
Examining Interactions of Uranyl(VI) Ions with Amino Acids in the Gas Phase


Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
3. Results and Discussion
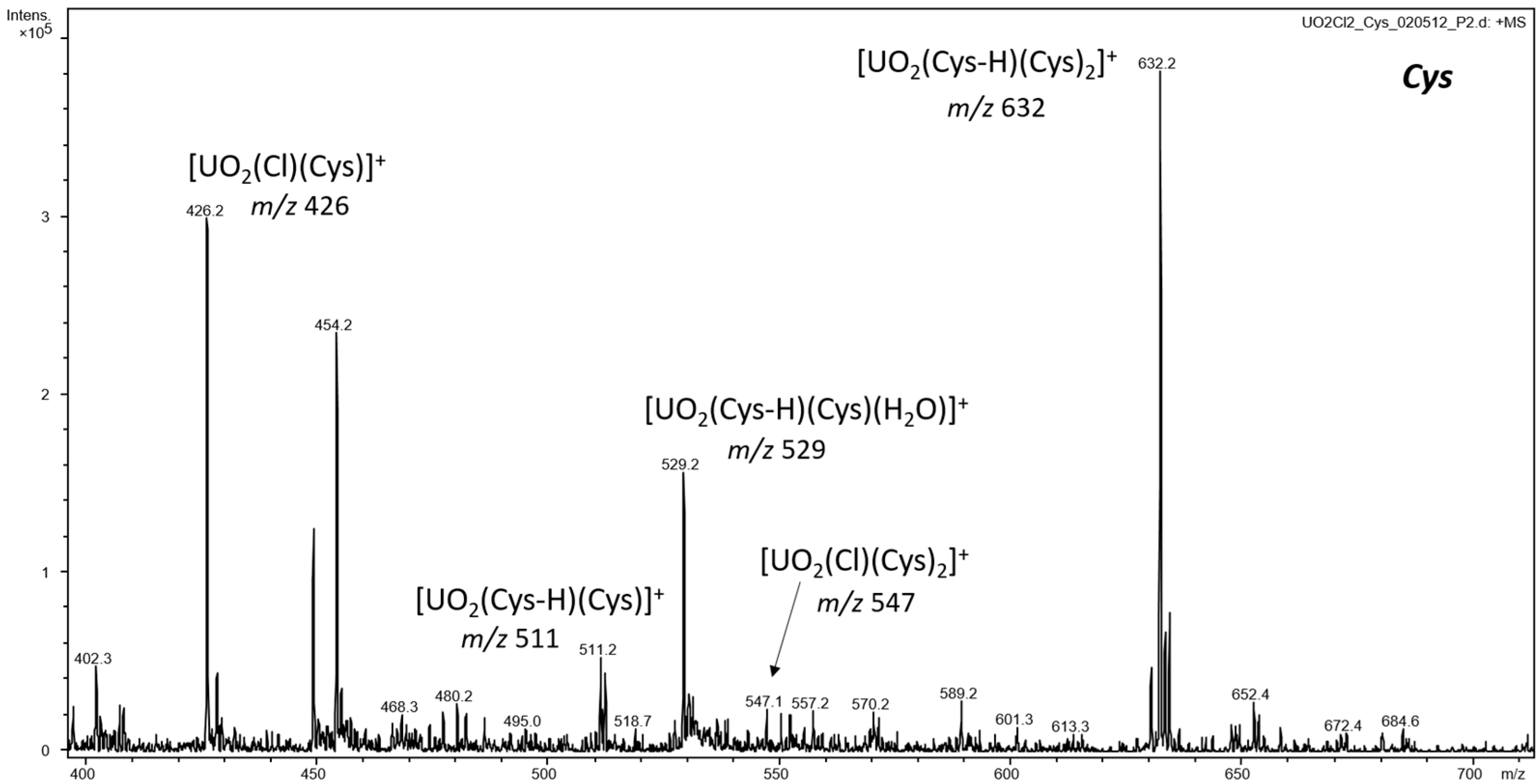
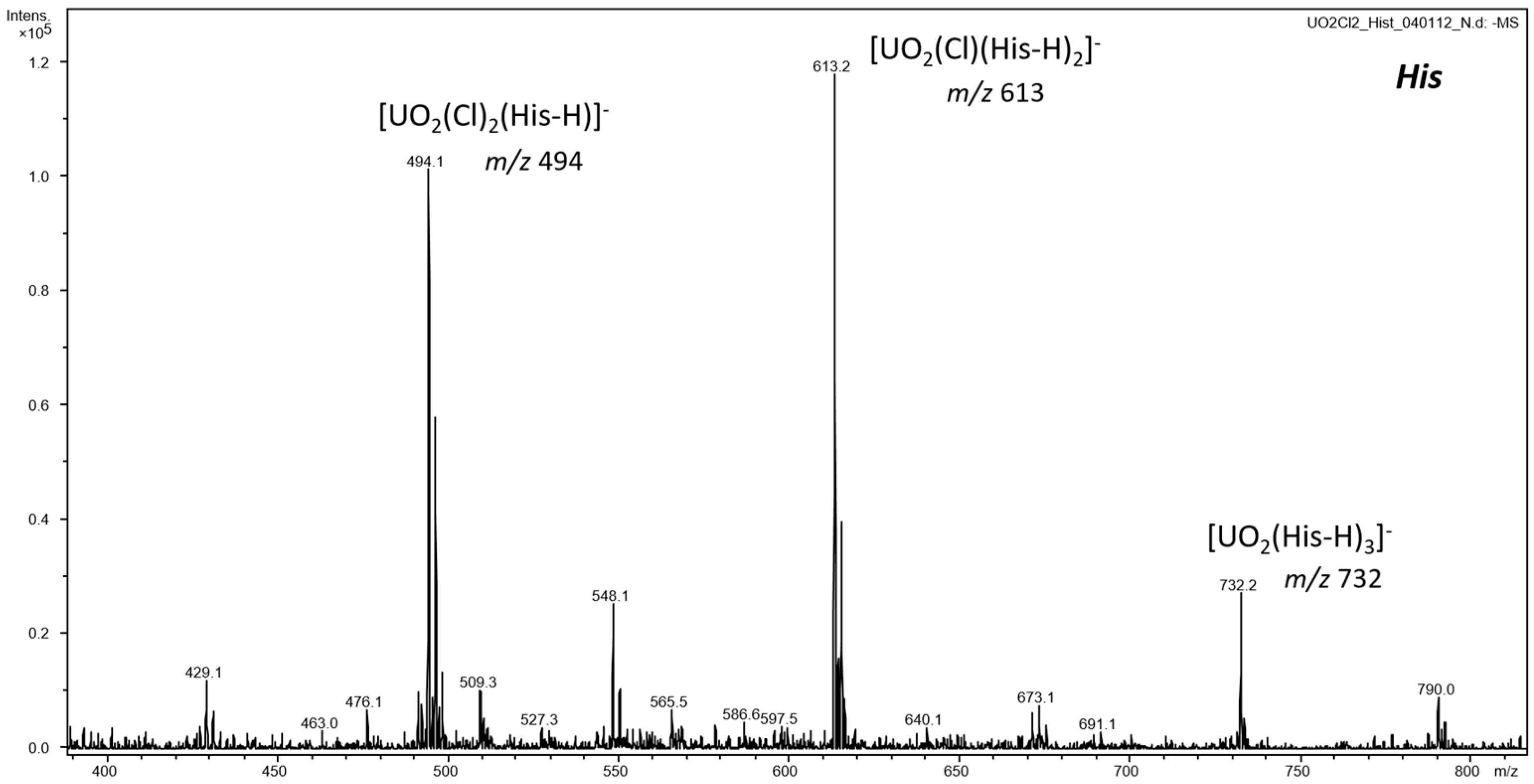
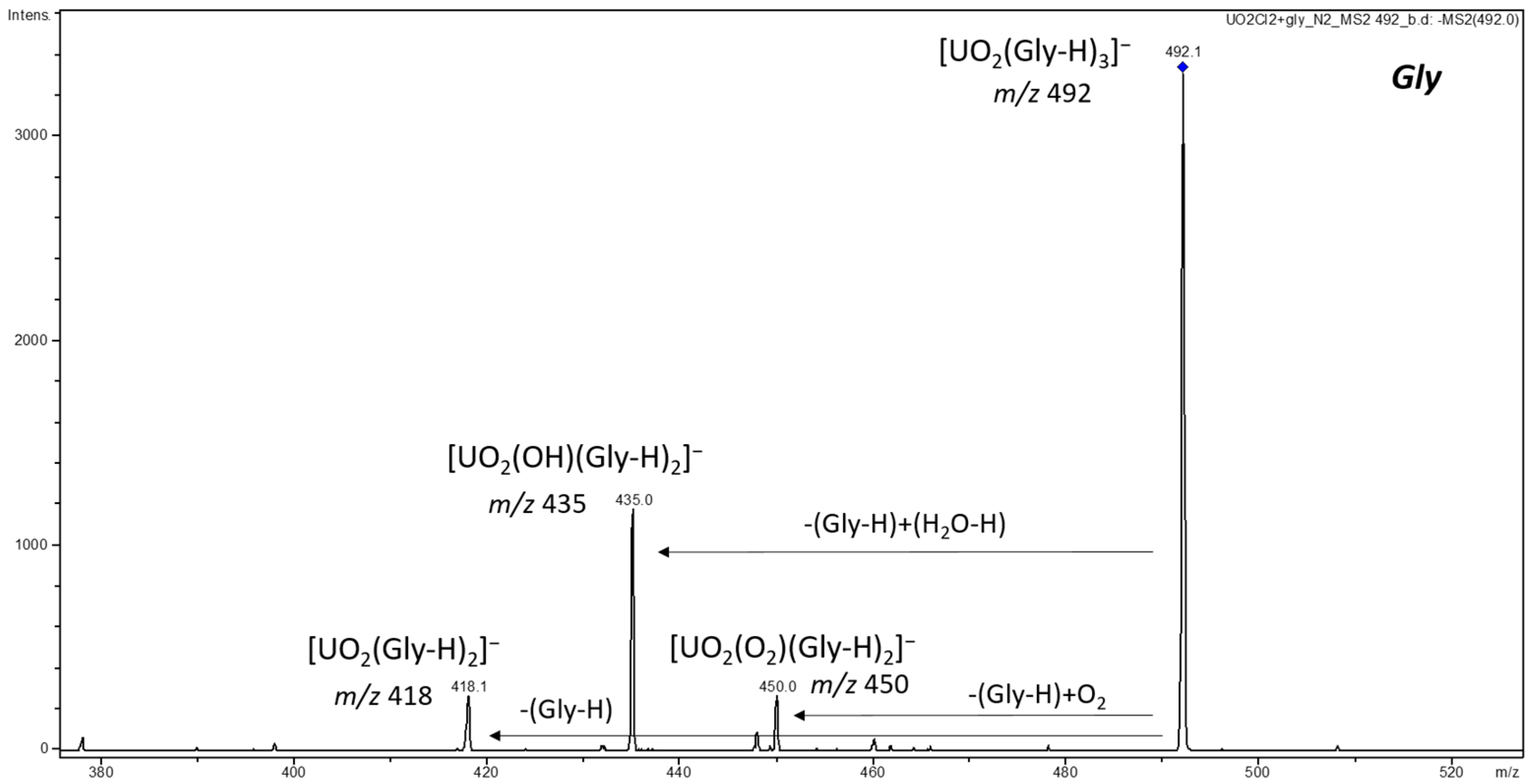
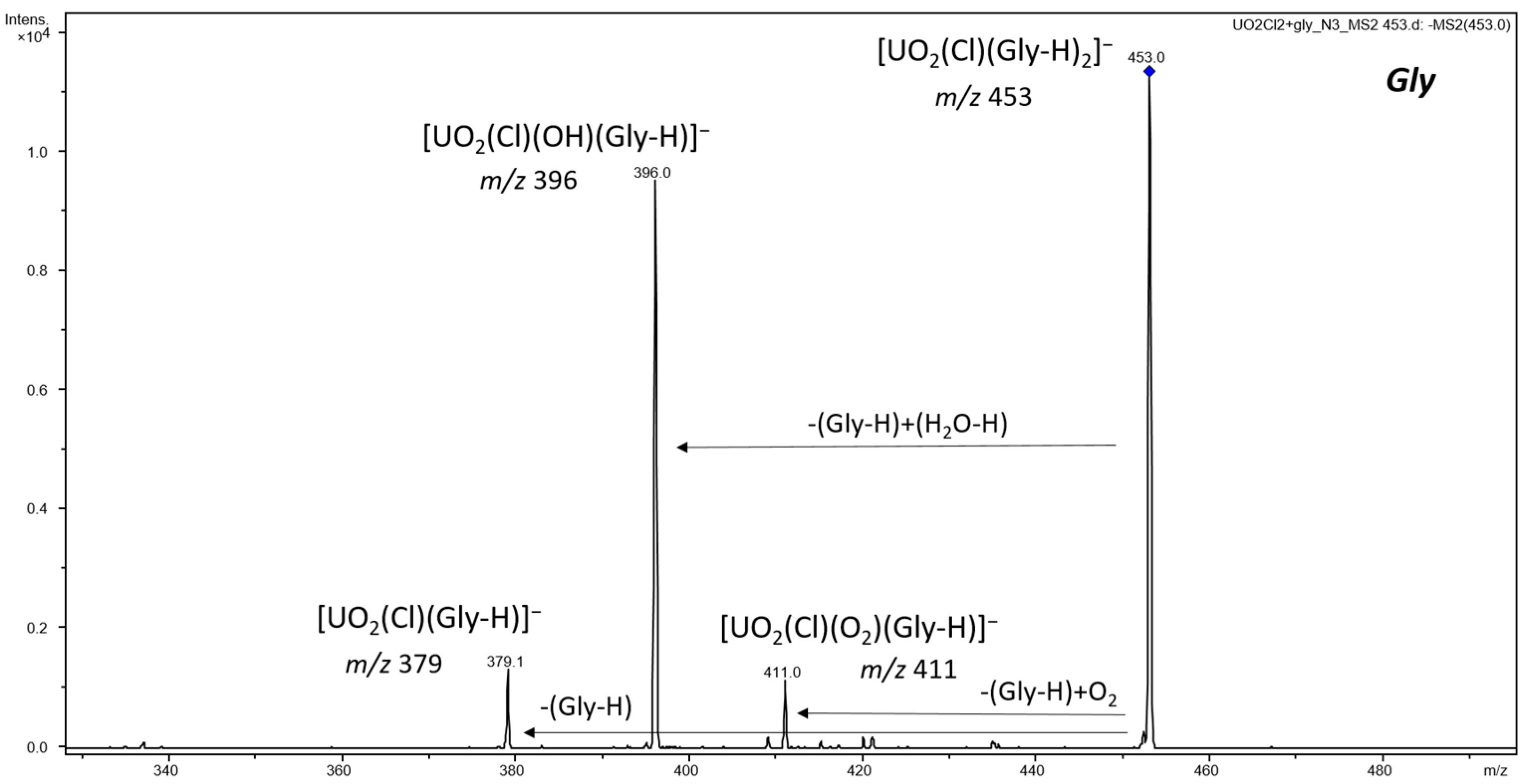
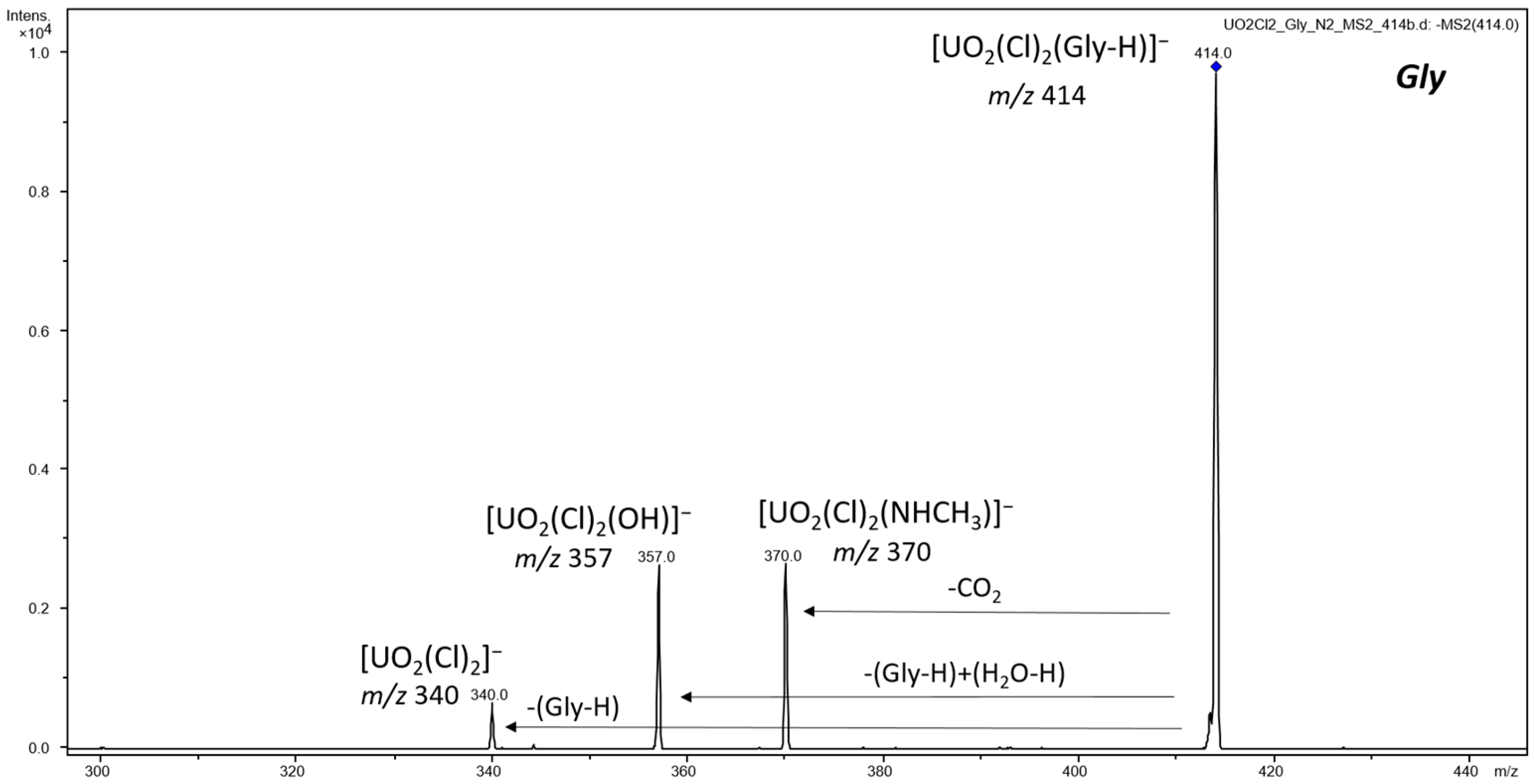
3.1. ESI-MS of Uranyl Complexes with Glycine, Aspartic Acid, Cysteine, and Histidine
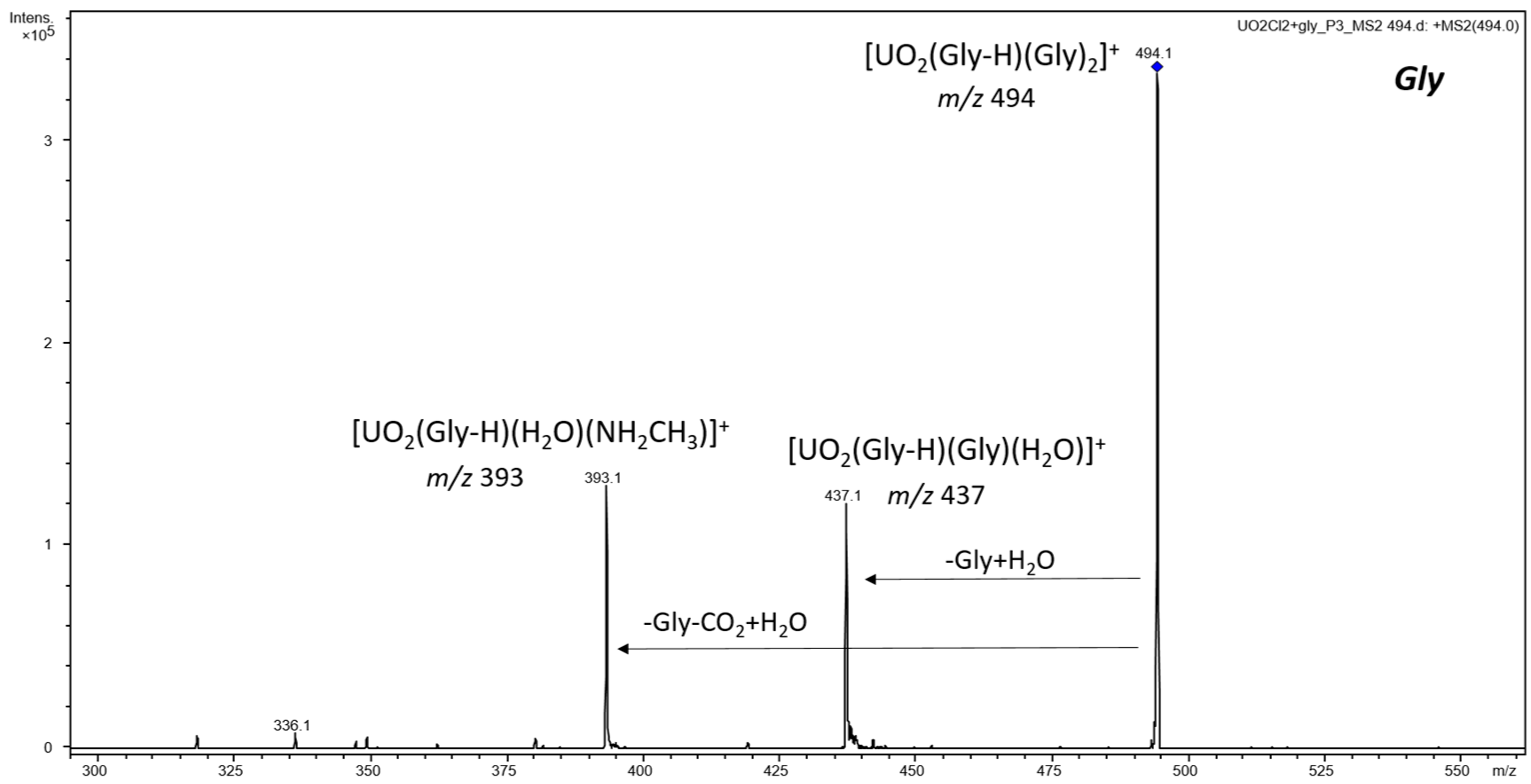
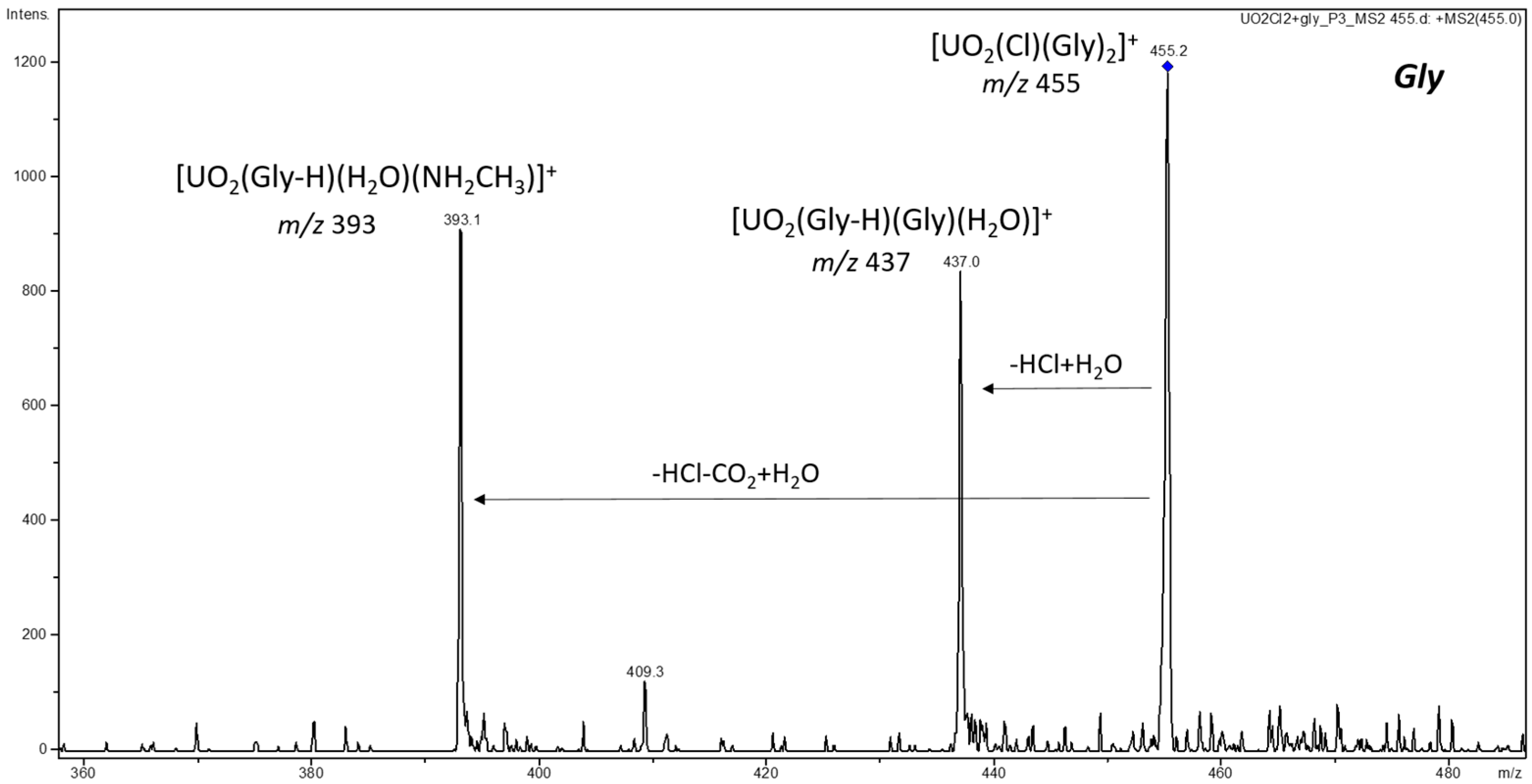
3.2. CID and Reactivity of Uranyl–Glycine, Uranyl–Aspartic Acid, Uranyl–Cysteine, and Uranyl–Histidine Complexes
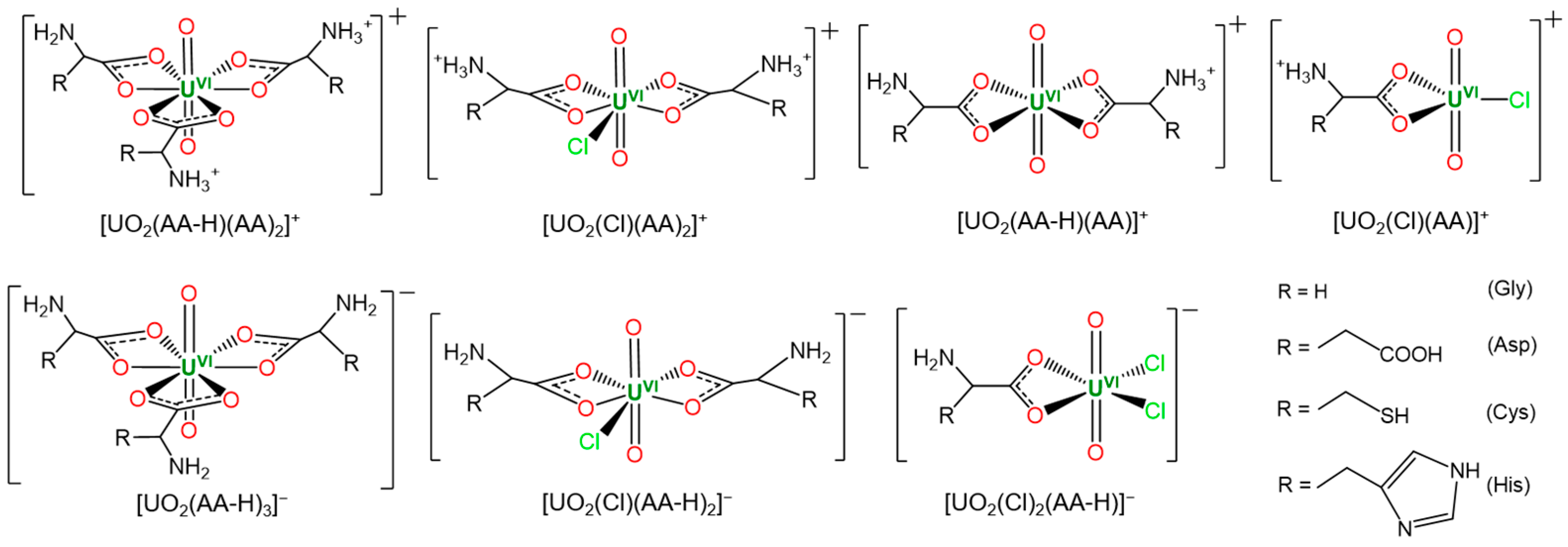
3.2.1. Cationic Uranyl–Amino Acid Complexes
3.2.2. Anionic Uranyl–Amino Acid Complexes
3.3. ESI-MS, Competitive CID, and Reactivity of Mixed Glycine, Aspartic Acid, Cysteine, and Histidine Complexes with Uranyl
3.3.1. Competitive CID and Reactivity of Cationic Uranyl Mixed Amino Acid Complexes
3.3.2. Competitive CID and Reactivity of Anionic Uranyl Mixed Amino Acid Complexes
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ma, M.; Wang, R.; Xu, L.; Xu, M.; Liu, S. Emerging health risks and underlying toxicological mechanisms of uranium contamination: Lessons from the past two decades. Environ. Int. 2020, 145, 106107. [Google Scholar] [CrossRef] [PubMed]
