
A Bioinformatics-Based Analysis of an Anoikis-Related Gene Signature Predicts the Prognosis of Patients with Low-Grade Gliomas

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Gene Expression and Clinical Data Acquisition
2.2. Acquisition of ANRGs
2.3. Functional Enrichment Analysis
2.4. Consensus Clustering
2.5. Selection of Characteristic Genes
2.6. Development and Validation of Prognostic Signatures
2.7. Estimation of the Tumor Immune Microenvironment of the Prognostic Signature
2.8. Tumor Immune Cycle and ICB Response
2.9. Construction and Evaluation of a Predictive Nomogram
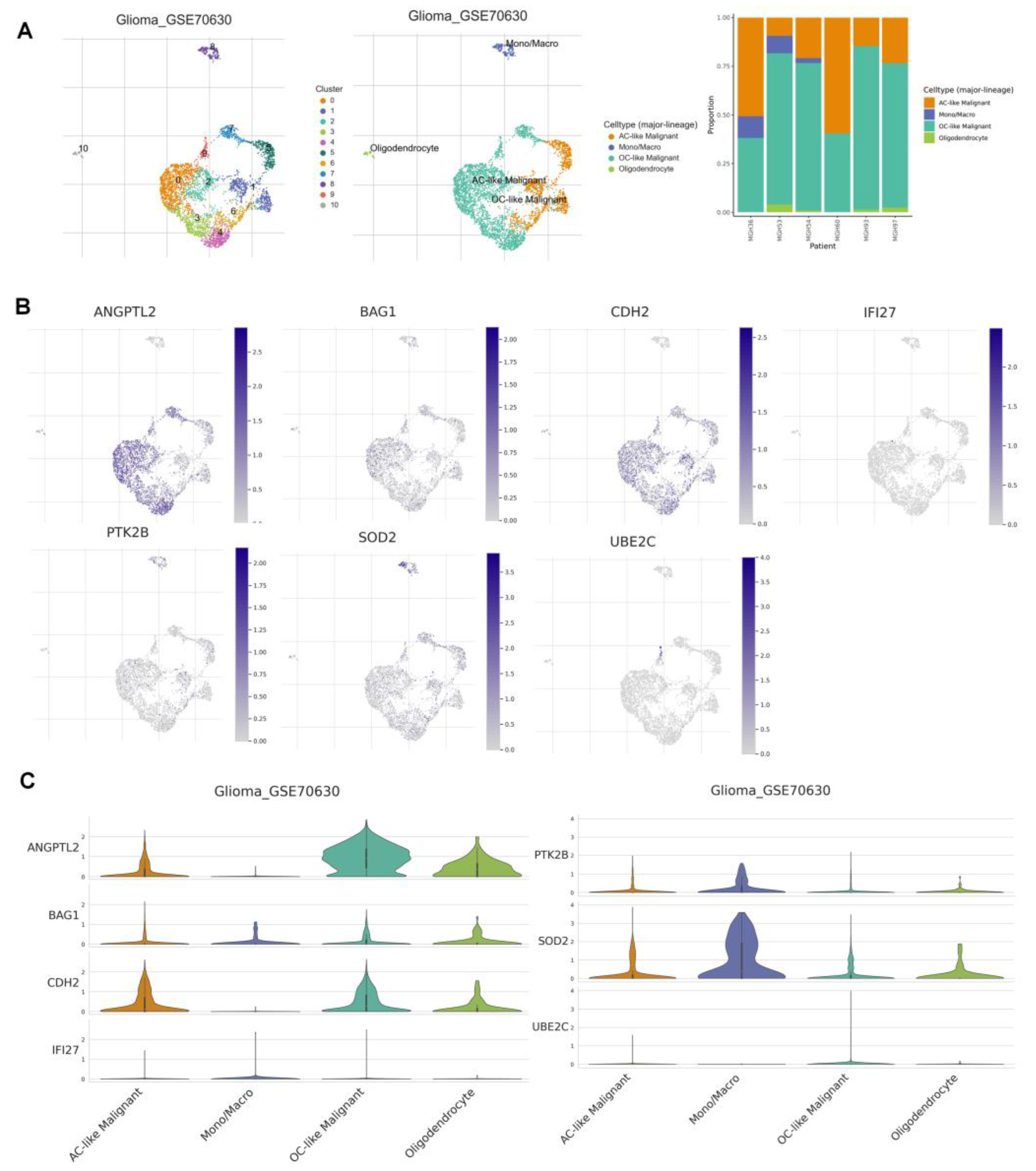
2.10. Tumor Immune Single Cell Hub Database
2.11. Statistical Analysis
3. Results
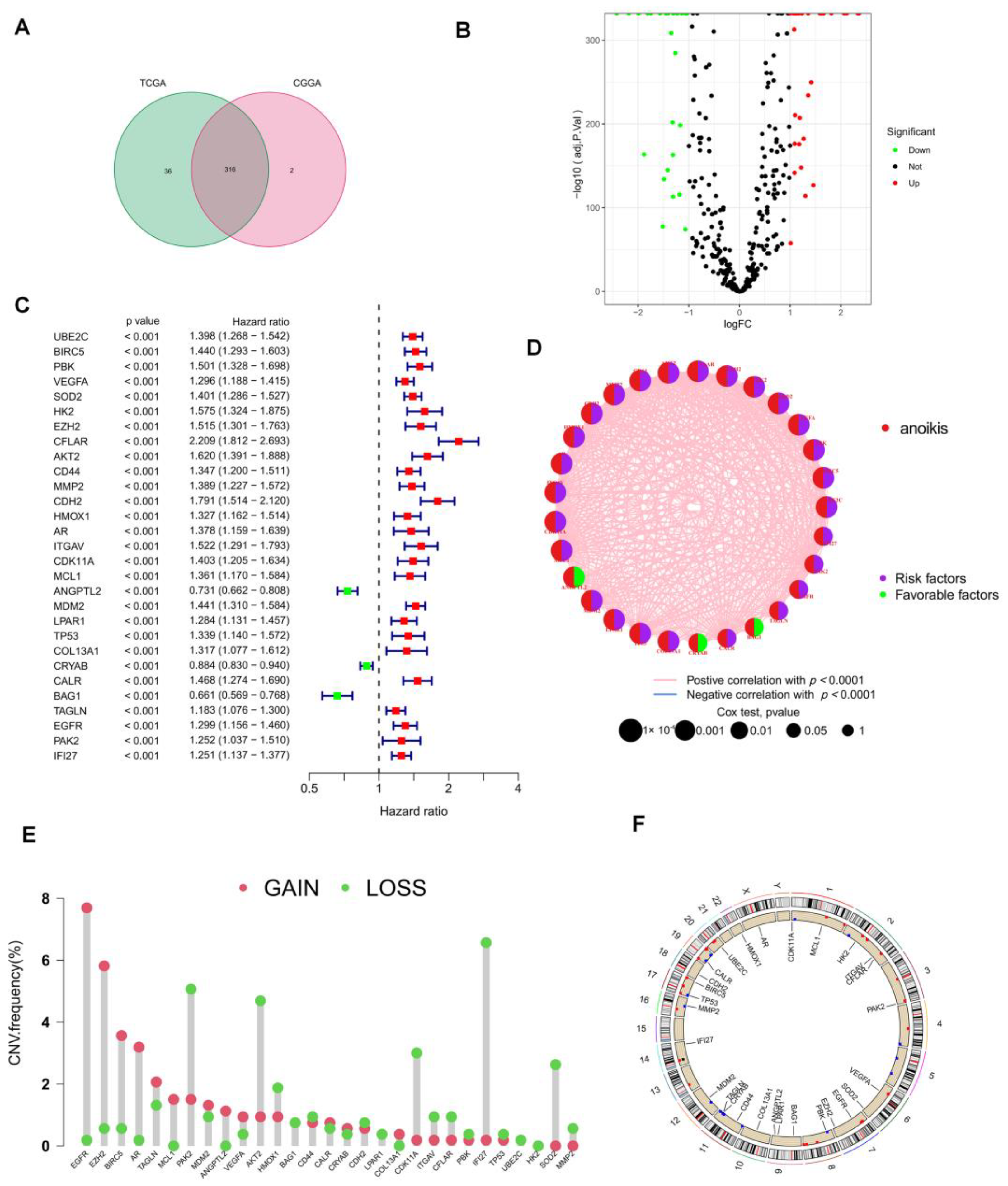
3.1. Acquisition of ANRGs
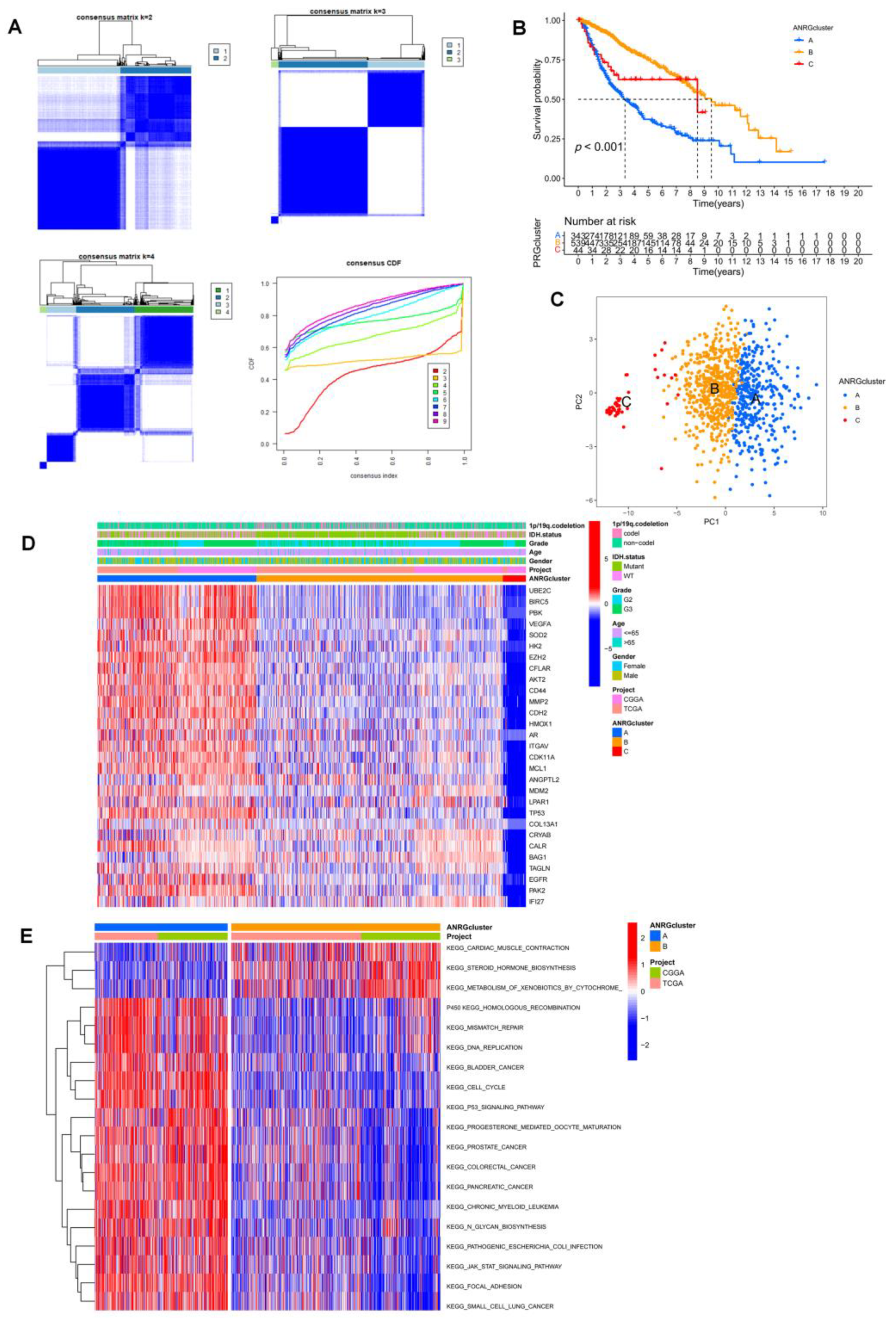
3.2. Consistent Clustering of 29 ANRGs in LGG
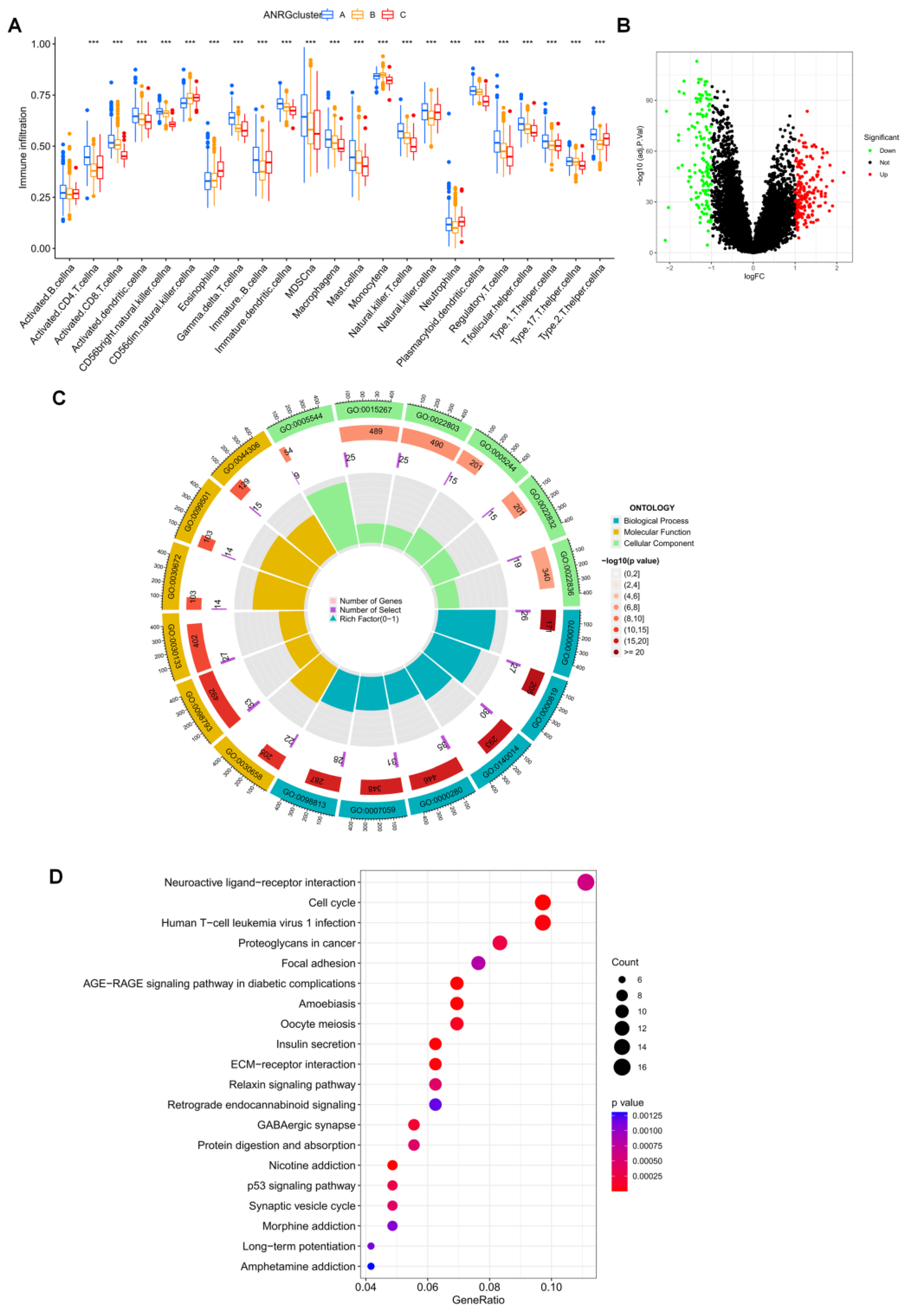
3.3. Immune Infiltration and Differential Gene Expression in the Two Subtype Clusters
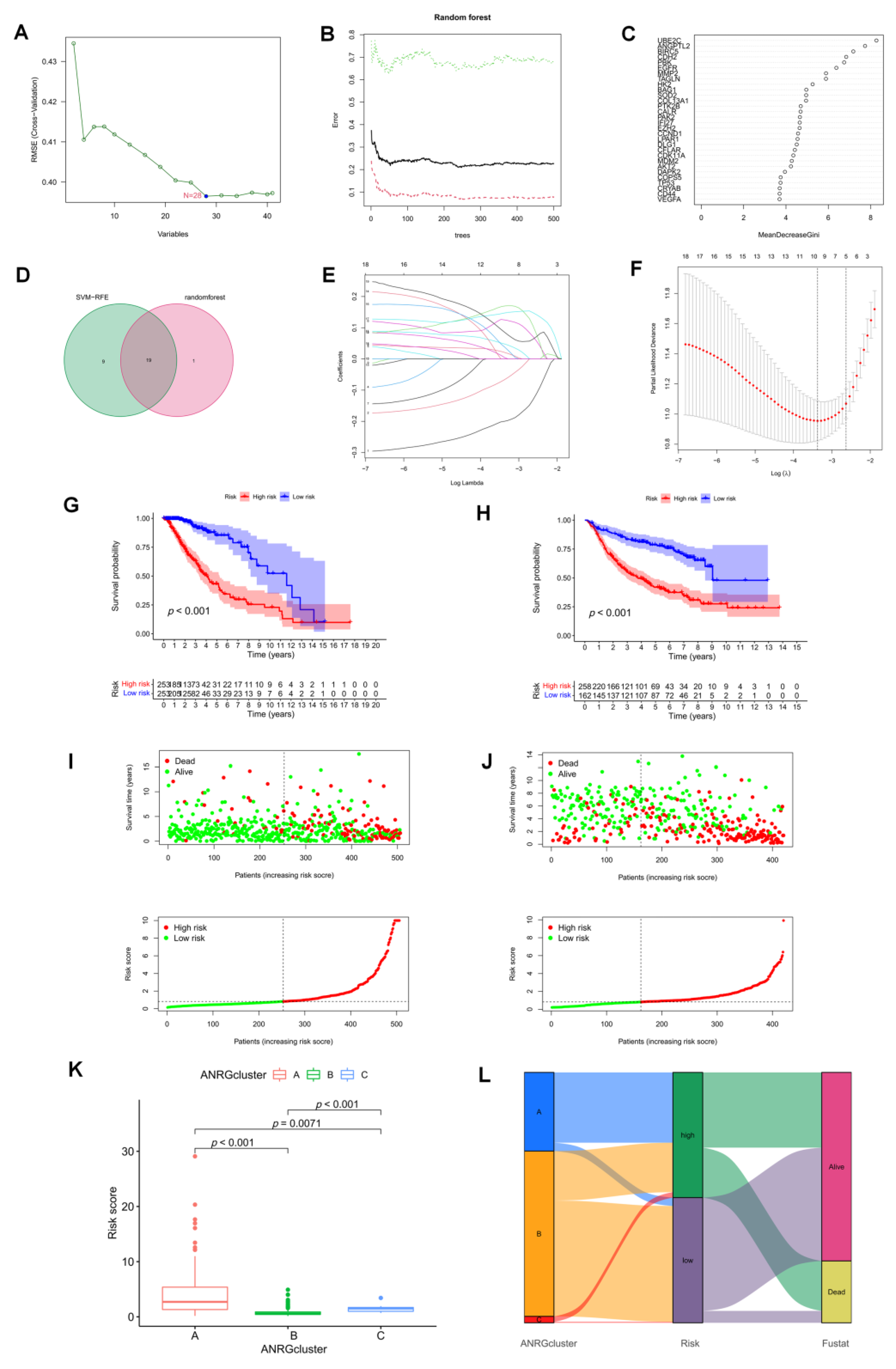
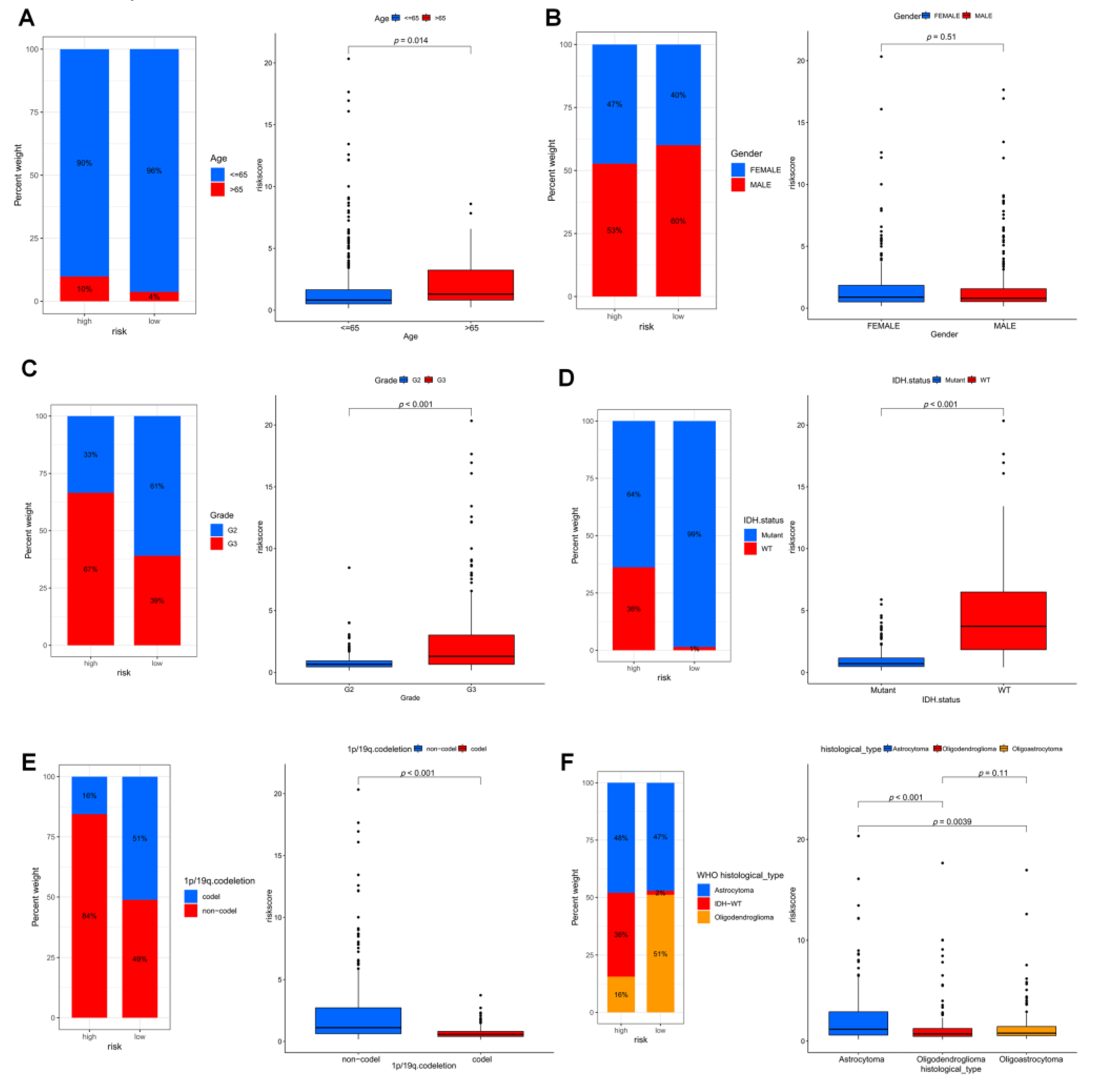
3.4. The Development and Validation of an Anoikis-Related Prognostic Signature
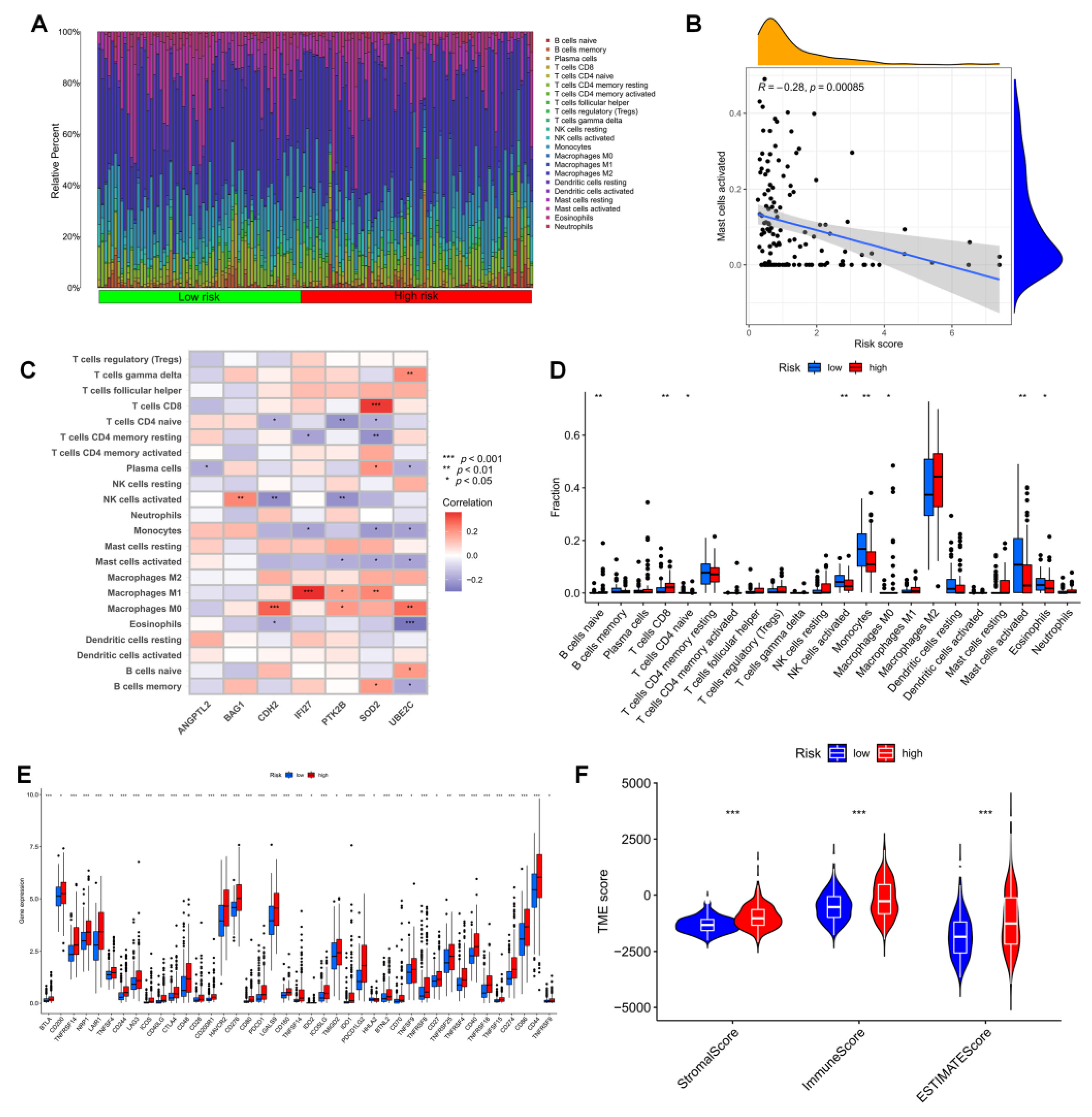
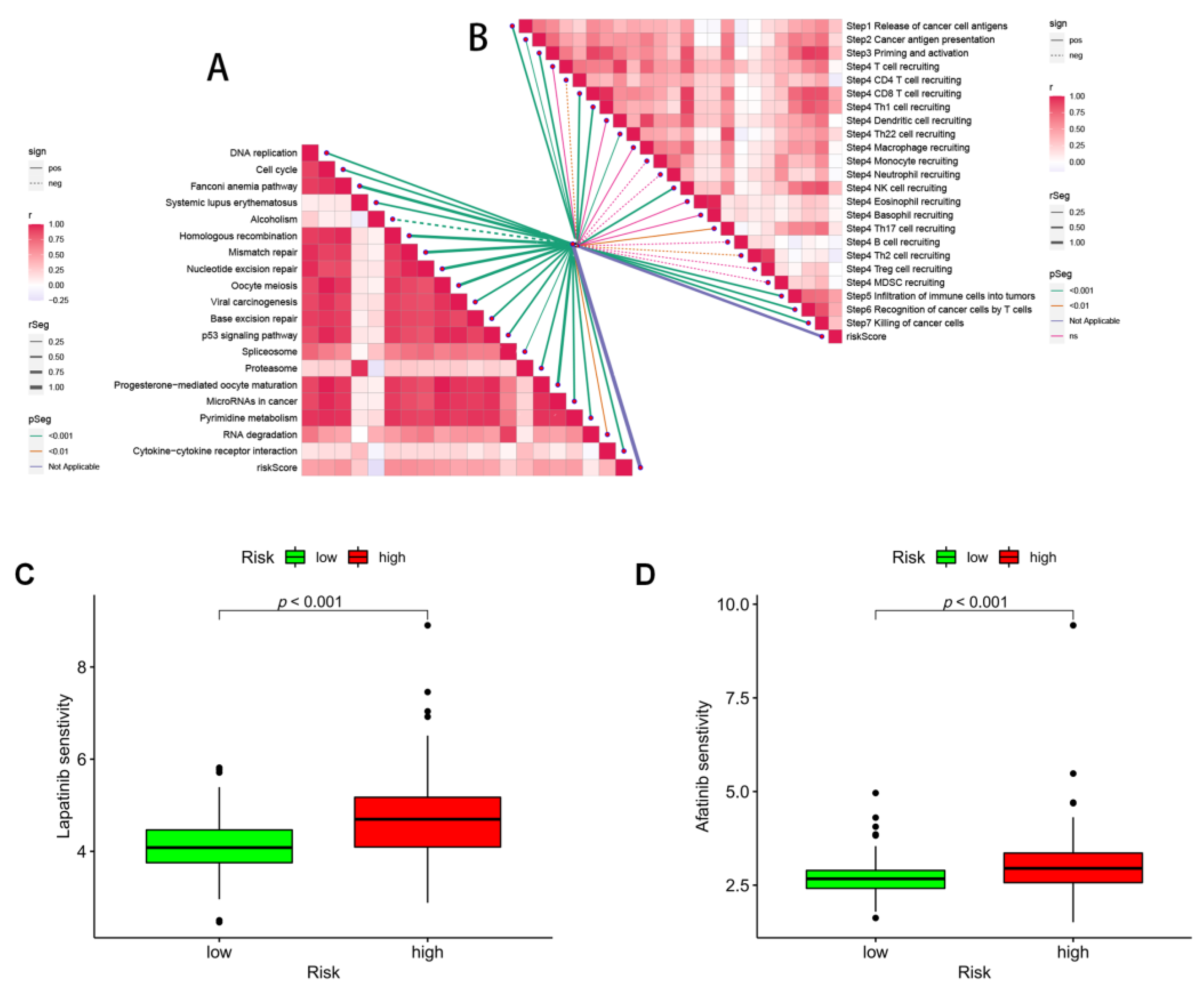
3.5. Immune Infiltration in Different Risk Groups
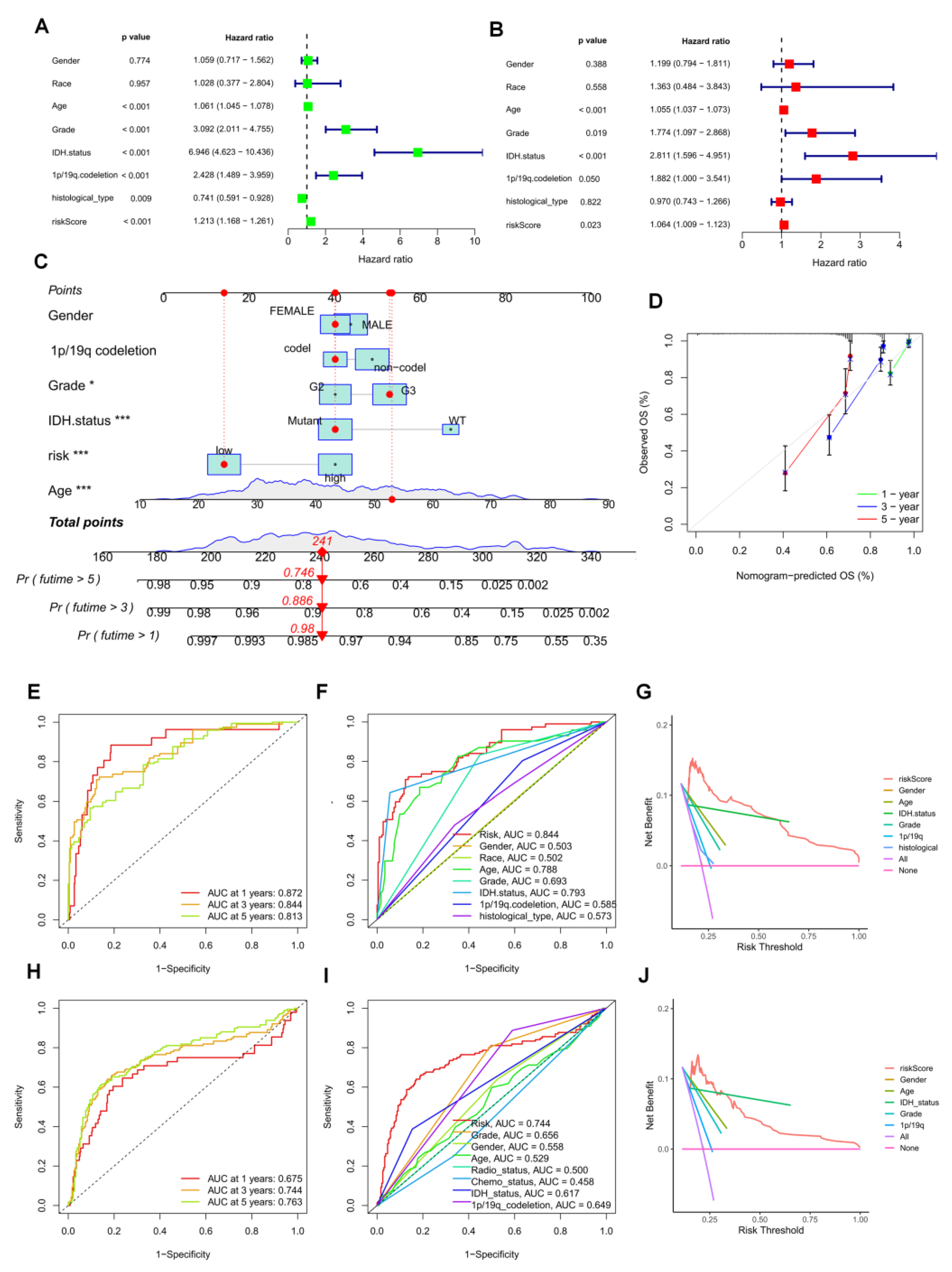
3.6. Establishment of a Prognostic Nomogram for LGG Patients
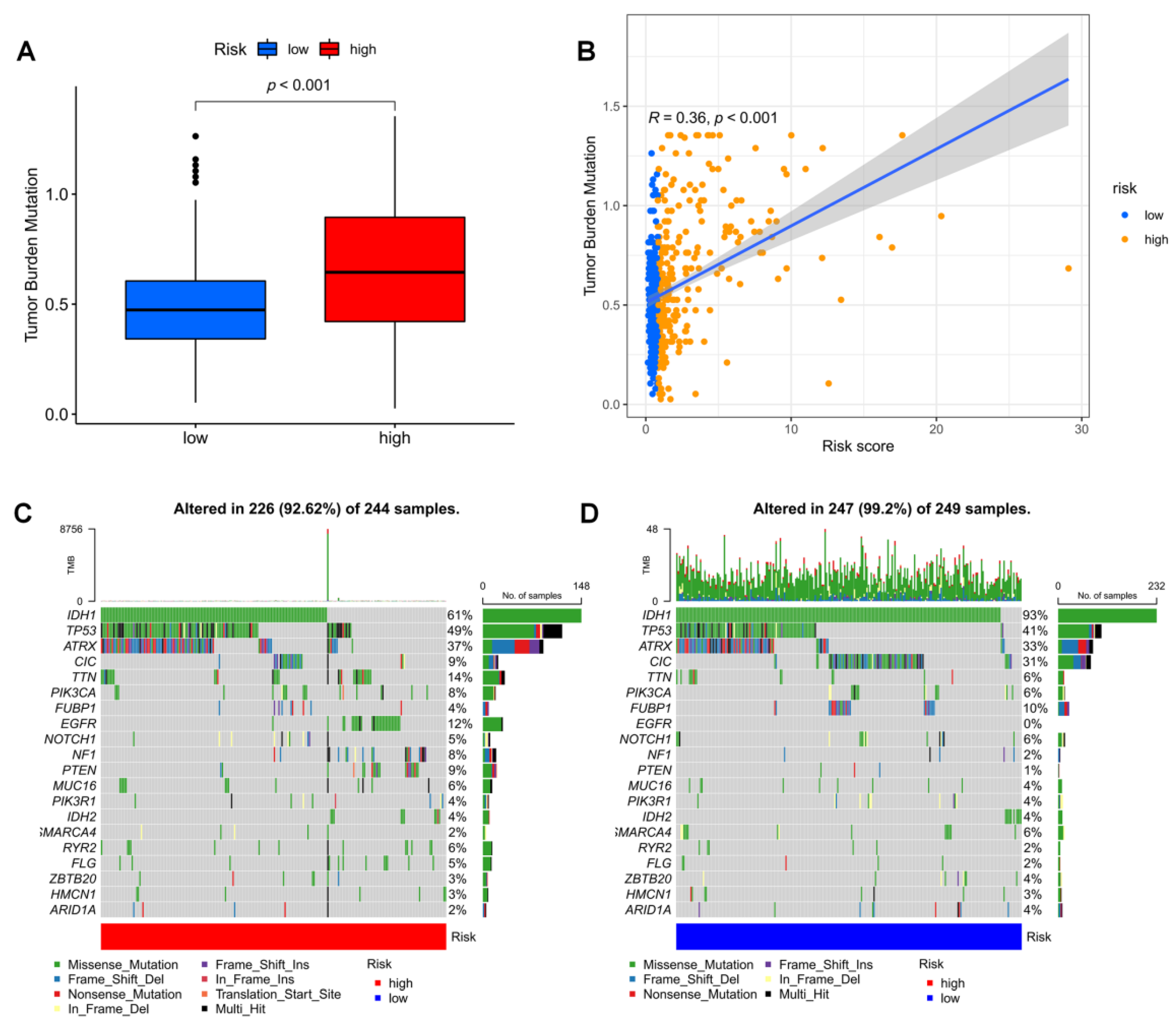
3.7. Mutation Landscape in Different Risk Groups
3.8. Immunotherapy and ICB Response
3.9. Correlation Analysis of ANRGs and Tumor Immune Microenvironment
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yang, Y.; Liu, X.; Cheng, L.; Li, L.; Wei, Z.; Wang, Z.; Han, G.; Wan, X.; Wang, Z.; Zhang, J.; et al. Tumor Suppressor microRNA-138 Suppresses Low-Grade Glioma Development and Metastasis via Regulating IGF2BP2. Oncol. Targets Ther. 2020, 13, 2247–2260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baumert, B.G.; Hegi, M.E.; van den Bent, M.J.; von Deimling, A.; Gorlia, T.; Hoang-Xuan, K.; Brandes, A.A.; Kantor, G.; Taphoorn, M.J.B.; Hassel, M.B.; et al. Temozolomide chemotherapy versus radiotherapy in high-risk low-grade glioma (EORTC 22033-26033): A randomised, open-label, phase 3 intergroup study. Lancet Oncol. 2016, 17, 1521–1532. [Google Scholar] [CrossRef] [Green Version]
