
Multiple Genetic Rare Variants in Autism Spectrum Disorders: A Single-Center Targeted NGS Study

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
- -
- Brain magnetic resonance imaging (MRI), 1.5 Tesla, without contrast enhancement (with sagittal, transverse, and coronal sequences, as well as FLAIR sequences of the whole brain and cerebellum);
- -
- Electroencephalogram (EEG);
- -
- A plasma amino acid assay using liquid chromatography–tandem mass spectrometry (ESI/MS/MS) and urinary organic acid assay using gas chromatography–mass spectrometry;
- -
- Standard karyotyping on peripheral blood by means of a lymphocyte culture and QFQ banding, with a resolution of 550 bands (number of metaphases analyzed: 16);
- -
- Molecular analysis for fragile X syndrome, using Southern blotting;
- -
- Array-comparative genomic hybridization.
Statistical Analysis
3. Results
3.1. Gene Panel
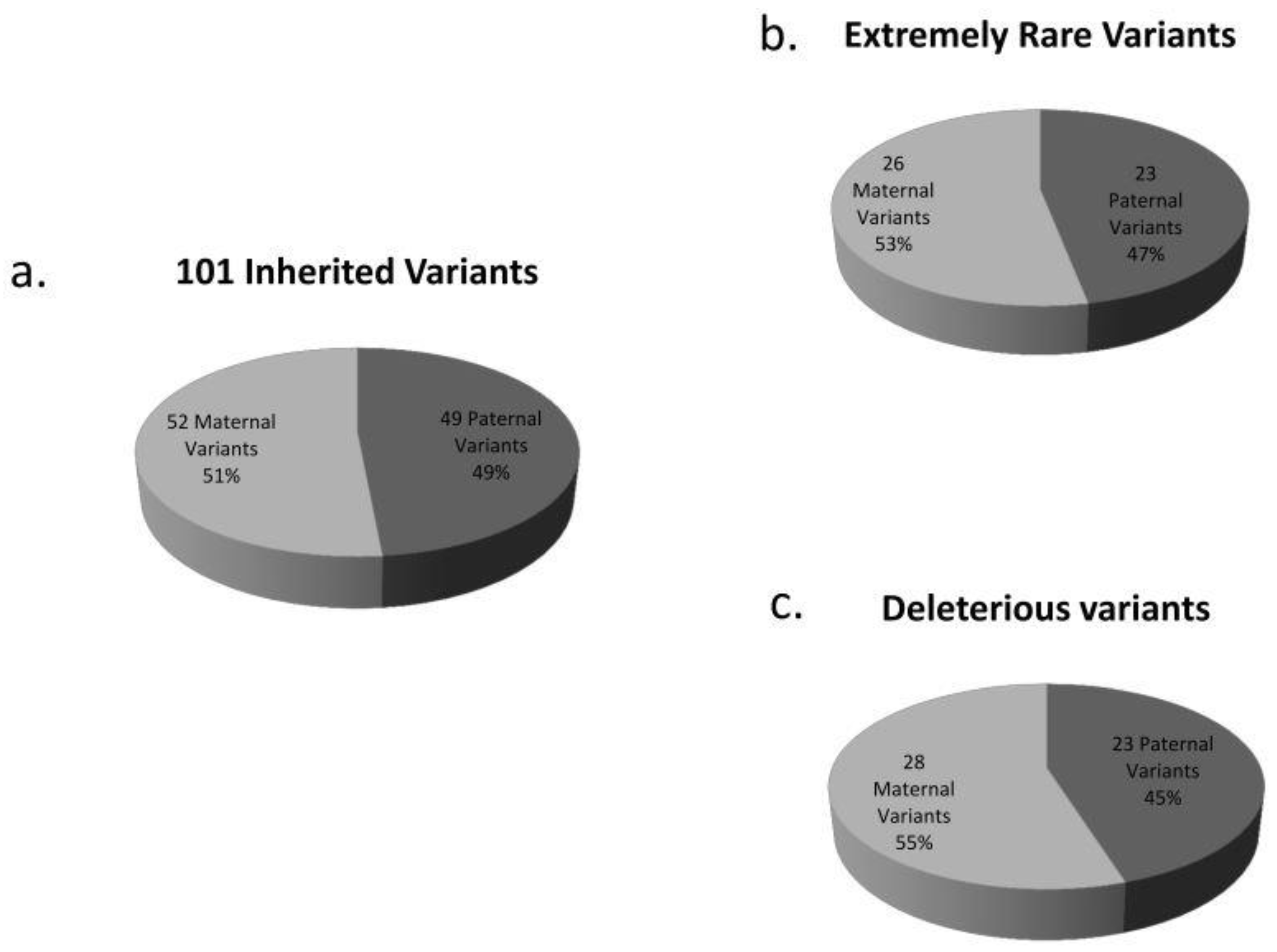
3.2. De Novo and Inherited Variants
3.3. Variants Already Associated with Autism
3.4. Variants Found More Than Once
3.5. More Variants in the Same Gene
3.6. Most Frequently Mutated Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, 5th ed.; American Psychiatric Association: Arlington, VA, USA, 2013. [Google Scholar]
- Baio, J.; Wiggins, L.; Christensen, D.L.; Maenner, M.J.; Daniels, J.; Warren, Z.; Kurzius-Spencer, M.; Zahorodny, W.; Robinson, C.; Rosenberg, C.R.; et al. Prevalence of Autism Spectrum Disorder Among Children Aged 8 Years—Autism and Developmental Disabilities Monitoring Network, 11 Sites, United States, 2014. MMWR. Surveill. Summ. 2018, 67, 1–23. [Google Scholar] [CrossRef]
- Ozonoff, S.; Young, G.S.; Carter, A.; Messinger, D.; Yirmiya, N.; Zwaigenbaum, L.; Bryson, S.; Carver, L.J.; Constantino, J.N.; Dobkins, K.; et al. Recurrence Risk for Autism Spectrum Disorders: A Baby Siblings Research Consortium Study. Pediatrics 2011, 128, e488–e495. [Google Scholar] [CrossRef] [Green Version]
- Ronald, A.; Hoekstra, R. Progress in Understanding the Causes of Autism Spectrum Disorders and Autistic Traits: Twin Studies from 1977 to the Present Day. In Behavior Genetics of Psychopathology; Springer Science and Business Media LLC: Berlin/Heidelberg, Germany, 2014; pp. 33–65. [Google Scholar]
- Rosenberg, R.; Law, J.K.; Yenokyan, G.; Mcgready, J.; Kaufmann, W.E.; Law, P.A. Characteristics and Concordance of Autism Spectrum Disorders Among 277 Twin Pairs. Arch. Pediatr. Adolesc. Med. 2009, 163, 907–914. [Google Scholar] [CrossRef] [Green Version]
- Sanders, S.J.; Murtha, M.T.; Gupta, A.R.; Murdoch, J.D.; Raubeson, M.J.; Willsey, A.J.; Ercan-Sencicek, A.G.; DiLullo, N.M.; Parikshak, N.N.; Stein, J.L.; et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 2012, 485, 237–241. [Google Scholar] [CrossRef]
- Sanders, S.; Ercan-Sencicek, A.G.; Hus, V.; Luo, R.; Murtha, M.T.; Moreno-De-Luca, D.; Chu, S.; Moreau, M.P.; Gupta, A.R.; Thomson, S.A.; et al. Multiple Recurrent De Novo CNVs, Including Duplications of the 7q11.23 Williams Syndrome Region, Are Strongly Associated with Autism. Neuron 2011, 70, 863–885. [Google Scholar] [CrossRef] [Green Version]
- O’Roak, B.J.; Vives, L.; Girirajan, S.; Karakoc, E.; Krumm, N.; Coe, B.P.; Levy, R.; Ko, A.; Lee, C.; Smith, J.A.B.; et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 2012, 485, 246–250. [Google Scholar] [CrossRef] [Green Version]
