
The Interaction of Methyl Formate with Proton-Bound Solvent Clusters in the Gas Phase and the Unimolecular Chemistry of the Reaction Products

Abstract
:1. Introduction
2. Materials and Methods
2.1. Tandem Mass Spectrometry
2.2. Computational Methods
3. Results
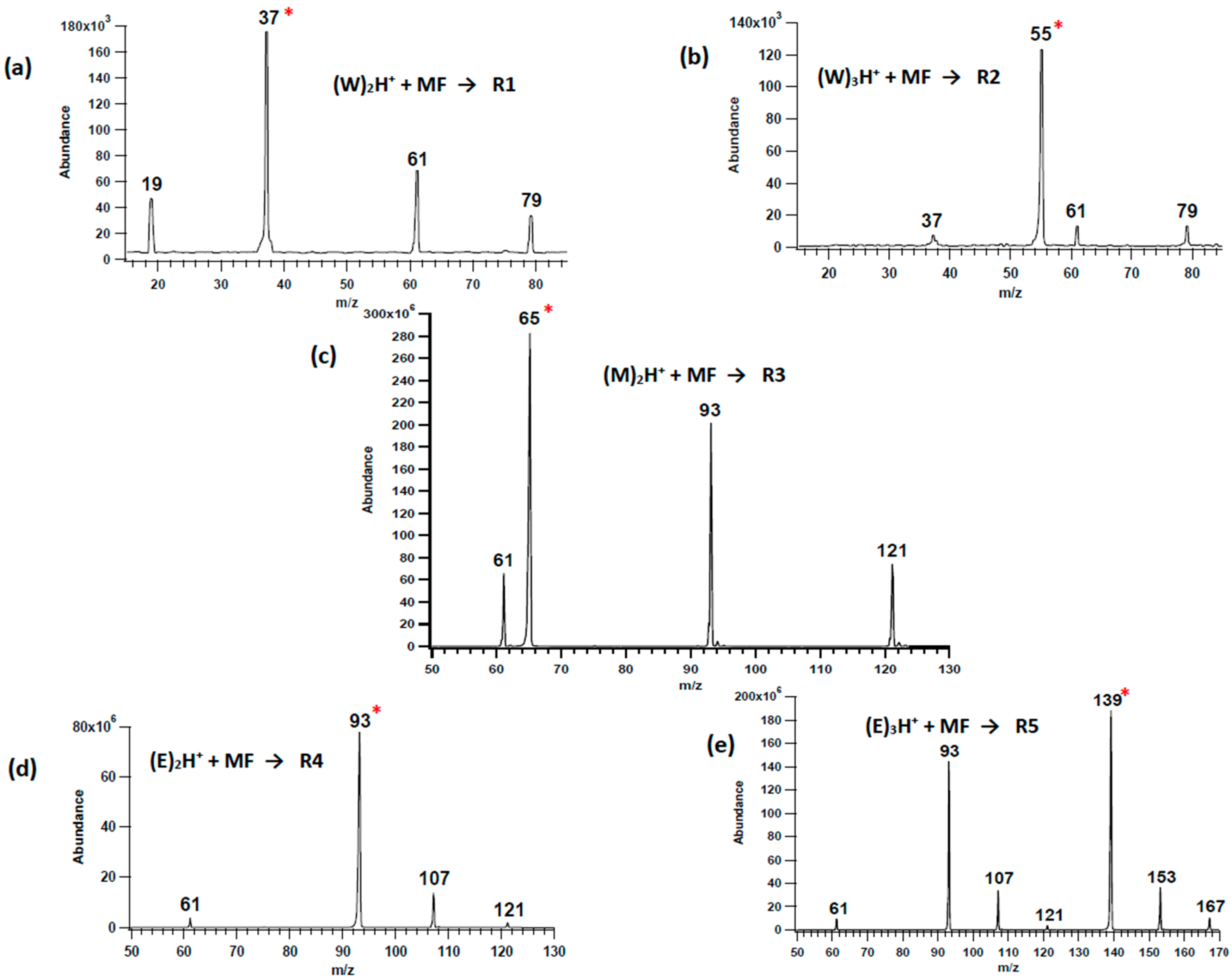
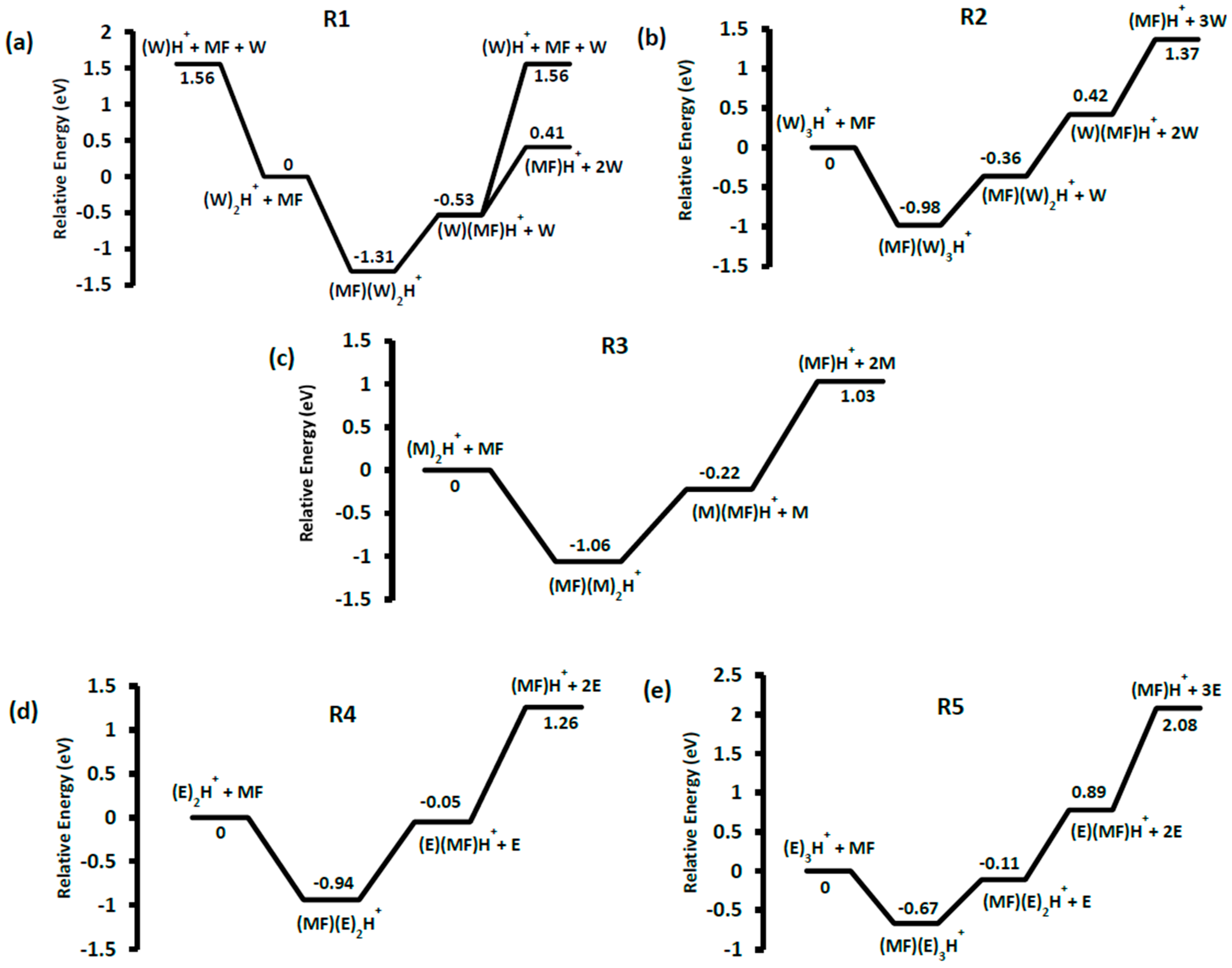
3.1. Solvent Cluster Ion/Methyl Formate Reactions
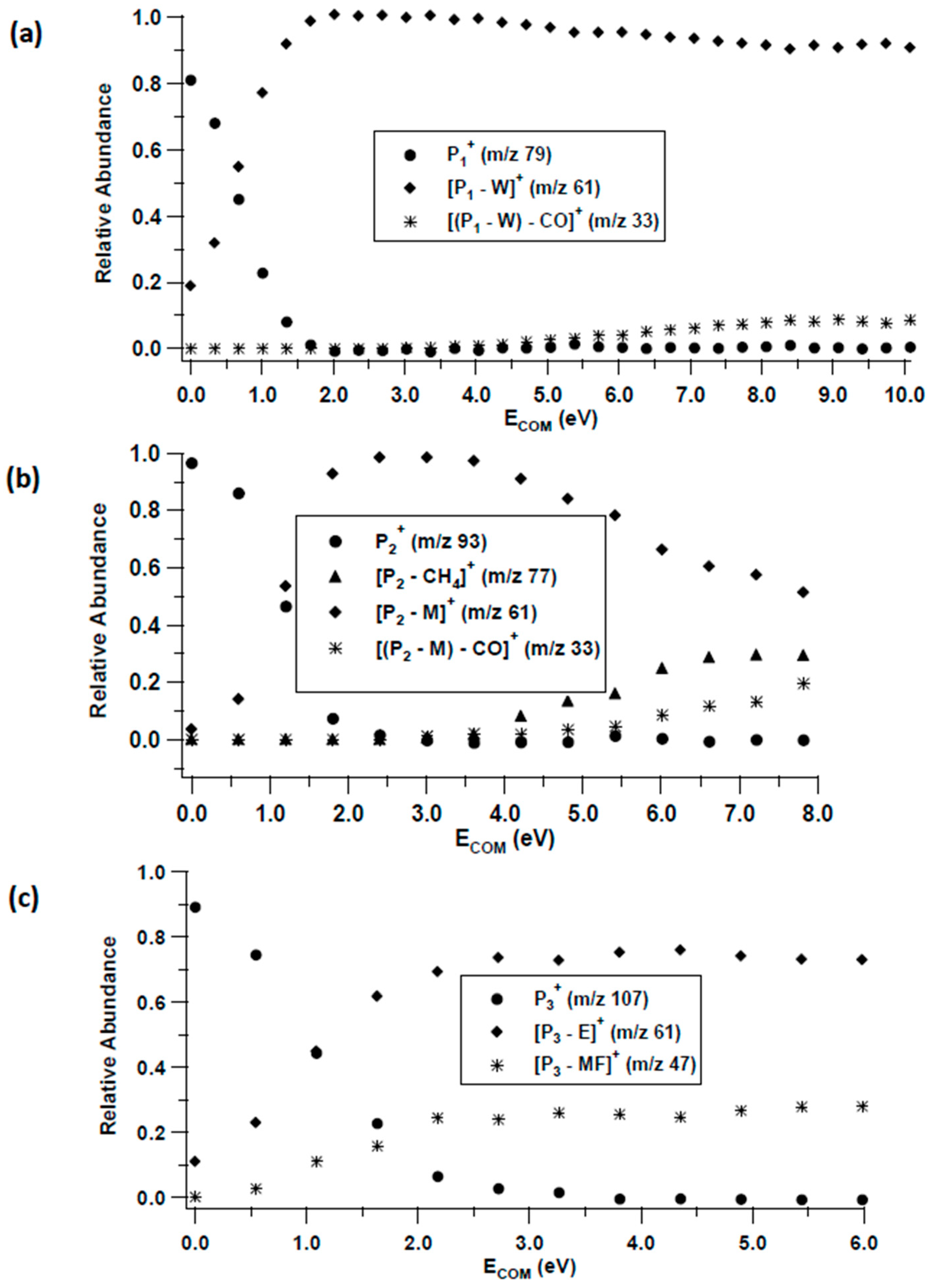
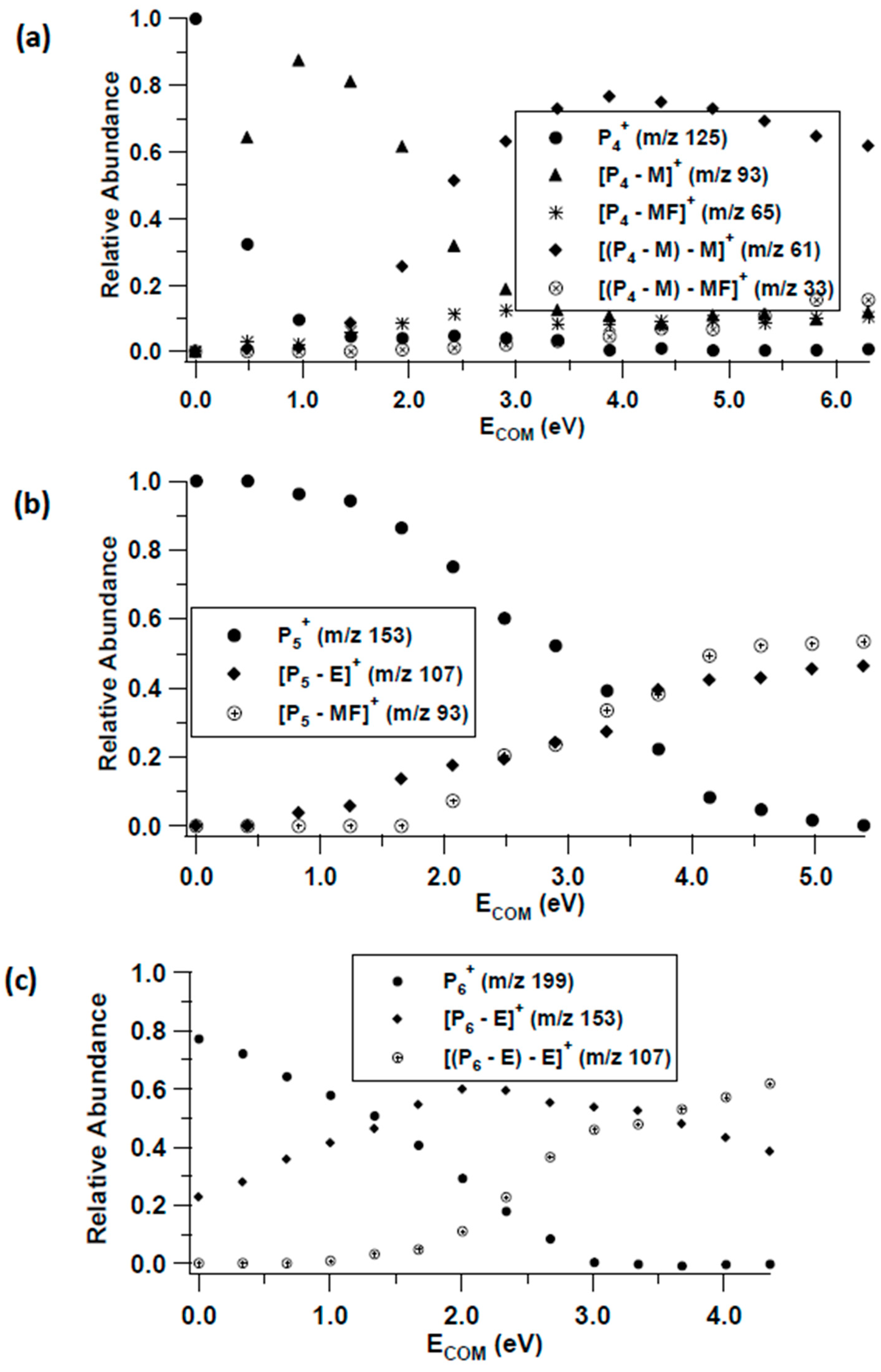
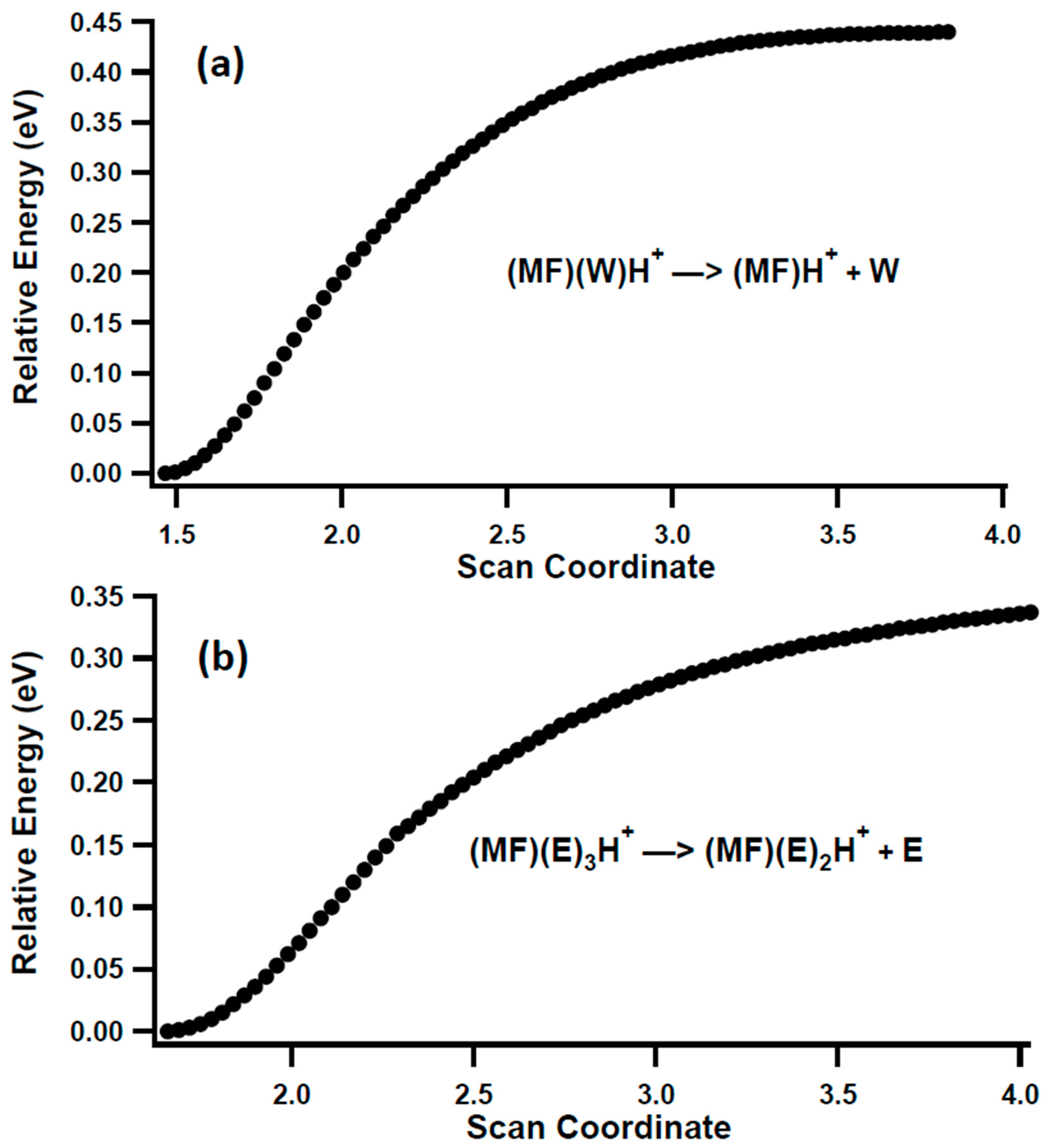
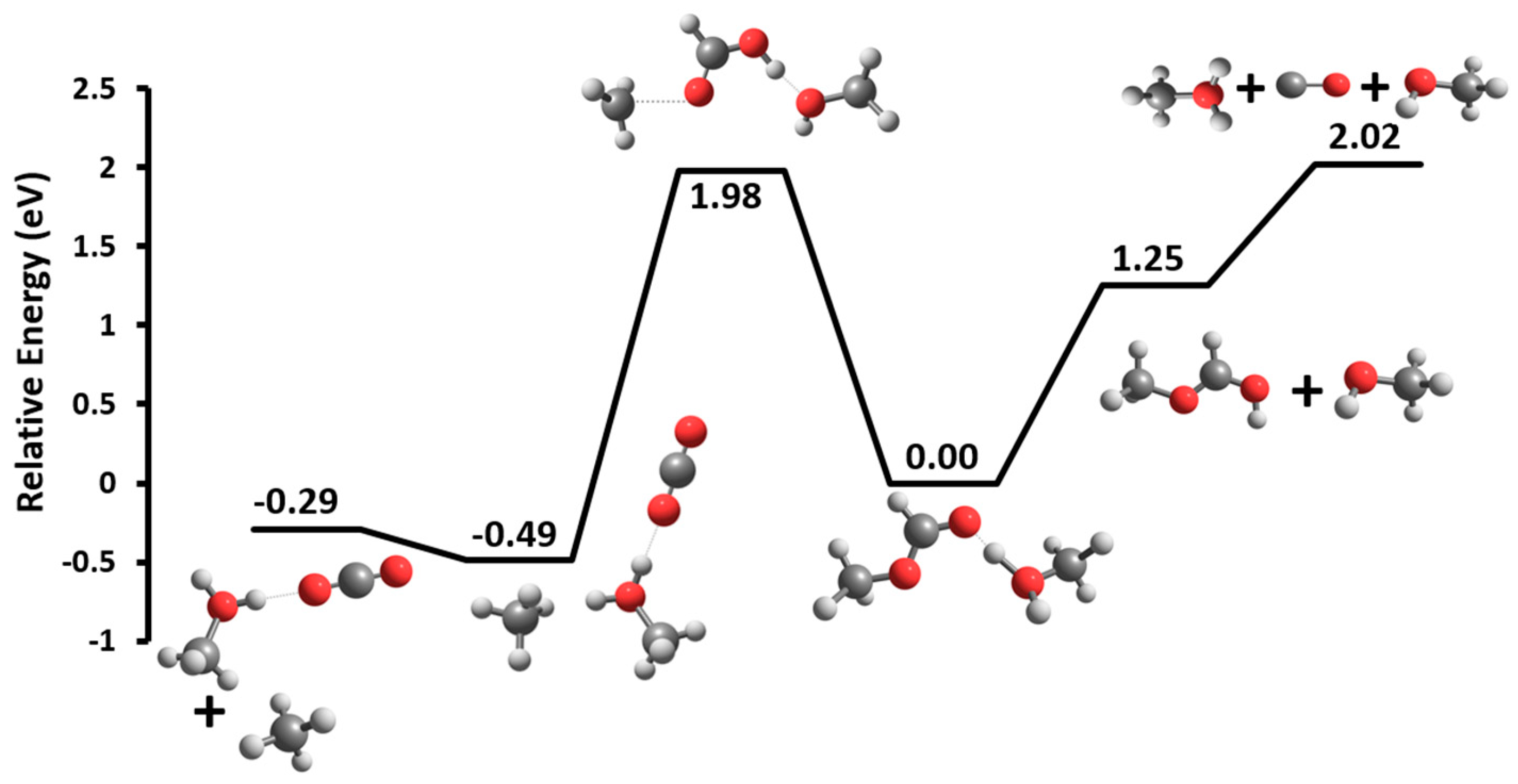
3.2. Unimolecular Reactions of Proton-Bound Solvent-MF Clusters
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rivett, A.C.; Martin, D.; Gray, D.J.; Price, C.S.; Nickless, G.; Simmonds, P.G.; O’Doherty, S.J.; Greally, B.R.; Knights, A.; Shallcross, D.E. The