- Tom, M.C.; Cahill, D.P.; Buckner, J.C.; Dietrich, J.; Parsons, M.W.; Yu, J.S. Management for Different Glioma Subtypes: Are All Low-Grade Gliomas Created Equal? Am. Soc. Clin. Oncol. Educ. Book 2019, 39, 133–145. [Google Scholar] [CrossRef] [PubMed]
- Wesseling, P.; Capper, D. WHO 2016 Classification of gliomas. Neuropathol. Appl. Neurobiol. 2018, 44, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Solheim, K.; Polley, M.Y.; Lamborn, K.R.; Page, M.; Fedoroff, A.; Rabbitt, J.; Butowski, N.; Prados, M.; Chang, S.M. Quality of life in low-grade glioma patients receiving temozolomide. Neuro Oncol. 2009, 11, 59–68. [Google Scholar] [CrossRef]
- Hersh, D.S.; Harder, B.G.; Roos, A.; Peng, S.; Heath, J.E.; Legesse, T.; Kim, A.J.; Woodworth, G.F.; Tran, N.L.; Winkles, J.A. The TNF receptor family member Fn14 is highly expressed in recurrent glioblastoma and in GBM patient-derived xenografts with acquired temozolomide resistance. Neuro Oncol. 2018, 20, 1321–1330. [Google Scholar] [CrossRef]
- Delgado-Martin, B.; Medina, M.A. Advances in the Knowledge of the Molecular Biology of Glioblastoma and Its Impact in Patient Diagnosis, Stratification, and Treatment. Adv. Sci. 2020, 7, 1902971. [Google Scholar] [CrossRef]
- Chiarugi, P.; Giannoni, E. Anoikis: A necessary death program for anchorage-dependent cells. Biochem. Pharmacol. 2008, 76, 1352–1364. [Google Scholar] [CrossRef]
- Boudreau, N.J.; Jones, P.L. Extracellular matrix and integrin signalling: The shape of things to come. Biochem. J. 1999, 339, 481–488. [Google Scholar] [CrossRef]
- Frisch, S.M.; Ruoslahti, E. Integrins and anoikis. Curr. Opin. Cell Biol. 1997, 9, 701–706. [Google Scholar] [CrossRef]