- Levy, D.; Ronemus, M.; Yamrom, B.; Lee, Y.-H.; Leotta, A.; Kendall, J.; Marks, S.; Lakshmi, B.; Pai, D.; Ye, K.; et al. Rare De Novo and Transmitted Copy-Number Variation in Autistic Spectrum Disorders. Neuron 2011, 70, 886–897. [Google Scholar] [CrossRef] [Green Version]
- Satterstrom, F.K.; Kosmicki, J.A.; Wang, J.; Breen, M.S.; De Rubeis, S.; An, J.-Y.; Peng, M.; Collins, R.; Grove, J.; Klei, L.; et al. Large-Scale Exome Sequencing Study Implicates Both Developmental and Functional Changes in the Neurobiology of Autism. Cell 2020, 180, 568–584.e23. [Google Scholar] [CrossRef]
- Beaudet, A.L. The Utility of Chromosomal Microarray Analysis in Developmental and Behavioral Pediatrics. Child Dev. 2013, 84, 121–132. [Google Scholar] [CrossRef] [Green Version]
- Jeste, S.S.; Geschwind, D.H. Disentangling the heterogeneity of autism spectrum disorder through genetic findings. Nat. Rev. Neurol. 2014, 10, 74–81. [Google Scholar] [CrossRef] [Green Version]
- Schaefer, G.B.; Professional Practice and Guidelines Committee; Mendelsohn, N.J. Clinical genetics evaluation in identifying the etiology of autism spectrum disorders: 2013 guideline revisions. Genet. Med. 2013, 15, 399–407. [Google Scholar] [CrossRef] [Green Version]
- Miller, D.T.; Adam, M.P.; Aradhya, S.; Biesecker, L.G.; Brothman, A.R.; Carter, N.P.; Church, D.M.; Crolla, J.A.; Eichler, E.E.; Epstein, C.J.; et al. Consensus Statement: Chromosomal Microarray Is a First-Tier Clinical Diagnostic Test for Individuals with Developmental Disabilities or Congenital Anomalies. Am. J. Hum. Genet. 2010, 86, 749–764. [Google Scholar] [CrossRef]
- Vicari, S.; Napoli, E.; Cordeddu, V.; Menghini, D.; Alesi, V.; Loddo, S.; Novelli, A.; Tartaglia, M. Copy number variants in autism spectrum disorders. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2019, 92, 421–427. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.; Rutter, M.; Le Couteur, A. Autism Diagnostic Interview-Revised: A revised version of a diagnostic interview for caregivers of individuals with possible pervasive developmental disorders. J. Autism Dev. Disord. 1994, 24, 659–685. [Google Scholar] [CrossRef]
- Luyster, R.; Gotham, K.; Guthrie, W.; Coffing, M.; Petrak, R.; Pierce, K.; Bishop, S.; Esler, A.; Hus, V.; Oti, R.; et al. The Autism Diagnostic Observation Schedule—Toddler Module: A New Module of a Standardized Diagnostic Measure for Autism Spectrum Disorders. J. Autism Dev. Disord. 2009, 39, 1305–1320. [Google Scholar] [CrossRef] [Green Version]
- Wechsler, D. Wechsler Intelligence Scale for Children, 4th ed.; American Psychological Association: Washington, DC, USA, 2012. [Google Scholar]
- Griffiths, R. The Griffiths Mental Development Scales from Birth to 2 Years, Manual, the 1996 Revision; Association for Research in Infant and Child Development, Test agency: Henley, UK, 1996. [Google Scholar]
- Green, E.; Stroud, L.; Bloomfield, S.; Cronje, J.; Foxcroft, C.; Hurter, K. Griffith Scale of Child Development, 3rd ed.; Hogrefe Verlag: Göttingen, Germany, 2016. [Google Scholar]
- Sutcliffe, A.G.; Soo, A.; Barnes, J. Predictive Value of Developmental Testing in the Second Year for Cognitive Development at Five Years of Age. Pediatr. Rep. 2010, 2, 48–50. [Google Scholar] [CrossRef] [Green Version]