- Garai, A.; Delangle, P. Recent advances in uranyl binding in proteins thanks to biomimetic peptides. J. Inorg. Biochem. 2020, 203, 110936. [Google Scholar] [CrossRef]
- Lin, Y.-W. Uranyl Binding to Proteins and Structural-Functional Impacts. Biomolecules 2020, 10, 457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Creff, G.; Zurita, C.; Jeanson, A.; Carle, G.; Vidaud, C.; Auwer, C.D. What do we know about actinides-proteins interactions? Radiochim. Acta 2019, 107, 993–1009. [Google Scholar] [CrossRef]
- Carugo, O. Structural features of uranium-protein complexes. J. Inorg. Biochem. 2018, 189, 1–6. [Google Scholar] [CrossRef]
- Pardoux, R.; Sauge-Merle, S.; Bremond, N.; Beccia, M.R.; Lemaire, D.; Battesti, C.; Delangle, P.; Solari, P.L.; Guilbaud, P.; Berthomieu, C. Optimized Coordination of Uranyl in Engineered Calmodulin Site 1 Provides a Subnanomolar Affinity for Uranyl and a Strong Uranyl versus Calcium Selectivity. Inorg. Chem. 2022, 61, 20480–20492. [Google Scholar] [CrossRef] [PubMed]
- Starck, M.; Laporte, F.A.; Oros, S.; Sisommay, N.; Gathu, V.; Solari, P.L.; Creff, G.; Roques, J.; Auwer, C.D.; Lebrun, C.; et al. Cyclic Phosphopeptides to Rationalize the Role of Phosphoamino Acids in Uranyl Binding to Biological Targets. Chem.—A Eur. J. 2017, 23, 5281–5290. [Google Scholar] [CrossRef]