- Kakavandi, E.; Shahbahrami, R.; Goudarzi, H.; Eslami, G.; Faghihloo, E. Anoikis resistance and oncoviruses. J. Cell. Biochem. 2018, 119, 2484–2491. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.N.; Koo, K.H.; Sung, J.Y.; Yun, U.J.; Kim, H. Anoikis resistance: An essential prerequisite for tumor metastasis. Int. J. Cell Biol. 2012, 2012, 306879. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Song, Y.; Cheng, L.; Chen, L.; Liu, B.; Lu, D.; Li, X.; Li, Y.; Lv, F.; Xing, Y. CircCEMIP promotes anoikis-resistance by enhancing protective autophagy in prostate cancer cells. J. Exp. Clin. Cancer Res. 2022, 41, 188. [Google Scholar] [CrossRef] [PubMed]
- Leek, J.T.; Johnson, W.E.; Parker, H.S.; Jaffe, A.E.; Storey, J.D. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 2012, 28, 882–883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rebhan, M.; Chalifa-Caspi, V.; Prilusky, J.; Lancet, D. GeneCards: Integrating information about genes, proteins and diseases. Trends Genet. 1997, 13, 163. [Google Scholar] [CrossRef]
- Rouillard, A.D.; Gundersen, G.W.; Fernandez, N.F.; Wang, Z.; Monteiro, C.D.; McDermott, M.G.; Ma’ayan, A. The harmonizome: A collection of processed datasets gathered to serve and mine knowledge about genes and proteins. Database 2016, 2016, baw100. [Google Scholar] [CrossRef]
- Hanzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef] [Green Version]
- Sanz, H.; Valim, C.; Vegas, E.; Oller, J.M.; Reverter, F. SVM-RFE: Selection and visualization of the most relevant features through non-linear kernels. BMC Bioinform. 2018, 19, 432. [Google Scholar] [CrossRef] [Green Version]
- Newman, A.M.; Liu, C.L.; Green, M.R.; Gentles, A.J.; Feng, W.; Xu, Y.; Hoang, C.D.; Diehn, M.; Alizadeh, A.A. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 2015, 12, 453–457. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Deng, C.; Pang, B.; Zhang, X.; Liu, W.; Liao, G.; Yuan, H.; Cheng, P.; Li, F.; Long, Z.; et al. TIP: A Web Server for Resolving Tumor Immunophenotype Profiling. Cancer Res. 2018, 78, 6575–6580. [Google Scholar] [CrossRef]
