|---|---|---|---|---|
| Babu et al. (1994) [75] | Wistar rats (n = 6) | MSG; 4 mg/g, s.c.; 10 days postnatally (PD 1–10) | ↑ LPO ↑ CAT activity ↓ sulfhydryl levels | (p < 0.01) (p < 0.025) (p < 0.05) |
| Ferger et al. (1998) [76] | Albino Wistar rats (n = 5) adults male (350 g) | L-Glu 50 mM, striatum microdialysis | ↑ OH• | ND |
| Singh et al. (2003) [77] | Wistar rats (n = 3) male (160–180 g) 3–4 months old | MSG 4 mg/g/day, i.p.; 6 consecutive days | ↓ Mn-SOD activity ↓ CAT activity (mitochondrial) ↓ GSH content ↑ GPx content ↑ LPO ↑ uric acid | (p < 0.001) (p < 0.01–0.001) (p < 0.05–0.001) (p < 0.05–0.001) (p < 0.02–0.001) (p < 0.05–0.01) |
| Rivera-Cervantes et al. (2004) [78] | Wistar rats (n = 6) | MSG; 4 mg/g, s.c.; PD 1, 3, 5, 7 | ↑ neurons’ histologic changes and degeneration ↑ Gliosis ↑ NR1 mRNA subunit of NMDAR ↑ GluR2 mRNA subunit of AMPAR ↑ ATF2 pp protein (for p38 MAPK activation) | (p < 0.001) ND (p < 0.001) (p < 0.001) (p < 0.001) |
| Chaparro-Huerta et al. (2005) [79] | Wistar rats (n = 8) | MSG: 4 mg/g, s.c.; PD 1, 3, 5, 7 | ↓ glial size and processes ↑ apoptosis ↑ TNF-α mRNA and protein ↑ IL-1ß mRNA and protein ↑ IL-6 mRNA and protein | ND (p < 0.001) (p < 0.05, and p < 0.001) (p < 0.001) (p < 0.001) |
| Mejía-Toiber et al. (2006) [80] | Wistar rats (n = 3–9) male (250–320 g) | 1 µmol/µL, intrastriatal injection; 0.5 µL/min for 2 min | ↑ striatal lesions ↓ ATP level (after 6 h but not at 3 h) ↑ LPO level (after 5 or 24 h) | (p < 0.0005) ND (p < 0.05) |
| Segura Torres et al. (2006) [81] | Wistar rats (n = 5) | MSG; 4 mg/g, s.c.; PD 1, 3, 5, 7 | ↑ GluR2 protein subunit of AMPAR at PD 8 ↓ GluR2 protein subunit of AMPAR at PD 14 ↑ REST mRNA at PD 8 and 14 ↑ Fas-L and Bcl-2 mRNA at PD 8 | ND ND (p ≤ 0.01–0.001) (p ≤ 0.01) |
| Chaparro-Huerta et al. (2008) [36] | Wistar rats (n = 8) | MSG; 4 mg/g, s.c.; PD 1, 3, 5, 7 | ↑ apoptosis ↑ nuclear material condensed ↑ TNF-α mRNA ↑ IL-1ß mRNA ↑ IL-6 mRNA | (p < 0.05, p < 0.001) ND (p < 0.05–p < 0.001) (p < 0.001) (p < 0.001) |
| Del Río and Massieu (2008) [82] | Wistar rats (n = 4–7) male (250–300 g) | 1 µL (500 nmoles), intrastriatal injections; rate of 0.5 µL/min | ↑ brain lesions ↑ calpain activation protein | ND (p ≤ 0.05) |
| Hashem et al. (2012) [83] | Albino rats (n = 10) adult male (150–200 g) 3–6 months old | MSG; 3 g/kg/day, p.o.; 14 days | Neurons’ morphological alterations Darkly stained cytoplasm of Purkinje cells Shrunken darkly stained nuclei ↑ neurons degeneration ↑ inflammatory cells ↑ GFAP immunoreactivity in the astrocytes of granular layer | ND |
| Shivasharan et al. (2013) [27] | Wistar rats (n = 6) adult female (190–220 g) | MSG; 2 g/kg/day, i.p.; 7 days | ↓ locomotor activity Decreased hippocampus layer, darkly stained shrunken cells, and mildly separated interconnected neuropil fibres ↑ NO− ↑ LPO level ↓ GSH level ↓ GST activity ↓ CAT activity ↓ total thiols level | (p < 0.001) ND (p < 0.001) (p < 0.001) (p < 0.001) (p < 0.001) (p < 0.05) (p < 0.01) |