- Redin, C.; Gérard, B.; Lauer, J.; Herenger, Y.; Muller, J.; Quartier, A.; Masurel-Paulet, A.; Willems, M.; Lesca, G.; El-Chehadeh, S.; et al. Efficient strategy for the molecular diagnosis of intellectual disability using targeted high-throughput sequencing. J. Med. Genet. 2014, 51, 724–736. [Google Scholar] [CrossRef]
- Cukier, H.N.; Dueker, N.D.; Slifer, S.H.; Lee, J.M.; Whitehead, P.L.; Lalanne, E.; Leyva, N.; Konidari, I.; Gentry, R.C.; Hulme, W.F.; et al. Exome sequencing of extended families with autism reveals genes shared across neurodevelopmental and neuropsychiatric disorders. Mol. Autism 2014, 5, 1. [Google Scholar] [CrossRef] [Green Version]
- Iossifov, I.; Ronemus, M.; Levy, D.; Wang, Z.; Hakker, I.; Rosenbaum, J.; Yamrom, B.; Lee, Y.-H.; Narzisi, G.; Leotta, A.; et al. De Novo Gene Disruptions in Children on the Autistic Spectrum. Neuron 2012, 74, 285–299. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Takumi, T. Genomic and genetic aspects of autism spectrum disorder. Biochem. Biophys. Res. Commun. 2014, 452, 244–253. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.-G.; Kishikawa, S.; Higgins, A.W.; Seong, I.-S.; Donovan, D.J.; Shen, Y.; Lally, E.; Weiss, L.A.; Najm, J.; Kutsche, K.; et al. Disruption of Neurexin 1 Associated with Autism Spectrum Disorder. Am. J. Hum. Genet. 2008, 82, 199–207. [Google Scholar] [CrossRef] [Green Version]
- Onay, H.; Kacamak, D.; Kavasoglu, A.; Akgun, B.; Yalcinli, M.; Köse, S.; Ozbaran, B. Mutation analysis of the NRXN1 gene in autism spectrum disorders. Balk. J. Med. Genet. 2016, 19, 17–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talkowski, M.; Mullegama, S.V.; Rosenfeld, J.A.; van Bon, B.W.; Shen, Y.; Repnikova, E.A.; Gastier-Foster, J.; Thrush, D.L.; Kathiresan, S.; Ruderfer, D.; et al. Assessment of 2q23.1 Microdeletion Syndrome Implicates MBD5 as a Single Causal Locus of Intellectual Disability, Epilepsy, and Autism Spectrum Disorder. Am. J. Hum. Genet. 2011, 89, 551–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonora, E.; The International Molecular Genetic Study of Autism Consortium (IMGSAC); Beyer, K.S.; Lamb, J.; Parr, J.R.; Klauck, S.M.; Benner, A.; Paolucci, M.; Abbott, A.; Ragoussis, I.; et al. Analysis of reelin as a candidate gene for autism. Mol. Psychiatry 2003, 8, 885–892. [Google Scholar] [CrossRef] [Green Version]
- Adamsen, D.; Meili, D.; Blau, N.; Thöny, B.; Ramaekers, V. Autism associated with low 5-hydroxyindolacetic acid in CSF and the heterozygous SLC6A4 gene Gly56Ala plus 5-HTTLPR L/L promoter variants. Mol. Genet. Metab. 2011, 102, 368–373. [Google Scholar] [CrossRef] [PubMed]
- Sutcliffe, J.S.; Delahanty, R.J.; Prasad, H.C.; McCauley, J.L.; Han, Q.; Jiang, L.; Li, C.; Folstein, S.E.; Blakely, R.D. Allelic Heterogeneity at the Serotonin Transporter Locus (SLC6A4) Confers Susceptibility to Autism and Rigid-Compulsive Behaviors. Am. J. Hum. Genet. 2005, 77, 265–279. [Google Scholar] [CrossRef] [Green Version]
- Coupry, I.; Roudaut, C.; Stef, M.; Delrue, M.A.; Marche, M.; Burgelin, I.; Taine, L.; Cruaud, C.; Lacombe, D.; Arveiler, B. Molecular analysis of the CBP gene in 60 patients with Rubinstein-Taybi syndrome. J. Med. Genet. 2002, 39, 415–421. [Google Scholar] [CrossRef] [Green Version]