role of volatile organic compounds in the polluted urban atmosphere of Bristol, UK. Atmos. Chem. Phys. Disc. 2003, 3, 769–796. [Google Scholar]
- Xiong, Y.; Bari, M.A.; Xing, Z.; Du, K. Ambient volatile organic compounds (VOCs) in two coastal cities in western Canada: Spatiotemporal variation, source apportionment, and health risk assessment. Sci. Total Environ. 2020, 706, 135970. [Google Scholar] [CrossRef] [PubMed]
- Wallington, T.J.; Hurley, M.D.; Maurer, T.; Barnes, I.; Becker, K.H.; Tyndall, G.S.; Orlando, J.J.; Pimentel, A.S.; Bilde, M. Atmospheric Oxidation Mechanism of Methyl Formate. J. Phys. Chem. A 2001, 105, 5146–5154. [Google Scholar] [CrossRef]
- Sinha, A.; Thomson, M.J. The chemical structures of opposed flow diffusion flames of C3 oxygenated hydrocarbons (isopropanol, dimethoxy methane, and dimethyl carbonate) and their mixtures. Combust. Flame 2004, 136, 548–556. [Google Scholar] [CrossRef]
- Daly, C.A.; Simmie, J.M.; Dagaut, P.; Cathonnet, M. Oxidation of dimethoxymethane in a jet-stirred reactor. Combust. Flame 2001, 125, 1106–1117. [Google Scholar] [CrossRef]
- Liu, I.; Cant, N.W.; Bromly, J.H.; Barnes, F.J.; Nelson, P.F.; Haynes, B.S. Formate species in the low-temperature oxidation of dimethyl ether. Chemosphere 2001, 42, 583–589. [Google Scholar] [CrossRef]
- Bertin, M.; Romanzin, C.; Michaut, X.; Jeseck, P.; Fillion, J.H. Adsorption of Organic Isomers on Water Ice Surfaces: A Study of Acetic Acid and Methyl Formate. J. Phys. Chem. C 2011, 115, 12920–12928. [Google Scholar] [CrossRef]
- Goken, E.G.; Castleman, A.W., Jr. Reactions of formic acid with protonated water clusters: Implications of cluster growth in the atmosphere. J. Geophys. Res. 2010, 115, D16203. [Google Scholar] [CrossRef]