- Qi, L.; Basset, C.; Averseng, O.; Quéméneur, E.; Hagège, A.; Vidaud, C. Characterization of UO22+binding to osteopontin, a highly phosphorylated protein: Insights into potential mechanisms of uranyl accumulation in bones. Metallomics 2014, 6, 166–176. [Google Scholar] [CrossRef]
- Deblonde, G.J.-P.; Sturzbecher-Hoehne, M.; Mason, A.B.; Abergel, R.J. Receptor recognition of transferrin bound to lanthanides and actinides: A discriminating step in cellular acquisition of f-block metals. Metallomics 2013, 5, 619–626. [Google Scholar] [CrossRef] [Green Version]
- Götzke, L.; Schaper, G.; März, J.; Kaden, P.; Huittinen, N.; Stumpf, T.; Kammerlander, K.K.; Brunner, E.; Hahn, P.; Mehnert, A.; et al. Coordination chemistry of f-block metal ions with ligands bearing bio-relevant functional groups. Coord. Chem. Rev. 2019, 386, 267–309. [Google Scholar] [CrossRef]
- Berto, S.; Crea, F.; Daniele, P.G.; Gianguzza, A.; Pettignano, A.; Sammartano, S. Advances in the investigation of dioxouranium(VI) complexes of interest for natural fluids. Coord. Chem. Rev. 2012, 256, 63–81. [Google Scholar] [CrossRef]
- Bresson, C.; Ansoborlo, E.; Vidaud, C. Radionuclide speciation: A key point in the field of nuclear toxicology studies. J. Anal. At. Spectrom. 2011, 26, 593–601. [Google Scholar] [CrossRef]
- Van Horn, J.D.; Huang, H. Uranium(VI) bio-coordination chemistry from biochemical, solution and protein structural data. Coord. Chem. Rev. 2006, 250, 765–775. [Google Scholar] [CrossRef]