| Swamy et al. (2013) [84] | Wistar albino rats (n = 6) (50–200 g) of either sex | MSG 2 g/kg/day, i.p.; 7 days | ↑ behavioural alterations and reduced locomotor activity Marked cerebral oedema, neuronal eosinophilia, nuclear pyknosis, and neuronal karyorrhexis ↑ Ca2+ ↑ Na+ ↓ K+ ↓ GABA ↓ GSH level ↓ SOD activity ↓ CAT activity ↑ LPO levels | (p < 0.05) ND (p < 0.05) (p < 0.05) (p < 0.05) (p < 0.05) (p < 0.05) (p < 0.05) (p < 0.05) (p < 0.05) |
| Dief et al. (2014) [33] | Wistar rats (n = 6) male (40–60 g) 5 weeks old | MSG; 2 g/kg/day, p.o.; 10 consecutive days MSG; 4 g/kg/day, s.c.; 10 alternate days | ↑ anxiety behaviour ↓ working memory ↓ AMPK protein ↑ Fas-L protein ↑ Aβ (1-42) | (p < 0.05) (p < 0.05) (p < 0.05) (p < 0.05) (p < 0.05) |
| Thonda et al. (2014) [85] | Wistar rats (n = 6) adult female (180–220 g) | MSG 2 g/kg/day, i.p.; 7 days | ↓ locomotor activity ↓ grip strength ↓ memory retention ↓ motor coordination and body balance Hippocampal pyramidal cells’ degeneration with intact neuropil fibres ↓ body weight ↑ LPO levels ↓ CAT activity ↓ SOD activity ↓ GSH level | (p < 0.001) (p < 0.01) (p < 0.01) (p < 0.001) ND (p < 0.01) (p < 0.001) (p < 0.01) (p < 0.001) (p < 0.01) |
| Rivera-Cervantes et al. (2015) [86] | Wistar rats (n = 4–5) | MSG; 4 mg/g, s.c.; PD 1, 3, 5, 7 | ↑ alterations and loss in the hippocampal neurons at PD 8, 10, 12, and 14 ↑ TUNEL-positive cells at PD 8, 10, and 14 ↑ NMDAR subunit NR1 mRNA at PD 10, 12, and 14 ↑ AMPAR subunit GluR1 mRNA at PD 12 and 14 ↓ AMPAR subunit GluR2 mRNA and protein at PD 10 and 14 ↑ NRSF mRNA at PD 8 and 14 ↑ ATF2pp protein | (p < 0.01) (p < 0.001) (p < 0.001) (p < 0.001) (p < 0.001) (p < 0.01) (p < 0.001) |
| Khalil and Khedr (2016) [87] | Wistar rats (n = 8) male (120–150 g) 12 weeks old | MSG; 4 mg/kg/day, p.o.; 4 weeks | ↑ L-Glu level ↑ AChE activity ↑ TNF-α level ↑ NMDA2B mRNA ↑ mGluR5 mRNA | (p < 0.01) (p < 0.001) (p < 0.001) (p < 0.001) NS |
| Sadek et al. (2016) [88] | Albino Wistar rats (n = 8) male (130–160 g) 2 months old | MSG; 5 mg/kg/day, s.c.; 4 weeks | ↑ LDH ↑ Na+ ↓ K+ ↑ LPO level ↑ GST activity and mRNA ↑ CAT activity and mRNA ↑ SOD activity ↓ GSH level ↑ AChE activity ↑ Bax mRNA ↓ Bcl-2 mRNA ↑ Serum ChE level ↑ CPK activity ↑ CPK-BB activity | (p < 0.05) (p < 0.05) (p < 0.05) (p < 0.05) (p < 0.05) (p < 0.05) (p < 0.05) (p < 0.05) (p < 0.05) (p < 0.05) (p < 0.05) (p < 0.05) (p < 0.05) (p < 0.05) |
| Hussein et al. (2017) [89] | Albino rats (n = 6) male (100–130 g) | MSG; 100 mg/kg/day, p.o.; 2 months | Pathological damage to brain tissue ↑ LPO level ↑ NO• ↓ SOD activity ↓ CAT activity ↓ GSH level ↑ Aβ (1-42) ↓ AChE activity ↑ serotonin level ↑ dopamine level ↑ L-Glu level ↑ Ca2+ ↑ Na+ ↓ K+ ↑ 8-OHdG in the brain DNA | ND (p < 0.01) (p < 0.001) (p < 0.001) (p < 0.001) (p < 0.01) (p < 0.001) (p < 0.01) (p < 0.001) (p < 0.001) (p < 0.001) (p < 0.05) (p < 0.05) (p < 0.05) (p < 0.01) |