- Mariathasan, S.; Turley, S.J.; Nickles, D.; Castiglioni, A.; Yuen, K.; Wang, Y.; Kadel, E.E., III; Koeppen, H.; Astarita, J.L.; Cubas, R.; et al. TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018, 554, 544–548. [Google Scholar] [CrossRef] [PubMed]
- Vickers, A.J.; Cronin, A.M.; Elkin, E.B.; Gonen, M. Extensions to decision curve analysis, a novel method for evaluating diagnostic tests, prediction models and molecular markers. BMC Med. Inform. Decis. Mak. 2008, 8, 53. [Google Scholar] [CrossRef] [Green Version]
- Sun, D.; Wang, J.; Han, Y.; Dong, X.; Ge, J.; Zheng, R.; Shi, X.; Wang, B.; Li, Z.; Ren, P.; et al. TISCH: A comprehensive web resource enabling interactive single-cell transcriptome visualization of tumor microenvironment. Nucleic Acids Res. 2021, 49, D1420–D1430. [Google Scholar] [CrossRef]
- Reiter, J.G.; Baretti, M.; Gerold, J.M.; Makohon-Moore, A.P.; Daud, A.; Iacobuzio-Donahue, C.A.; Azad, N.S.; Kinzler, K.W.; Nowak, M.A.; Vogelstein, B. An analysis of genetic heterogeneity in untreated cancers. Nat. Rev. Cancer 2019, 19, 639–650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis-Tuffin, L.J.; Rodriguez, F.; Giannini, C.; Scheithauer, B.; Necela, B.M.; Sarkaria, J.N.; Anastasiadis, P.Z. Misregulated E-cadherin expression associated with an aggressive brain tumor phenotype. PLoS ONE 2010, 5, e13665. [Google Scholar] [CrossRef] [PubMed]
- Tseng, T.; Uen, W.; Tseng, J.; Lee, S. Enhanced chemosensitization of anoikis-resistant melanoma cells through syndecan-2 upregulation upon anchorage independency. Oncotarget 2017, 8, 61528–61537. [Google Scholar] [CrossRef] [Green Version]
- Carneiro, B.R.; Pernambuco Filho, P.C.; Mesquita, A.P.; da Silva, D.S.; Pinhal, M.A.; Nader, H.B.; Lopes, C.C. Acquisition of anoikis resistance up-regulates syndecan-4 expression in endothelial cells. PLoS ONE 2014, 9, e116001. [Google Scholar] [CrossRef] [Green Version]
- Shen, Y.; Chi, H.; Xu, K.; Li, Y.; Yin, X.; Chen, S.; Yang, Q.; He, M.; Zhu, G.; Li, X. A Novel Classification Model for Lower-Grade Glioma Patients Based on Pyroptosis-Related Genes. Brain Sci. 2022, 12, 700. [Google Scholar] [CrossRef]