- Fattal, E.; Tsapis, N.; Phan, G. Novel drug delivery systems for actinides (uranium and plutonium) decontamination agents. Adv. Drug Deliv. Rev. 2015, 90, 40–54. [Google Scholar] [CrossRef]
- Sturzbecher-Hoehne, M.; Deblonde, G.J.-P.; Abergel, R.J. Solution thermodynamic evaluation of hydroxypyridinonate chelators 3,4,3-LI(1,2-HOPO) and 5-LIO(Me-3,2-HOPO) for UO2(VI) and Th(IV) decorporation. Radiochim. Acta 2013, 101, 359–366. [Google Scholar] [CrossRef]
- Durbin, P.W.; Lauriston, S. Taylor lecture: The quest for therapeutic actinide chelators. Health Phys. 2008, 95, 465–492. [Google Scholar] [CrossRef] [PubMed]
- Gorden, A.E.V.; Xu, J.; Raymond, K.N.; Durbin, P. Rational Design of Sequestering Agents for Plutonium and Other Actinides. Chem. Rev. 2003, 103, 4207–4282. [Google Scholar] [CrossRef]
- Zhang, L.; Jia, M.; Wang, X.; Gao, L.; Zhang, B.; Wang, L.; Kong, J.; Li, L. A novel fluorescence sensor for uranyl ion detection based on a dansyl-modified peptide. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2023, 292, 122403. [Google Scholar] [CrossRef]
- Wu, X.; Huang, Q.; Mao, Y.; Wang, X.; Wang, Y.; Hu, Q.; Wang, H.; Wang, X. Sensors for determination of uranium: A review. TrAC Trends Anal. Chem. 2019, 118, 89–111. [Google Scholar] [CrossRef]