| Abdel Moneim et al. (2018) [90] | Albino rats (n = 20) male (45–70 g) 5–6 weeks old | High; MSG 1.66 g/kg/day, p.o.; 30 days Low; MSG 0.83 g/kg/day, p.o.; 30 days | ↓ cognitive functions ↓ serotonin level (high-dose MSG) | (p < 0.001) (p < 0.001) |
| Fouad et al. (2018) [91] | Albino rats (n = 12) adult male (250–300 g) 2 months old | 2 g/kg/day, i.p.; 7 days | ↓ spontaneous alternation behaviour (spatial working memory) ↑ MEL in the MWM ↓ time spent in target quadrant (MWM) ↑ cytochrome c mRNA ↑ caspase-3 level ↑ LDH ↑ Aβ (1-42) | (p < 0.05) (p < 0.05) (p < 0.05) (p < 0.05) (p < 0.05) (p < 0.05) (p < 0.05) |
| Hazzaa et al. (2020) [92] | Wistar albino rats (n = 10) male (40 g) 1 month old | MSG 4 g/kg/day, i.p.; 7 days | ↓ locomotor activity ↓ spatial memory ↑ morphological alteration in hippocampus neurons ↑ LPO level ↑ caspase-3 protein ↓ SOD activity ↑ GFAP protein ↓ Ki-67 protein ↑ calretinin protein | (p < 0.001) (p < 0.05–0.001) (p < 0.05) (p < 0.05) ND (p < 0.05) (p < 0.05) (p < 0.05) (p < 0.05) |
| Greene and Greenamyre (1995) [94] | Sprague–Dawley rats (n = 5) male (200–250 g) | L-Glu 0.3 M intrastriatal injection; 2 µL of solution at 0.5 µL/min (0.6 µmoles) | ↑ lesion | (p < 0.01) |
| Yang et al. (1998) [95] | Sprague–Dawley rats (n = 10) male (280–350 g) | 1.5 or 15 mM L-Glu, cortex microdialysis; 2 μL/min 20–180 min | ↑ LPO at 1.5 and 15 mM | ND |
| Bodnár et al. (2001) [96] | Sprague–Dawley rats (n = 7–9) of both sexes | MSG; 4 mg/g, s.c.; PD 2, 4, 6, 8, 10 | ↑ degradation of TH-positive (dopaminergic) neurons of hypothalamic arcuate nucleus | (p < 0.05) |
| Kumar and Babu (2010) [97] | Sprague–Dawley rats (n = 6) male (300–350 g) 3 months old | 1 µmole/1 µL, cerebral cortex injection | ↓ pyramidal neurons’ size ↑ condensed nuclei ↑ Ca2+ ↑ LPO level ↑ ROS ↓ SOD activity ↓ CAT activity ↓ GSH level ↓ GR activity ↑ TNF-α level ↑ IFN-ɣ level ↑ NO• ↓ MMP | ND (p < 0.001) (p < 0.001) (p < 0.001) (p < 0.001) (p < 0.001) (p < 0.001) (p < 0.05) (p < 0.001) (p < 0.001) (p < 0.05) (p < 0.001) (p < 0.001) |
| Kumar et al. (2010) [98] | Sprague–Dawley rats (n = 6) male (300–350 g) 3 months old | 1 µmole/1 µL, cerebral cortex injection | ↓ pyramidal neurons’ size ↑ condensed nuclei ↑ ROS ↑ ONOO− ↓ MMP ↓ GSH ↑ Ca2+ ↑ nNOS mRNA ↑ iNOS mRNA ↑ caspase-3 mRNA ↑ caspase-9 mRNA ↓ Bcl-2 mRNA ↑ Bax mRNA ↓ Bcl-2/Bax ratio mRNA | ND (p < 0.001) (p < 0.001) (p < 0.001) (p < 0.001) (p < 0.001) (p < 0.001) (p < 0.001) (p < 0.001) (p < 0.001) (p < 0.001) ND ND (p < 0.001) |
| Nagesh Babu et al. (2011) [99] | Sprague–Dawley rats (n = 6) male (300–350 g) 3 months old | 1 µmole/1 µL, cerebral cortex injection | ↑ LPO level ↑ ROS ↓ SOD activity ↓ CAT activity ↓ GSH level ↓ GR activity ↑ TNF-α level ↑ IFN-ɣ level ↑ NO• ↓ MMP ↑ Ca2+ ↑ caspase-3 mRNA ↑ caspase-9 mRNA ↑ iNOS mRNA ↑ nNOS mRNA | (p < 0.001) (p < 0.001) (p < 0.05) (p < 0.05) (p < 0.05) (p < 0.05) (p < 0.001) (p < 0.001) (p < 0.001) (p < 0.001) (p < 0.001) ND ND ND ND |