- Kelleher, R.J., 3rd; Geigenmüller, U.; Hovhannisyan, H.; Trautman, E.; Pinard, R.; Rathmell, B.; Carpenter, R.; Margulies, D. High-Throughput Sequencing of mGluR Signaling Pathway Genes Reveals Enrichment of Rare Variants in Autism. PLoS ONE 2012, 7, e35003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez-Mora, M.I.; Escalona, R.C.; Navarro, O.P.; Madrigal, I.; Quintela, I.; Amigo, J.; Martinez-Elurbe, D.; Linder-Lucht, M.; Lain, G.A.; Carracedo, A.; et al. Comprehensive molecular testing in patients with high functioning autism spectrum disorder. Mutat. Res. Mol. Mech. Mutagen. 2016, 784–785, 46–52. [Google Scholar] [CrossRef]
- Kalsner, L.; Twachtman-Bassett, J.; Tokarski, K.; Stanley, C.; Dumont-Mathieu, T.; Cotney, J.; Chamberlain, S. Genetic testing including targeted gene panel in a diverse clinical population of children with autism spectrum disorder: Findings and implications. Mol. Genet. Genom. Med. 2017, 6, 171–185. [Google Scholar] [CrossRef]
- De Rubeis, S.; He, X.; Goldberg, A.P.; Poultney, C.S.; Samocha, K.; Cicek, A.E.; Kou, Y.; Liu, L.; Fromer, M.; Walker, S.; et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 2014, 515, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Iossifov, I.; O’Roak, B.J.; Sanders, S.J.; Ronemus, M.; Krumm, N.; Levy, D.; Stessman, H.A.; Witherspoon, K.T.; Vives, L.; Patterson, K.E.; et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature 2014, 515, 216–221. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Dietrich, U.M.; Geng, J.-G.; Bicknell, R.; Esko, J.D.; Wang, L. Repulsive axon guidance molecule Slit3 is a novel angiogenic factor. Blood 2009, 114, 4300–4309. [Google Scholar] [CrossRef] [Green Version]
- Iossifov, I.; Levy, D.; Allen, J.; Ye, K.; Ronemus, M.; Lee, Y.-H.; Yamrom, B.; Wigler, M. Low load for disruptive mutations in autism genes and their biased transmission. Proc. Natl. Acad. Sci. USA 2015, 112, E5600–E5607. [Google Scholar] [CrossRef] [Green Version]
- Gee, H.Y.; Sadowski, C.E.; Aggarwal, P.K.; Porath, J.D.; Yakulov, T.A.; Schueler, M.; Lovric, S.; Ashraf, S.; Braun, D.A.; Halbritter, J.; et al. FAT1 mutations cause a glomerulotubular nephropathy. Nat. Commun. 2016, 7, 10822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neale, B.M.; Kou, Y.; Liu, L.; Ma’Ayan, A.; Samocha, K.E.; Sabo, A.; Lin, C.-F.; Stevens, C.; Wang, L.-S.; Makarov, V.; et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature 2012, 485, 242–245. [Google Scholar] [CrossRef]
- Puppo, F.; Dionnet, E.; Gaillard, M.-C.; Gaildrat, P.; Castro, C.; Vovan, C.; Bertaux, K.; Bernard, R.; Attarian, S.; Goto, K.; et al. Identification of Variants in the 4q35 GeneFAT1in Patients with a Facioscapulohumeral Dystrophy-Like Phenotype. Hum. Mutat. 2015, 36, 443–453. [Google Scholar] [CrossRef] [PubMed]
- Caruso, N.; Herberth, B.; Bartoli, M.; Puppo, F.; Dumonceaux, J.; Zimmermann, A.; Denadai, S.; Lebossé, M.; Roche, S.; Geng, L.; et al. Deregulation of the Protocadherin Gene FAT1 Alters Muscle Shapes: Implications for the Pathogenesis of Facioscapulohumeral Dystrophy. PLoS Genet. 2013, 9, e1003550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, L.G.; Ramaswami, D.; Chan, T.A. The FAT epidemic: A gene family frequently mutated across multiple human cancer types. Cell Cycle 2013, 12, 1011–1012. [Google Scholar] [CrossRef]