- Safi, S.; Jeanson, A.; Roques, J.; Solari, P.L.; Charnay-Pouget, F.; Auwer, C.D.; Creff, G.; Aitken, D.J.; Simoni, E. Thermodynamic and Structural Investigation of Synthetic Actinide–Peptide Scaffolds. Inorg. Chem. 2016, 55, 877–886. [Google Scholar] [CrossRef]
- Zhou, L.; Bosscher, M.; Zhang, C.; Özçubukçu, S.; Zhang, L.; Zhang, W.; Li, C.J.; Liu, J.; Jensen, M.P.; Lai, L.; et al. A protein engineered to bind uranyl selectively and with femtomolar affinity. Nat. Chem. 2014, 6, 236–241. [Google Scholar] [CrossRef] [PubMed]
- Odoh, S.O.; Bondarevsky, G.D.; Karpus, J.; Cui, Q.; He, C.; Spezia, R.; Gagliardi, L. UO22+ Uptake by Proteins: Understanding the Binding Features of the Super Uranyl Binding Protein and Design of a Protein with Higher Affinity. J. Am. Chem. Soc. 2014, 136, 17484–17494. [Google Scholar] [CrossRef]
- Lebrun, C.; Starck, M.; Gathu, V.; Chenavier, Y.; Delangle, P. Engineering Short Peptide Sequences for Uranyl Binding. Chem.—A Eur. J. 2014, 20, 16566–16573. [Google Scholar] [CrossRef] [PubMed]
- Carlton, D.D.; Schug, K.A. A review on the interrogation of peptide–metal interactions using electrospray ionization-mass spectrometry. Anal. Chim. Acta 2011, 686, 19–39. [Google Scholar] [CrossRef]
- Rodgers, M.T.; Armentrout, P.B. A Thermodynamic “Vocabulary” for Metal Ion Interactions in Biological Systems. Acc. Chem. Res. 2004, 37, 989–998. [Google Scholar] [CrossRef]