| Morales and Rodriguez (2012) [100] | Sprague–Dawley rats (n = 5, 7, 8) male (300–350 g) | L-Glu in Ringer solution of 2 µL/min, striatum microdialysis; 60 min | ↑ astrogliosis ↑ L-Glu level ↑ alanine level ↓ glutamine level | (p < 0.001) (p < 0.001) (p < 0.001) (p < 0.001) |
| Kim et al. (2013) [101] | Sprague–Dawley rats (n = 6) male (300–350 g) 3 months old | 1 µmol/1 µL, cerebral cortex injection | ↑ LPO level ↑ Ca2+ ↑ ROS ↓ GSH level ↓ SOD activity ↓ CAT activity ↓ GPx level ↓ GR level ↑ TNF-α level ↑ IFN-ɣ level ↑ NO− ↑ NADPH oxidase activity ↑ nNOS mRNA ↓ MMP ↑ p-ERK1/2 mRNA ↑ caspase-3 mRNA | ND ND ND ND ND ND ND ND ND ND ND ND ND ND ND ND |
| Shah et al. (2015) [102] | Sprague–Dawley male rats (n = 10) male (18 g) pups at PD 7 | 5 mg/kg, s.c.; 4 or 12 h, PD 7 10 mg/kg, s.c.; 4 or 12 h, PD 7 | ↑ L-Glu level ↑ Bax protein ↓ Bcl-2 protein ↑ Bax/Bcl-2 ratio ↑ cytochrome c protein ↑ cleaved caspase-3 protein and level ↑ p-AMPK protein (high dose) ↑ FJB positive neurons (high dose) ↑ cleaved PARP-1 protein | (p < 0.01–0.0001) ND ND (p < 0.01–0.0001) (p < 0.01–0.0001) (p < 0.01–0.0001) (p < 0.0001) (p < 0.0001) (p < 0.01–0.0001) |
| Calis et al. (2016) [103] | Sprague–Dawley rats (n = 7) Female (250–300 g) 3–4 months old | MSG; 2 g/kg/day, i.p.; 7 days | No neuron degeneration in pyramidal and granular neurons in the brain cortex No effect on SOD level ↓ LPO level | ND NS (p < 0.001) |
| Shah et al. (2016) [56] | Sprague–Dawley rats (n = 5) (18 g) pups at PD 7 | 10 mg/kg, i.p.; 2, 3, 4 h | ↑ DNA fragmentation ↑ L-Glu ↑ AMPAR protein ↑ p-AMPK protein ↑ p-NF-kB protein ↓ Nrf2 protein ↑ CaMKII protein ↓ GSH level ↓GSH/GSSG ratio ↑ GFAP ↑ microglia Iba-1 ↑ ROS ↑ COX-2 protein ↑ TNF-α protein ↑ caspase-3 protein ↓ HO-1 protein | (p < 0.001) (p < 0.05–0.001) (p < 0.05–0.001) (p < 0.05–0.001) (p < 0.05–0.001) (p < 0.05–0.01) (p < 0.001) (p < 0.001) (p < 0.001) (p < 0.001) (p < 0.001) (p < 0.001) (p < 0.001) (p < 0.001) (p < 0.001) (p < 0.001) |
| Yang et al. (2017) [104] | Sprague–Dawley rats (n = 6) adult male (270–320 g) | 1 M/1 μL, cerebral cortex injection | ↑ TUNEL-positive cells ↑ caspase-3 protein and activity ↑ calpain protein and activity ↑ Bax protein ↓ Bcl-2 protein ↓ Bcl-2/Bax ratio ↑ ROS ↑ NO ↑ TNF-α level ↑ IFN-ɣ level ↑ IL-1β level ↑ NOX activity ↑ LPO level ↓ SOD activity ↓ GR activity ↓ GSH level ↓ CAT activity ↑ iNOS protein ↑ nNOS protein ↓ Nrf2 protein ↓ HO-1 protein ↓ GCLC protein ↓ ATP level ↓ Na+-K+-ATPase level ↓ cytochrome c oxidase activity | ND ND ND ND ND ND ND ND ND ND ND ND ND ND ND ND ND ND ND ND ND ND ND ND ND |