- Kolehmainen, J.; Black, G.; Saarinen, A.; Chandler, K.; Clayton-Smith, J.; Träskelin, A.-L.; Perveen, R.; Kivitie-Kallio, S.; Norio, R.; Warburg, M.; et al. Cohen Syndrome Is Caused by Mutations in a Novel Gene, COH1, Encoding a Transmembrane Protein with a Presumed Role in Vesicle-Mediated Sorting and Intracellular Protein Transport. Am. J. Hum. Genet. 2003, 72, 1359–1369. [Google Scholar] [CrossRef] [Green Version]
- Loviglio, M.N.; Beck, C.R.; White, J.J.; Leleu, M.; Harel, T.; Guex, N.; Niknejad, A.; Bi, W.; Chen, E.S.; Crespo, I.; et al. Identification of a RAI1-associated disease network through integration of exome sequencing, transcriptomics, and 3D genomics. Genome Med. 2016, 8, 105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huguet, G.; Benabou, M.; Bourgeron, T. The Genetics of Autism Spectrum Disorders. In Research and Perspectives in Endocrine Interactions; Springer: Berlin/Heidelberg, Germany, 2016; pp. 101–129. [Google Scholar]


{kind=link}
| AGAP1 | DHCR7 | IQSEC2 | PCDH19 | SLIT3 |
| ANK2 | DLGAP2 | JARID2 | PCDH9 | SMG6 |
| ANKRD11 | DMD | KATNAL2 | PHF6 | SNRPN |
| AP1S2 | DOCK4 | KCNMA1 | PHF8 | SOX5 |
| ARX | DPP10 | KCTD13 | POGZ | SPAST |
| ASH1L | DST | KDM5C | PRICKLE1 | STXBP5 |
| ASTN2 | DYRK1A | KDM6B | PTCHD1 | ST7 |
| ATRX | EHMT1 | KIRREL3 | PTEN | SYNGAP1 |
| AUTS2 | EN2 | LAMC3 | PTPN11 | SYN1 |
| AVPR1A | FAT1 | MBD5 | RAB39B | TBR1 |
| BDNF | FGD1 | MECP2 | RAI1 | TCF4 |
| BRAF | FMR1 | MED12 | RBFOX1 | TRIMM33 |
| CACNA1A | FOXG1 | MED13L | RELN | TSC1 |
| CACNA1C | FOXP1 | MEF2C | RIMS1 | TSC2 |
| CACNA1H | FOXP2 | MET | RPL10 | UBE3A |
| CADPS2 | GABRB3 | NF1 | SATB2 | VPS13B |
| CASK | GNA14 | NIPBL | SCN1A | WNT2 |
| CDKL5 | GRIN2B | NLGN3 | SCN2A | YWHAE |
| CEP290 | GRIN2A | NLGN4X | SHANK2 | ZEB2 |
| CHD8 | GRM5 | NRXN1 | SHANK3 | ZNF804A |
| CNTNAP2 | GRPR | NRXN3 | SLC16A2 | |
| CNTN4 | HCFC1 | NSD1 | SLC6A3 | |
| CREBBP | HOXA1 | NTNG1 | SLC6A4 | |
| CSMD1 | IL1RAPL1 | OPHN1 | SLC9A6 | |
| CUL4B | IMMP2L | OXTR | SLC9A9 |
| Group | Allele Frequency (AF) |
|---|---|
| *** | (AF) < 1/10,000 (0.01%) |
| ** | 1/10,000 (0.01%) < AF < 1/5000 (0.02%) |
| * | 1/5000 (0.02%) < AF < 1/1000 (0.1%) |
| - | 1/1000 (0.1%) < AF < 1/500 (0.2%) |
| f | 1/500 (0.2%) < AF < 1/100 (1%) |
| Gene | SFARI Score | Number of Variants | Patients | Same Variants | ASD Frequency in Our Cohort | GnomAD Frequency | Statistical Significance Exact p-Value (1-Tailed) |
|---|---|---|---|---|---|---|---|
| FAT1 | 3S | 12 | 11 | none | 0.15 | 0.000061 | exact p < 0.0001 |
| VPS13B | S | 12 | 10 | 1 | 0.15 | 0.000062 | exact p < 0.0001 |
| ANK2 | 1 | 6 | 6 | none | 0.075 | 0.000046 | exact p < 0.0001 |
| RAI1 | 3S | 6 | 6 | 1 | 0.075 | 0.000049 | exact p < 0.0001 |
| SHANK3 | 1S | 5 | 5 | none | 0.0625 | 0.000056 | exact p < 0.0001 |
| ANKRD11 | 2S | 5 | 5 | none | 0.0625 | 0.000040 | exact p < 0.0001 |
| CSMD1 | 4 | 5 | 5 | none | 0.0625 | 0.000038 | exact p < 0.0001 |
| DLGAP2 | 4 | 5 | 5 | none | 0.0625 | 0.000024 | exact p < 0.0001 |
| ZNF804A | 3 | 5 | 4 | none | 0.0625 | 0.000052 | exact p < 0.0001 |