- Zheng, Z.; Pavlov, J.; Attygalle, A.B. Fortuitous Ion–Molecule Reaction Enables Enumeration of Metal–Hydrogen Bonds Present in Gaseous Ions. ACS Omega 2019, 4, 3965–3972. [Google Scholar] [CrossRef] [Green Version]
- Osburn, S.; Ryzhov, V. Ion–Molecule Reactions: Analytical and Structural Tool. Anal. Chem. 2013, 85, 769–778. [Google Scholar] [CrossRef]
- Vaida, V. Perspective: Water cluster mediated atmospheric chemistry. J. Chem. Phys. 2011, 135, 020901. [Google Scholar] [CrossRef]
- Španěl, P.; Pavlik, M.; Smith, D. Reactions of H3O+ and OH− ions with some organic molecules; applications to trace gas analysis in air. Int. J. Mass Spectrom. Ion Proc. 1995, 145, 177–186. [Google Scholar] [CrossRef]
- Adams, N.G.; Smith, D. The selected ion flow tube (SIFT); A technique for studying ion-neutral reactions. Int. J. Mass Spectrom. Ion Phys. 1976, 21, 349–359. [Google Scholar] [CrossRef]
- Smith, D.; Španěl, P. Selected ion flow tube mass spectrometry (SIFT-MS) for on-line trace gas analysis. Mass Spectrom. Rev. 2005, 24, 661–700. [Google Scholar] [CrossRef]
- Curtis, S.; DiMuzio, J.; Mungham, A.; Roy, J.; Hassan, D.; Renaud, J.; Mayer, P.M. Reactions of Atomic Metal Anions in the Gas phase: Competition between Electron Transfer, Proton Abstraction and Bond Activation. J. Phys. Chem. A 2011, 115, 14006–14012. [Google Scholar] [CrossRef] [PubMed]
- Cooks, R.G. Collision Spectroscopy; Springer: New York, NY, USA, 1977. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Rev. B.01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [Green Version]