- Rodgers, M.T.; Armentrout, P.B. Cationic Noncovalent Interactions: Energetics and Periodic Trends. Chem. Rev. 2016, 116, 5642–5687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polfer, N.C.; Oomens, J. Vibrational spectroscopy of bare and solvated ionic complexes of biological relevance. Mass Spectrom. Rev. 2009, 28, 468–494. [Google Scholar] [CrossRef]
- Dunbar, R.C. Spectroscopy of Metal-Ion Complexes with Peptide-Related Ligands. In Gas-Phase IR Spectroscopy and Structure of Biological Molecules; Rijs, A., Oomens, J., Eds.; Topics in Current Chemistry; Springer: Cham, Switzerland, 2015; Volume 364, pp. 183–223. [Google Scholar] [CrossRef]
- Jašíková, L.; Roithová, J. Infrared Multiphoton Dissociation Spectroscopy with Free-Electron Lasers: On the Road from Small Molecules to Biomolecules. Chem.—A Eur. J. 2018, 24, 3374–3390. [Google Scholar] [CrossRef] [Green Version]
- Jones, R.M.; Nilsson, T.; Walker, S.; Armentrout, P.B. Potassium Binding Interactions with Aliphatic Amino Acids: Thermodynamic and Entropic Effects Analyzed via a Guided Ion Beam and Computational Study. J. Am. Soc. Mass Spectrom. 2022, 33, 1427–1442. [Google Scholar] [CrossRef]
- IAEA Live Chart of Nuclides. Available online: https://www-nds.iaea.org/relnsd/vcharthtml/VChartHTML.html (accessed on 26 February 2023).