| Patient | Total Variants | Paternal Variants | Maternal Variants | De Novo Variants | Variants Already Associated with ASD | Very Rare Variants (***, **) | Variants in Most Mutated Genes | Variants Found More than Once | Variants with at Least One Deleterious Prediction |
|---|---|---|---|---|---|---|---|---|---|
| 1 (H16/18) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 2 (E10/24) | 4 | U | U | U | 0 | 3 | 1 (DLGAP2,***) | 0 | 3(***,***,**) |
| 3 (O12/11) | 5 | 2 (***,f) | 3 (***,**,f) | 0 | 1 (M, f) | 3 (P,M,M) | 1 (ANKRD11, P, f) | 0 | 1 (M,f) |
| 4 (C14/16) | 2 | U | U | U | 0 | 2 | 1 (VPS13B,***) | 0 | 1 (***) |
| 5 (C.P.P) | 5 | 2 (*,***) | 3 (f,*,***) | 0 | 0 | 2 (P,M) | 2 (DLGAP2, M,*)(RAI1, M, f) | 1 (f,M) | 3 (P,*)(M,*)(P,***) |
| 6 (C.L.) | 7 | U | U | U | 1 (*) | 3 | 2 (FAT1,***)(SHANK3,***) | 1 (f) | 3 (*)(***)(f) |
| 7 (A14/06) | 6 | 3 (*,f,f) | 3 (***,***,f) | 0 | 1 (P,f) | 2 (M,M) | 3 (ZNF804A,P,*)(VPS13B,M,f)(RAI1,P,f) | 1 (P,f) | 4 (M,***)(M,***)(P,*)(P,f) |
| 8 (D.F.M.) | 1 | 1(**) | 0 | 0 | 0 | 1 (P) | 0 | 0 | 0 |
| 9 (E13/58) | 2 | U | U | U | 0 | 2 | 0 | 0 | 0 |
| 10 (A14/55) | 2 | 0 | 0 | 2 (-, ***) | 0 | 1 (D.N.) | 1 (ANKRD11, D.N., -) | 0 | 1 (D.N., -) |
| 11 (G14/62) | 7 | 2 (***,***) | 5 (***,***,*,*,f) | 0 | 0 | 4 (P,P,M,M) | 3 (DLGAP2, M,***)(RAI1, M,***) (ZNF804A, M, *)(ZNF804A, M, *) | 1 (M,f) | 4 (P,***)(M,***)(M,*)(M.f) |
| 12 (G.F.M.) | 2 | 1 (*) | 1 (***) | 0 | 0 | 1 (M) | 0 | 0 | 1 (M,***) |
| 13 (IB1A) | 5 | 4 (***,***,*,***) | 1 (**) | 0 | 0 | 4 (P,P,P,M) | 1 (RAI1,P,***) | 0 | 4 (P,***)(M,**)(P,***)(P,***) |
| 14 (IB17A) | 2 | U | 1 (f) | U | 0 | 1 | 0 | 1 (M,f) | 2 (U,***)(M,f) |
| 15 (A15/18) | 9 | U | U | U | 0 | 6 | 6 (ZNF804A,***)(VPS13B,***)(VPS13B,***) (SHANK3,**)(SHANK3,f)(FAT1,**) | 0 | 3 (***)(***)(**) |
| 16 (E11/61) | 4 | U | U | U | 0 | 0 | 1 (FAT1,-) | 0 | 3 (-)(-)(f) |
| 17 (F11/04) | 3 | 2 (***,f) | 1 (***) | 0 | 0 | 2 (P,M) | 0 | 0 | 3 (P,***)(M,***)(P,f) |
| 18 (F12/67) | 2 | 2 (***,*) | 0 | 0 | 0 | 1 (P) | 2 (FAT1,P,***)(FAT1,P,*) | 0 | 2 (P,***)(P,*) |
| 19 (D15/81) | 4 | 3 (f,f,f) | 1 (***) | 0 | 1 (P,f) | 1 (M) | 1 (VPS13B,P,f) | 1 (P,f) | 1 (P,f) |
| 20 (F14/70) | 5 (1 Homo) | 2 (***,***) | 4 (***,***,***,-) | 0 | 0 | 4 (P, P/M,M,M) | 0 | 1(M,***) | 3 (P,***)(M,***)(P/M,***) |
| 21 (M.L.B.) | 1 | U | U | U | 0 | 1 | 1(ANK2,***) | 0 | 0 |
| 22 (N13/03) | 4 | 2 (***,f) | 2 (*,f) | 0 | 0 | 1 (P) | 1 (DLGAP2,P,f) | 1 (M,f) | 3 (P,***)(M,*)(M,f) |
| 23 (L12/26) | 5 | 4 (***,*,-,f) | 1 (***) | 0 | 0 | 2 (P,M) | 1 (ZNF804A,P,*) | 0 | 3 (P,***)(M,***)(P,f) |
| 24 (B12/69) | 4 | 1(***) | 3 (**,-,f) | 0 | 0 | 2 (P,M) | 2 (ANK2,M,-)(CSMD1,M,f) | 1 (P,***) | 3 (P,***)(M,-)(M,**) |
| 25 (D10/14) | 3 | U | U | U | 1 (f) | 0 | 2 (VPS13B,-)(VPS13B,*) | 0 | 2 (f)(-) |
| 26 (C11/79) | 4 | 3 (***,***,***) | 1(***) | 0 | 0 | 4 (P,P,P,M) | 1 (ANK2,P,***) | 0 | 1 (P,***) |
| 27 (H11/97) | 2 | U | U | U | 0 | 1 | 1 (FAT1,f) | 0 | 1 (f) |
| 28 (C15/87) | 5 | 1 (*) | 4 (***,*,*,f) | 0 | 0 | 1(M) | 3 (FAT1,P,*)(DLGAP2,M,1)(CSMD1,M,f) | 0 | 5 (M,***)(P,*)(M,*)(M,*)(M,f) |
| 29 (C14/83) | 4 | U | U | U | 0 | 2 | 1 (VPS13B,f) | 0 | 3 (***)(*)(f) |
| 30 (M14/99) | 2 | U | U | U | 0 | 1 | 1 (FAT1,f) | 0 | 2 (**)(f) |
| 31 (L14/95) | 5 | 0 | 4 (*,**,f,f) | 1 (f) | 1 (D.N.,f) | 1 (M) | 2 (ANK2,M,*)(VPS13B,M,f) | 0 | 2 (M,**)(M,f) |