| Firgany and Sarhan (2020) [105] | Sprague–Dawley rats (n = 10) male (365 g) 18 months old | MSG; 4.0 g/kg/day, s.c.; 10 days | Remarkable morphological alteration in motoneurons and neuroglia ↑ caspase-3 activity ↑ LPO level ↑ IL-1β level ↑ IL-6 level ↑ TNF-α level ↑ IFN-ɣ level ↓ IL-10 level ↓ SOD activity ↓ CAT activity ↓ GFAP level ↑ ATF2pp protein | ND (p < 0.001) (p < 0.001) (p < 0.001) (p < 0.001) (p < 0.001) (p < 0.001) (p < 0.001) (p < 0.001) NS (p < 0.001) (p < 0.001) |
| Hamza et al. (2019) [106] | Rats (n = 8) adult male (200–250 g) | MSG high; 17.5 mg/kg/day, p.o.; 30 days MSG low; 6 mg/kg/day, p.o.; 30 days | MSG high dose: Large area of haemorrhage and necrotic areas of the brain with the congested area with degeneration in some glial ↓ catecholamine level ↓ dopamine level ↓ serotonin level ↓ AChE activity ↓ thiol level ↓ SOD activity ↓ CAT activity ↓ GPx activity ↓ GSH level ↓ BDNF level ↑ COX-2 activity ↑ PGE2 level MSG low dose: Moderate area of haemorrhage and necrosis in the brains | ND (p ≤ 0.05) (p ≤ 0.05) (p ≤ 0.05) (p ≤ 0.05) (p ≤ 0.05) (p ≤ 0.05) (p ≤ 0.05) (p ≤ 0.05) (p ≤ 0.05) (p ≤ 0.05) (p ≤ 0.05) (p ≤ 0.05) ND |
| Burde et al. (1971) [93] | Wistar rat 4 day old and Swiss albino mice 10 day old (n = 2–9) | MSG; 1 mg/g, 4 mg/g, p.o., or s.c.; 5 h MSG; 4 mg/g, 2 mg/g, p.o., or s.c.; 5 h | ↑ lesions in arcuate of the hypothalamus Lesions more extensive in rat than mice ↑ necrosis of neuron of arcuate Perikaryon swelling loss of cytoplasmic density and nuclear pyknosis | ND |
| Onaolapo et al. (2016) [28] | Swiss mice (n = 10) adult male (20–22 g) | MSG; 10, 20, 40, and 80 mg/kg/day, p.o.; 28 days L-Glu; 10 mg/kg/day, p.o.; 28 days | MSG: ↑ brain weight (40 and 80 mg/kg) ↑ neurons’ morphological alteration (MSG and L-Glu) ↑ glial cell number ↑ L-Glu plasma level (40 and 80 mg/kg) ↑ glutamine plasma level (40 and 80 mg/kg) ↓ SOD level (20, 40, and 80 mg/kg) ↓ CAT level (40 and 80 mg/kg) ↑ NO• (all doses) | (p < 0.01) (p < 0.05) (p < 0.05) (p < 0.001) (p < 0.01) (p < 0.01) (p < 0.001) (p < 0.002) |
| Mohan et al. (2017) [107] | Swiss albino mice (n = 5 for some experiments) of either sex (18–22 g) | MSG; 1000 mg/kg/day, p.o.; 14 days | ↓ onset of immobility delayed ↓ total immobility period ↓ brain weight Neurodegeneration characterised by deformed brain layers, pyknosis, and neuronal cell vacuolisation ↓ SOD activity ↓ CAT activity ↓ RGSH ↑ LPO level | (p < 0.05) (p < 0.05) (p < 0.05) ND (p < 0.05) (p < 0.05) (p < 0.05) (p < 0.05) |
| Estrada-Sánchez et al. (2009) [108] | Wild-type mice (n = 3–4) female 10–14 weeks old | 500 nM/0.5 μL, intrastriatal injection; 3 and 24 h | ↑ striatal lesions at 10 and 14 weeks ↑ FJB-positive degenerating neurons | (p ≤ 0.05) (p ≤ 0.05) |
| Estrada-Sánchez et al. (2010) [109] | Wild-type mice (n = 3–6) female 10 weeks old | 500 nM/0.5 µL, intrastriatal injection; 24 h | ↑ lesions | (p ≤0.05) |