- Jones, C.M.; Bernier, M.; Carson, E.; Colyer, K.E.; Metz, R.; Pawlow, A.; Wischow, E.D.; Webb, I.; Andriole, E.J.; Poutsma, J.C. Gas-phase acidities of the 20 protein amino acids. Int. J. Mass Spectrom. 2007, 267, 54–62. [Google Scholar] [CrossRef]
- Smith, J.R.; Kim, J.B.; Lineberger, W.C. High-resolution threshold photodetachment spectroscopy of OH−. Phys. Rev. A 1997, 55, 2036–2043. [Google Scholar] [CrossRef] [Green Version]
- Di Marco, V.B.; Bombi, G.G. Electrospray mass spectrometry (ESI-MS) in the study of metal–ligand solution equilibria. Mass Spectrom. Rev. 2006, 25, 347–379. [Google Scholar] [CrossRef] [PubMed]
- Tsierkezos, N.G.; Roithová, J.; Schröder, D.; Ončák, M.; Slavíček, P. Can Electrospray Mass Spectrometry Quantitatively Probe Speciation? Hydrolysis of Uranyl Nitrate Studied by Gas-Phase Methods. Inorg. Chem. 2009, 48, 6287–6296. [Google Scholar] [CrossRef]
- Keith-Roach, M.J. A review of recent trends in electrospray ionisation–mass spectrometry for the analysis of metal–organic ligand complexes. Anal. Chim. Acta 2010, 678, 140–148. [Google Scholar] [CrossRef] [PubMed]
- Indelicato, S.; Bongiorno, D.; Ceraulo, L. Recent Approaches for Chemical Speciation and Analysis by Electrospray Ionization (ESI) Mass Spectrometry. Front. Chem. 2021, 8, 625945. [Google Scholar] [CrossRef] [PubMed]
- Haynes, W.M.; Lide, D.R.; Bruno, T.J. (Eds.) CRC Handbook of Chemistry and Physics, 97th ed.; CRC Press: Boca Raton, FL, USA, 2017. [Google Scholar]
- Groenewold, G.S.; de Jong, W.A.; Oomens, J.; Van Stipdonk, M.J. Variable denticity in carboxylate binding to the uranyl coordination complexes. J. Am. Soc. Mass Spectrom. 2010, 21, 719–727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dookeran, N.N.; Yalcin, T.; Harrison, A.G. Fragmentation Reactions of Protonated α-Amino Acids. J. Mass Spectrom. 1996, 31, 500–508. [Google Scholar] [CrossRef]
- Rogalewicz, F.; Hoppilliard, Y.; Ohanessian, G. Fragmentation mechanisms of α-amino acids protonated under electrospray ionization: A collisional activation and ab initio theoretical study. Int. J. Mass Spectrom. 2000, 195–196, 565–590. [Google Scholar] [CrossRef]
- Zhang, P.; Chan, W.; Ang, I.L.; Wei, R.; Lam, M.M.T.; Lei, K.M.K.; Poon, T.C.W. Revisiting Fragmentation Reactions of Protonated α-Amino Acids by High-Resolution Electrospray Ionization Tandem Mass Spectrometry with Collision-Induced Dissociation. Sci. Rep. 2019, 9, 6453. [Google Scholar] [CrossRef] [Green Version]
- Armentrout, P.B. Energetics and mechanisms for decomposition of cationized amino acids and peptides explored using guided ion beam tandem mass spectrometry. Mass Spectrom. Rev. 2021, 1–26. [Google Scholar] [CrossRef]
- O’Hair, R.A.J.; Rijs, N.J. Gas Phase Studies of the Pesci Decarboxylation Reaction: Synthesis, Structure, and Unimolecular and Bimolecular Reactivity of Organometallic Ions. Acc. Chem. Res. 2015, 48, 329–340. [Google Scholar] [CrossRef] [PubMed]
- Dau, P.D.; Rios, D.; Gong, Y.; Michelini, M.C.; Marçalo, J.; Shuh, D.K.; Mogannam, M.; Van Stipdonk, M.J.; Corcovilos, T.A.; Martens, J.K.; et al. Synthesis and Hydrolysis of Uranyl, Neptunyl, and Plutonyl Gas-Phase Complexes Exhibiting Discrete Actinide–Carbon Bonds. Organometallics 2016, 35, 1228–1240. [Google Scholar] [CrossRef] [Green Version]
- van Stipdonk, M.J.; Tatosian, I.J.; Iacovino, A.C.; Bubas, A.R.; Metzler, L.J.; Sherman, M.C.; Somogyi, A. Gas-Phase Deconstruction of UO22+: Mass Spectrometry Evidence for Generation of [OUVICH]+ by Collision-Induced Dissociation of [UVIO2(C≡CH)]+. J. Am. Soc. Mass Spectrom. 2019, 30, 796–805. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Z.; Chen, X.; Gong, Y. Mass spectrometric and theoretical study on the formation of uranyl hydride from uranyl carboxylate. Phys. Chem. Chem. Phys. 2021, 23, 20073–20079. [Google Scholar] [CrossRef] [PubMed]