- Lai, G.; Li, K.; Deng, J.; Liu, H.; Xie, B.; Zhong, X. Identification and Validation of a Gene Signature for Lower-Grade Gliomas Based on Pyroptosis-Related Genes to Predict Survival and Response to Immune Checkpoint Inhibitors. J. Healthc. Eng. 2022, 2022, 8704127. [Google Scholar] [CrossRef]
- Jiang, L.; Chen, S.; Zhao, D.; Yan, J.; Chen, J.; Yang, C.; Zheng, G. MNX1 reduces sensitivity to anoikis by activating TrkB in human glioma cells. Mol. Med. Rep. 2018, 18, 3271–3279. [Google Scholar] [CrossRef]
- Schiff, D.; Van den Bent, M.; Vogelbaum, M.A.; Wick, W.; Miller, C.R.; Taphoorn, M.; Pope, W.; Brown, P.D.; Platten, M.; Jalali, R.; et al. Recent developments and future directions in adult lower-grade gliomas: Society for Neuro-Oncology (SNO) and European Association of Neuro-Oncology (EANO) consensus. Neuro Oncol. 2019, 21, 837–853. [Google Scholar] [CrossRef] [PubMed]
- Bakir, B.; Chiarella, A.M.; Pitarresi, J.R.; Rustgi, A.K. EMT, MET, Plasticity, and Tumor Metastasis. Trends Cell Biol. 2020, 30, 764–776. [Google Scholar] [CrossRef] [PubMed]
- Taddei, M.L.; Giannoni, E.; Fiaschi, T.; Chiarugi, P. Anoikis: An emerging hallmark in health and diseases. J. Pathol. 2012, 226, 380–393. [Google Scholar] [CrossRef] [PubMed]
- Lah, T.T.; Novak, M.; Breznik, B. Brain malignancies: Glioblastoma and brain metastases. Semin. Cancer Biol. 2020, 60, 262–273. [Google Scholar] [CrossRef]
- Ohsawa, S.; Vaughen, J.; Igaki, T. Cell Extrusion: A Stress-Responsive Force for Good or Evil in Epithelial Homeostasis. Dev. Cell 2018, 44, 284–296. [Google Scholar] [CrossRef] [Green Version]
- Tiwari, N.; Gheldof, A.; Tatari, M.; Christofori, G. EMT as the ultimate survival mechanism of cancer cells. Semin. Cancer Biol. 2012, 22, 194–207. [Google Scholar] [CrossRef]
- Kesanakurti, D.; Chetty, C.; Rajasekhar Maddirela, D.; Gujrati, M.; Rao, J.S. Functional cooperativity by direct interaction between PAK4 and MMP-2 in the regulation of anoikis resistance, migration and invasion in glioma. Cell Death Dis. 2012, 3, e445. [Google Scholar] [CrossRef] [Green Version]