| 32 (s.s.73499) | 1 | 1 (f) | 0 | 0 | 0 | 0 | 1 (FAT1,P,f) | 0 | 1 (P,f) |
| 33 (r.g.74126) | 5 | 2 (f,-) | 3 (*,f,f) | 0 | 1 (M, f) | 0 | 3 (FAT1,M,f)(CSMD1,M,f)(RAI1,P,f) | 2 (P,-)(P,f) | 4 (M,f)(M,f)(M,*)(P,f) |
| 34 (a.d.81815) | 5 | 2 (***,f) | 3 (***,***,-) | 0 | 0 | 3 (P,M,M) | 3 (FAT1,M,***)(CSMD1,P,***)(RAI1,P,f) | 2 (M,-)(P,f) | 2 (M,***)(P,f) |
| 35 (g.w.g.01350) | 2 | U | U | U | 0 | 2 | 1 (CSMD1,***) | 0 | 2 (***)(***) |
| 36 (p.a.59939) | 4 | 2 (**,f) | 2 (**,1) | 0 | 0 | 2 (P,M) | 2 (ANK2,P,**)(VPS13B,M,*) | 0 | 3 (P,**)(M,**)(M,*) |
| 37 (o.f.76867) | 2 | 2 (***,f) | 0 | 0 | 0 | 1 (P) | 1 (ANK2,P,***) | 0 | 0 |
| 38 (H.S.M.) | 4 | 2 (*,-) | 2 (**,*) | 0 | 1 (M,*) | 1 (M) | 1 (ANKRD11,P,*) | 0 | 1 (P,-) |
| 39 (LDM160) | 4 | 1 (***) | 3 (***,***,**) | 0 | 0 | 4 (P,M,M,M) | 2 (VPS13B,m,***)(SHANK3,P,***) | 0 | 3 (M,***)(P,***)(M,***) |
| 40 (LDM167) | 4 | 2 (***,f) | 1(f) | 1 (***) | 0 | 2 (P,D.N.) | 2 (FAT1,P,f)(VPS13B,M,f) | 1 (M,f) | 3 (D.N.,***)(P,***)(P,f) |
| Patient | Gene | Gene SFARI Score | Variant | Inheritance | Predictions | Reference | Allele Frequency (GnomAD) |
|---|---|---|---|---|---|---|---|
| A14/55 | ANKRD11 | 2S | c.890C>T, p.Thr297Met | De novo | S damaging, P probably damaging | / | 0.0012 (1/800) |
| NF1 | S | c.1985A>G, p.Lys662Arg | De novo | S tolerated, P benign | / | 0.00002 (1/50,000) | |
| L14/95 | SLIT3 | no rating | c.1886G>A, p.Ser629Asn | De novo | S tolerated, P benign | Cukier et al., 2014 [23] | 0.007 (1/140) |
| LDM167 | PTEN | 1S | c.1131_1132dupTA, p.Arg378IlefsTer39 | De novo | S/, P/ | / | not present |
| Patient | Gene | Gene SFARI Score | Variant | Inheritance | Predictions | Reference | Allele Frequency (GnomAD) |
|---|---|---|---|---|---|---|---|
| O12/11 | NRXN1 | 2 | c.2242C>A, p.Leu748Ile | Mother | Tolerated (S), Possibly damaging (P) | Kim et al., 2008 [26]; Onay et al., 2017 [27] | 0.004 (1/250) (f) |
| C.L. | MBD5 | 3S | c.236G>A, p.Gly79Glu | Unknown | Deleterious (S), Probably damaging (P) | Talkowski et al., 2011 [28] | 0.0005 (1/2000) (*) |
| A14/06 | RELN | 1 | c.5156C>T, p.Ser1719Leu | Father | Deleterious (S), Probably damaging (P) | Bonora et al., 2003 [29] | 0.006 (1/150) (f) |
| D15/81 | SLC6A4 | 4 | c.167G>C, p.Gly56Ala | Father | Tolerated (S), Benign (P) | Adamsen et al., 2011 [30] Sutcliffe et al., 2005 [31]; | 0.01 (1/100) (f) |
| D10/14 | CREBBP | 5 | c.2941G>A, p.Ala981Thr | Unknown | unknown (S), unknown (P) | Coupry et al., 2002 [32] | 0.003 (1/300) (f) |
| L14/95 | SLIT3 | no rating | c.1886G>A, p.Ser629Asn | De Novo | Tolerated (S), Benign (P) | Cukier et al., 2014 [23] | 0.007 (1/140) (f) |
| r.g.74126 | FAT1 | 4 | c.12653 A>G, p.Asp4218Gly | Mother | unknown (S), Possibly damaging (P) | Cukier et al., 2014 [23] | 0.01 (1/100) (f) |
| H.S.M. | TSC1 | S | c.1960 C>G, p.Gln654Glu | Mother | Tolerated (S), Benign (P) | Kelleher et al., 2012 [33] | 0.0007 (1/1400) (*) |
| Gene | SFARI Score | Variant | Chr Position (GRCh37) | Predictions (S-P) | GnomAD | Patient | Transmission |
|---|---|---|---|---|---|---|---|
| NSD1 | S | c.7367T>C; p.Met2456Thr | 5:176,721,736 | S deleterious–low confidence, P benign | 0.00006 (1/16,000) | F14/70 | Maternal |
| B12/69 | Paternal | ||||||