- Lucena, A.F.; Carretas, J.M.; Marçalo, J.; Michelini, M.C.; Gong, Y.; Gibson, J.K. Gas-Phase Reactions of Molecular Oxygen with Uranyl(V) Anionic Complexes—Synthesis and Characterization of New Superoxides of Uranyl(VI). J. Phys. Chem. A 2015, 119, 3628–3635. [Google Scholar] [CrossRef]
- Groenewold, G.S.; Cossel, K.C.; Gresham, G.L.; Gianotto, A.K.; Appelhans, A.D.; Olson, J.E.; Van Stipdonk, M.J.; Chien, W. Binding of Molecular O2 to Di- and Triligated [UO2]+. J. Am. Chem. Soc. 2006, 128, 3075–3084. [Google Scholar] [CrossRef]
- Bryantsev, V.S.; de Jong, W.A.; Cossel, K.C.; Diallo, M.S.; Goddard, I.W.A.; Groenewold, G.S.; Chien, W.; Van Stipdonk, M.J. Two-Electron Three-Centered Bond in Side-On (η2) Uranyl(V) Superoxo Complexes. J. Phys. Chem. A 2008, 112, 5777–5780. [Google Scholar] [CrossRef]
- Leavitt, C.M.; Bryantsev, V.S.; de Jong, W.A.; Diallo, M.S.; Iii, W.A.G.; Groenewold, G.S.; Van Stipdonk, M.J. Addition of H2O and O2 to Acetone and Dimethylsulfoxide Ligated Uranyl(V) Dioxocations. J. Phys. Chem. A 2009, 113, 2350–2358. [Google Scholar] [CrossRef]
- Rios, D.; Michelini, M.C.; Lucena, A.F.; Marçalo, J.; Bray, T.H.; Gibson, J.K. Gas-Phase Uranyl, Neptunyl, and Plutonyl: Hydration and Oxidation Studied by Experiment and Theory. Inorg. Chem. 2012, 51, 6603–6614. [Google Scholar] [CrossRef]
- Hunter, E.P.L.; Lias, S.G. Evaluated Gas Phase Basicities and Proton Affinities of Molecules: An Update. J. Phys. Chem. Ref. Data 1998, 27, 413–656. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Precursor Complex | Neutral Losses (%) | |||
|---|---|---|---|---|
| Gly | Asp | Cys | His | |
| [UO2(Cl)(AA)2]+ | -HCl (50) -HCl-CO2 (50) | -HCl (100) | -HCl (50) -HCl-NH3 (50) | -HCl (100) |
| [UO2(AA-H)(AA)2]+ | -AA (50) -AA-CO2 (50) | -CO2 (10) -AA (75) -AA-CO2 (15) | -NH3 (55) -AA (25) -AA-NH3 (20) | -AA (100) |
| Precursor Complex | Neutral Losses (%) | |||
|---|---|---|---|---|
| Gly | Asp | Cys | His | |
| [UO2(Cl)2(AA-H)]− | -(AA-H) (55) -CO2 (45) | -HCl (100) | -HCl (100) | -HCl (100) |
| [UO2(Cl)(AA-H)2]− | -(AA-H) (100) | -HCl (100) | -HCl (100) | -AA (25) -HCl (75) |
| [UO2(AA-H)3]− | -(AA-H) (100) | -AA (100) | -AA (100) | -AA (100) |
| Precursor Complex | Neutral Losses (%) | |||||
|---|---|---|---|---|---|---|
| AA/AA′ | ||||||
| Asp/Gly | Cys/Gly | His/Gly | Asp/Cys | Asp/His | His/Cys | |
| [UO2(AA-H)(AA′)2]+ | -AA′ (100) | -NH3 (10) -AA′ (50) -AA′-NH3 (25) -AA′-NH3-CO2 (15) | -AA′ (55) -AA′-CO2 (45) | -AA′ (100) | -AA (100) | -AA′ (100) |
| [UO2(AA-H)(AA)(AA′)]+ | -AA′ (85) -AA (15) | -NH3 (30) -AA (30) -AA′ (20) -AA′-NH3 (20) | -AA′ (100) | -AA′ (100) | -AA (100) | -AA′ (100) |
| Precursor Complex | Neutral Losses (%) | |||||
|---|---|---|---|---|---|---|
| AA/AA′ | ||||||
| Asp/Gly | Cys/Gly | His/Gly | Asp/Cys | Asp/His | His/Cys | |
| [UO2(Cl)(AA-H)(AA′-H)]− | -HCl (90) -AA′ (10) | -HCl (45) -AA′ (55) | -HCl (50) -AA′ (50) | -HCl (100) | -HCl (100) | -HCl (100) |
| [UO2(AA-H)2(AA′-H)]− | -AA′ (100) | -AA′ (100) | -AA (35) -AA′ (65) | -AA (10) -AA′ (90) | -AA (45) -AA′ (55) | -AA (85) -AA′ (15) |
| [UO2(AA-H)(AA′-H)2]− | -AA′ (100) | -AA′ (100) | -AA′ (100) | -AA′ (100) | -AA′ (100) | -AA (15) -AA′ (85) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lucena, A.F.; Maria, L.; Gibson, J.K.; Marçalo, J. Examining Interactions of Uranyl(VI) Ions with Amino Acids in the Gas Phase. Appl. Sci. 2023, 13, 3834. https://doi.org/10.3390/app13063834
Lucena AF, Maria L, Gibson JK, Marçalo J. Examining Interactions of Uranyl(VI) Ions with Amino Acids in the Gas Phase. Applied Sciences. 2023; 13(6):3834. https://doi.org/10.3390/app13063834
Chicago/Turabian StyleLucena, Ana F., Leonor Maria, John K. Gibson, and Joaquim Marçalo. 2023. "Examining Interactions of Uranyl(VI) Ions with Amino Acids in the Gas Phase" Applied Sciences 13, no. 6: 3834. https://doi.org/10.3390/app13063834