- Horiguchi, H.; Kadomatsu, T.; Yumoto, S.; Masuda, T.; Miyata, K.; Yamamura, S.; Sato, M.; Morinaga, J.; Ohtsuki, S.; Baba, H.; et al. Tumor cell-derived ANGPTL2 promotes beta-catenin-driven intestinal tumorigenesis. Oncogene 2022, 41, 4028–4041. [Google Scholar] [CrossRef]
- Takeshita, Y.; Motohara, T.; Kadomatsu, T.; Doi, T.; Obayashi, K.; Oike, Y.; Katabuchi, H.; Endo, M. Angiopoietin-like protein 2 decreases peritoneal metastasis of ovarian cancer cells by suppressing anoikis resistance. Biochem. Biophys. Res. Commun. 2021, 561, 26–32. [Google Scholar] [CrossRef]
- Ozfiliz, P.; Kizilboga, T.; Demir, S.; Alkurt, G.; Palavan-Unsal, N.; Arisan, E.D.; Dinler-Doganay, G. Bag-1 promotes cell survival through c-Myc-mediated ODC upregulation that is not preferred under apoptotic stimuli in MCF-7 cells. Cell Biochem. Funct. 2015, 33, 293–307. [Google Scholar] [CrossRef]
- Lin, T.; Cheng, H.; Liu, D.; Wen, L.; Kang, J.; Xu, L.; Shan, C.; Chen, Z.; Li, H.; Lai, M.; et al. A Novel Six Autophagy-Related Genes Signature Associated With Outcomes and Immune Microenvironment in Lower-Grade Glioma. Front. Genet. 2021, 12, 698284. [Google Scholar] [CrossRef] [PubMed]
- Ha, C.T.; Cheng, C.Y.; Zheng, M.Y.; Hsu, T.H.; Miao, C.C.; Lee, C.J.; Wang, H.D.; Pan, S.T.; Chou, Y.T. ID4 predicts poor prognosis and promotes BDNF-mediated oncogenesis of colorectal cancer. Carcinogenesis 2021, 42, 951–960. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, S.; Li, L.; Puliyappadamba, V.T.; Guo, G.; Hatanpaa, K.J.; Mickey, B.; Souza, R.F.; Vo, P.; Herz, J.; Chen, M.R.; et al. Constitutive and ligand-induced EGFR signalling triggers distinct and mutually exclusive downstream signalling networks. Nat. Commun. 2014, 5, 5811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tasian, S.K.; Loh, M.L.; Hunger, S.P. Philadelphia chromosome-like acute lymphoblastic leukemia. Blood 2017, 130, 2064–2072. [Google Scholar] [CrossRef] [Green Version]