- Baer, T.; Mayer, P.M. Statistical RRKM/QET Calculations in mass spectrometry. J. Am. Soc. Mass Spectrom. 1997, 8, 103–115. [Google Scholar]
- Baer, T.; Hase, W.L. Unimolecular Reaction Dynamics, Theory and Experiments; Oxford University Press: New York, NY, USA, 1996. [Google Scholar]
- Beyer, T.; Swinehart, D.R. Number of Multiply-Restricted Partitions [A1] (Algorithm 448). ACM Commun. 1973, 16, 379. [Google Scholar] [CrossRef]
- Mayer, P.M.; Martineau, E. Gas-phase binding energies for non-covalent A[β]-40 peptide/small molecule complexes from CID mass spectrometry and RRKM theory. Phys. Chem. Chem. Phys. 2011, 13, 5178–5186. [Google Scholar] [CrossRef]
- Renaud, J.B.; Martineau, E.; Mironov, G.G.; Berezovski, M.V.; Mayer, P.M. The collaborative role of molecular conformation and energetics in the binding of gas-phase non-covalent polymer/amine complexes. Phys. Chem. Chem. Phys. 2012, 14, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Diedhiou, M.; Mayer, P.M. Fate of Protonated Formates in the Gas Phase. J. Phys. Chem. A 2021, 125, 5096–5102. [Google Scholar] [CrossRef] [PubMed]
- Hunter, E.P.L.; Lias, S.G. Evaluated Gas Phase Basicities and Proton Affinities of Molecules: An Update. J. Phys. Chem. Ref. Data 1998, 27, 413–656. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reaction | RRKM E0 | Theoretical E0 |
|---|---|---|
| MF(W)H+ → MFH+ + W | 0.34 | 0.94 |
| MF(M)H+ → MFH+ + M → M(CO2)H+ + CH4 MF(E)H+ → MFH+ + E → EH+ + MF MF(M)2H+ → MF(M)H+ + M → (M)2H+ + MF MF(E)2H+ → MF(E)H+ + E → (E)2H+ + MF MF(E)3H+ → MF(E)2H+ + E | 0.58 0.90 0.53 0.56 0.77 0.85 0.87 0.88 0.64 | 1.25 1.98 1.21 1.49 0.84 1.06 0.89 0.94 0.56 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Diedhiou, M.; Mayer, P.M. The Interaction of Methyl Formate with Proton-Bound Solvent Clusters in the Gas Phase and the Unimolecular Chemistry of the Reaction Products. Appl. Sci. 2023, 13, 1339. https://doi.org/10.3390/app13031339
Diedhiou M, Mayer PM. The Interaction of Methyl Formate with Proton-Bound Solvent Clusters in the Gas Phase and the Unimolecular Chemistry of the Reaction Products. Applied Sciences. 2023; 13(3):1339. https://doi.org/10.3390/app13031339
Chicago/Turabian StyleDiedhiou, Malick, and Paul M. Mayer. 2023. "The Interaction of Methyl Formate with Proton-Bound Solvent Clusters in the Gas Phase and the Unimolecular Chemistry of the Reaction Products" Applied Sciences 13, no. 3: 1339. https://doi.org/10.3390/app13031339