| Guemez-Gamboa et al. (2011) [110] | Wild-type mice (n = 3–7) | MSG; 500 nM/0.5 µL, intrastriatal injection; rate of 0.175 µL/min | ↑ microglia activation ↑ NT-positive cells (NT; index of oxidative damage) ↑ striatal lesions ↑ FJB-positive degenerating neurons ↑ ROS ↑ NADPH oxidase activity ↑ calpain protein | ND (p < 0.05) (p < 0.001) (p < 0.001) (p < 0.001) (p < 0.05) (p < 0.05) |
| Wang et al. (2015) [111] | Wild-type mice (n = 3) male 4–6 months old | 10 mM at a flow rate of 1 µL/h, left lateral ventricle, brain infusion cannula; 7 days | ↑ motor neurons clumping or fragmented nuclei ↑ mitochondrial fragmentation ↓ MFN2 protein ↑ cleaved caspase-3-positive neurons | ND (p < 0.001) (p < 0.05) ND |
| Zou et al. (2016) [112] | Wild-type mice (n = 6) (25–30 g) 4–5 months old | MSG; 0.25 M, 0.2 μL injected on each side of the parietal cortex | ↑ lesion | (p < 0.01) |
| Yu et al. (2006) [113] | Kunming mice (n = 8–10) female, pregnant 7 weeks old | MSG; 1, 2, 4 g/kg/day, i.g.; at days 17–19 days of pregnancy | ↑ hyperactivity from open field test ↓ the memory retention and Y-maze discrimination learning capacities ↑ hippocampal lesions ↑ [3H]-Glu uptake ↓ Bcl-2 protein ↑ caspase-3 protein | (p < 0.0001- 0.0004) (p < 0.0001) ND ND ND ND |
| Ma et al. (2007) [114] | Kunming mice (n = 11–13) adults 8 weeks old | MSG; 1, 2, 4 g/kg/day, i.g.; 10 days | ↓ discrimination learning and memory using Y-maze test Hippocampal lesions’ intracellular oedema, degeneration and necrosis of neurons, and hyperplasia | (p = 0.0006) ND |
| Penugonda and Ercal (2011) [115] | CD-1 mice (n = 4) adult male (38–40 g) | 2000 mg/kg/day, p.o.; 1 week | ↓ GSH level ↑ LPO level ↑ PLA2s activity | NS (p < 0.05–0.005) (p < 0.05–0.001) |
| Chu et al. (2020) [116] | Caenorhabditis elegans (wild-type) nematodes (n = 30) | 20 mM, animals living media; 24 h | ↑ damaged locomotory ability ↑ ROS ↑ O2−• ↓ GSH level | (p < 0.05) ND ND ND |
| Spangenberg et al. (2004) [117] | Ephyrae of Aurelia aurita (n = 2–4) | MSG; 5 mM, animals living media (artificial sea water); 1–24 h | ↑ impaired pulsing and swimming motility ↓ pulsing rates ↑ Ca2+ ↑ ROS ↑ NO• | (p < 0.05) (p < 0.05) (p < 0.05) (p < 0.05) ND |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
AL-Nasser, M.N.; Mellor, I.R.; Carter, W.G. Is L-Glutamate Toxic to Neurons and Thereby Contributes to Neuronal Loss and Neurodegeneration? A Systematic Review. Brain Sci. 2022, 12, 577. https://doi.org/10.3390/brainsci12050577
AL-Nasser MN, Mellor IR, Carter WG. Is L-Glutamate Toxic to Neurons and Thereby Contributes to Neuronal Loss and Neurodegeneration? A Systematic Review. Brain Sciences. 2022; 12(5):577. https://doi.org/10.3390/brainsci12050577
Chicago/Turabian StyleAL-Nasser, Maryam N., Ian R. Mellor, and Wayne G. Carter. 2022. "Is L-Glutamate Toxic to Neurons and Thereby Contributes to Neuronal Loss and Neurodegeneration? A Systematic Review" Brain Sciences 12, no. 5: 577. https://doi.org/10.3390/brainsci12050577