- Sousa, B.; Pereira, J.; Marques, R.; Grilo, L.F.; Pereira, S.P.; Sardao, V.A.; Schmitt, F.; Oliveira, P.J.; Paredes, J. P-cadherin induces anoikis-resistance of matrix-detached breast cancer cells by promoting pentose phosphate pathway and decreasing oxidative stress. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165964. [Google Scholar] [CrossRef]
- Kim, Y.S.; Gupta Vallur, P.; Jones, V.M.; Worley, B.L.; Shimko, S.; Shin, D.H.; Crawford, L.C.; Chen, C.W.; Aird, K.M.; Abraham, T.; et al. Context-dependent activation of SIRT3 is necessary for anchorage-independent survival and metastasis of ovarian cancer cells. Oncogene 2020, 39, 1619–1633. [Google Scholar] [CrossRef]
- Kamarajugadda, S.; Cai, Q.; Chen, H.; Nayak, S.; Zhu, J.; He, M.; Jin, Y.; Zhang, Y.; Ai, L.; Martin, S.S.; et al. Manganese superoxide dismutase promotes anoikis resistance and tumor metastasis. Cell Death Dis. 2013, 4, e504. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.S.; Tang, P.W.; Welles, J.E.; Pan, W.; Javed, Z.; Elhaw, A.T.; Mythreye, K.; Kimball, S.R.; Hempel, N. HuR-dependent SOD2 protein synthesis is an early adaptation to anchorage-independence. Redox Biol. 2022, 53, 102329. [Google Scholar] [CrossRef]
- Ma, R.; Kang, X.; Zhang, G.; Fang, F.; Du, Y.; Lv, H. High expression of UBE2C is associated with the aggressive progression and poor outcome of malignant glioma. Oncol. Lett. 2016, 11, 2300–2304. [Google Scholar] [CrossRef] [Green Version]
- Guo, L.; Ding, Z.; Huang, N.; Huang, Z.; Zhang, N.; Xia, Z. Forkhead Box M1 positively regulates UBE2C and protects glioma cells from autophagic death. Cell Cycle 2017, 16, 1705–1718. [Google Scholar] [CrossRef]
- Chiocca, E.A.; Yu, J.S.; Lukas, R.V.; Solomon, I.H.; Ligon, K.L.; Nakashima, H.; Triggs, D.A.; Reardon, D.A.; Wen, P.; Stopa, B.M.; et al. Regulatable interleukin-12 gene therapy in patients with recurrent high-grade glioma: Results of a phase 1 trial. Sci. Transl. Med. 2019, 11, aaw5680. [Google Scholar] [CrossRef]