| SCN2A | 1 | c.2723A>G; p.Lys908Arg | 2:166,201,225 | S tolerated, P possibly damaging | 0.004 (1/250) | C.L. | Unknown |
| IB17A | Maternal | ||||||
| CEP290 | S | c.6401T>C; p.Ile2134Thr | 12:88,454,728 | S damaging, P probably damaging | 0.007 (1/150) | G14/62 | Maternal |
| N13/03 | Maternal | ||||||
| VPS13B | S | c.2485G>A; p.Ala829Thr | 8:100,205,255 | S tolerated, P benign | 0.0085 (1/115) | D15/81 | Paternal |
| LDM167 | Maternal | ||||||
| RAI1 | 3S | c.1142C>T; p.Ala381Val | 17:17,697,404 | S deleterious, P benign | 0.004 (1/250) | r.g.74126 | Paternal |
| a.d.81815 | Paternal | ||||||
| C.P.P | Maternal | ||||||
| ZEB2 | / | c.2230A>G; p.Ile744Val | 2:145,156,524 | S tolerated, P benign | 0.002 (1/500) | r.g.74126 | Paternal |
| a.d.81815 | Maternal |
| Gene | SFARI Score | Variant | Chr Position (GRCh37) | Predictions (S-P) | GnomAD | Patient | Transmission |
|---|---|---|---|---|---|---|---|
| ZNF804A | 3 | c.735 _737delATT; p.Phe246del | 2:185,800,857 | S/, P/ | 0.0005 (1/2000) | G14/62 | Maternal |
| c.1490T>C; p.Leu497Pro | 2:185,801,613 | S deleterious, P benign | 0.0004 (1/2500) | G14/62 | Maternal | ||
| VPS13B | S | c.611A>G; p.Asn204Ser | 8:100,123,356 | S tolerated, P possibly damaging | 0.00009 (1/11,000) | A15/18 | Unknown |
| c.6304C>T; p.Arg2102Cys | 8:100,711,935 | S tolerated, P benign | 0.000008 (1/125,000) | A15/18 | Unknown | ||
| GNA14 | 6 | c.215C>T; p.Thr72Met | 9:80,144,079 | S tolerated, P possibly damaging | 0.0013 (1/750) | E11/61 | Unknown |
| c.97C>T; p.Arg33Cys | 9:80,262,613 | S deleterious, P benign | 0.005 (1/200) | E11/61 | Unknown | ||
| FAT1 | 4 | c.9457G>A; p.Asp3153Asn | 4:187,534,269 | S deleterious, P probably damaging | not present | F12/67 | Paternal |
| c.7957G>A; p.Gly2653Ser | 4:187,539,783 | S deleterious, P probably damaging | 0.0006 (1/1500) | F12/67 | Paternal | ||
| VPS13B | S | c.11270G>A; p.Arg3757Gln | 8:100,874,154 | S tolerated, P possibly damaging | 0.0017 (1/600) | D10/14 | Unknown |
| c.11825_ 11827dupATG; p.Asp3942dup | 8:100,887,648 | S/, P/ | 0.00045 (1/2500) | D10/14 | Unknown | ||
| MET | 2 | c.3571A>G; p.Thr1191Ala | 7:116,419,006 | S tolerated, P probably damaging | 0.000008 (1/120,000) | F14/70 | Maternal |
| c.3571A>G; p.Thr1191Ala | 7:116,419,006 | S tolerated, P probably damaging | 0.000008 (1/120,000) | F14/70 | Paternal |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reale, C.; Tessarollo, V.; Bulgheroni, S.; Annunziata, S.; Legati, A.; Riva, D.; Pantaleoni, C.; Garavaglia, B.; D’Arrigo, S. Multiple Genetic Rare Variants in Autism Spectrum Disorders: A Single-Center Targeted NGS Study. Appl. Sci. 2021, 11, 8096. https://doi.org/10.3390/app11178096
Reale C, Tessarollo V, Bulgheroni S, Annunziata S, Legati A, Riva D, Pantaleoni C, Garavaglia B, D’Arrigo S. Multiple Genetic Rare Variants in Autism Spectrum Disorders: A Single-Center Targeted NGS Study. Applied Sciences. 2021; 11(17):8096. https://doi.org/10.3390/app11178096
Chicago/Turabian StyleReale, Chiara, Valeria Tessarollo, Sara Bulgheroni, Silvia Annunziata, Andrea Legati, Daria Riva, Chiara Pantaleoni, Barbara Garavaglia, and Stefano D’Arrigo. 2021. "Multiple Genetic Rare Variants in Autism Spectrum Disorders: A Single-Center Targeted NGS Study" Applied Sciences 11, no. 17: 8096. https://doi.org/10.3390/app11178096