- Bangalore Yogananda, C.G.; Shah, B.R.; Vejdani-Jahromi, M.; Nalawade, S.S.; Murugesan, G.K.; Yu, F.F.; Pinho, M.C.; Wagner, B.C.; Mickey, B.; Patel, T.R.; et al. A novel fully automated MRI-based deep-learning method for classification of IDH mutation status in brain gliomas. Neuro Oncol. 2020, 22, 402–411. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.; Liu, Z.; Ye, W.; Liu, F. Immune Infiltration-Related Signature Predicts Risk Stratification and Immunotherapy Efficacy in Grade II and III Gliomas. Front Cell Dev. Biol. 2021, 9, 756005. [Google Scholar] [CrossRef] [PubMed]
- Chan, T.A.; Yarchoan, M.; Jaffee, E.; Swanton, C.; Quezada, S.A.; Stenzinger, A.; Peters, S. Development of tumor mutation burden as an immunotherapy biomarker: Utility for the oncology clinic. Ann. Oncol. 2019, 30, 44–56. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Qin, B.; Moyer, A.M.; Nowsheen, S.; Tu, X.; Dong, H.; Boughey, J.C.; Goetz, M.P.; Weinshilboum, R.; Lou, Z.; et al. Regulation of sister chromatid cohesion by nuclear PD-L1. Cell Res 2020, 30, 590–601. [Google Scholar] [CrossRef]
- Atreya, C.E.; Turnbaugh, P.J. Probing the tumor micro(b)environment. Science 2020, 368, 938–939. [Google Scholar] [CrossRef]
- Denk, D.; Greten, F.R. Inflammation: The incubator of the tumor microenvironment. Trends Cancer 2022, S2405-8033, 00154-6. [Google Scholar] [CrossRef]











Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, S.; Chi, H.; Ji, W.; He, Q.; Lai, G.; Peng, G.; Zhao, X.; Cheng, C. A Bioinformatics-Based Analysis of an Anoikis-Related Gene Signature Predicts the Prognosis of Patients with Low-Grade Gliomas. Brain Sci. 2022, 12, 1349. https://doi.org/10.3390/brainsci12101349
Zhao S, Chi H, Ji W, He Q, Lai G, Peng G, Zhao X, Cheng C. A Bioinformatics-Based Analysis of an Anoikis-Related Gene Signature Predicts the Prognosis of Patients with Low-Grade Gliomas. Brain Sciences. 2022; 12(10):1349. https://doi.org/10.3390/brainsci12101349
Chicago/Turabian StyleZhao, Songyun, Hao Chi, Wei Ji, Qisheng He, Guichuan Lai, Gaoge Peng, Xiaoyu Zhao, and Chao Cheng. 2022. "A Bioinformatics-Based Analysis of an Anoikis-Related Gene Signature Predicts the Prognosis of Patients with Low-Grade Gliomas" Brain Sciences 12, no. 10: 1349. https://doi.org/10.3390/brainsci12101349