Genome-Wide Association Study for Reproductive Traits in a Duroc Pig Population

 , , ,
, , ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Material and Methods
2.1. Ethics Statement
2.2. Population and Phenotyping
2.3. Genomic DNA Extraction and Genotyping
2.4. Data Quality Control
2.5. Statistical Analysis
2.6. Identification of Candidate Genes
2.7. Functional Enrichment Analysis
3. Results
3.1. Description of Phenotypes and Genotypes
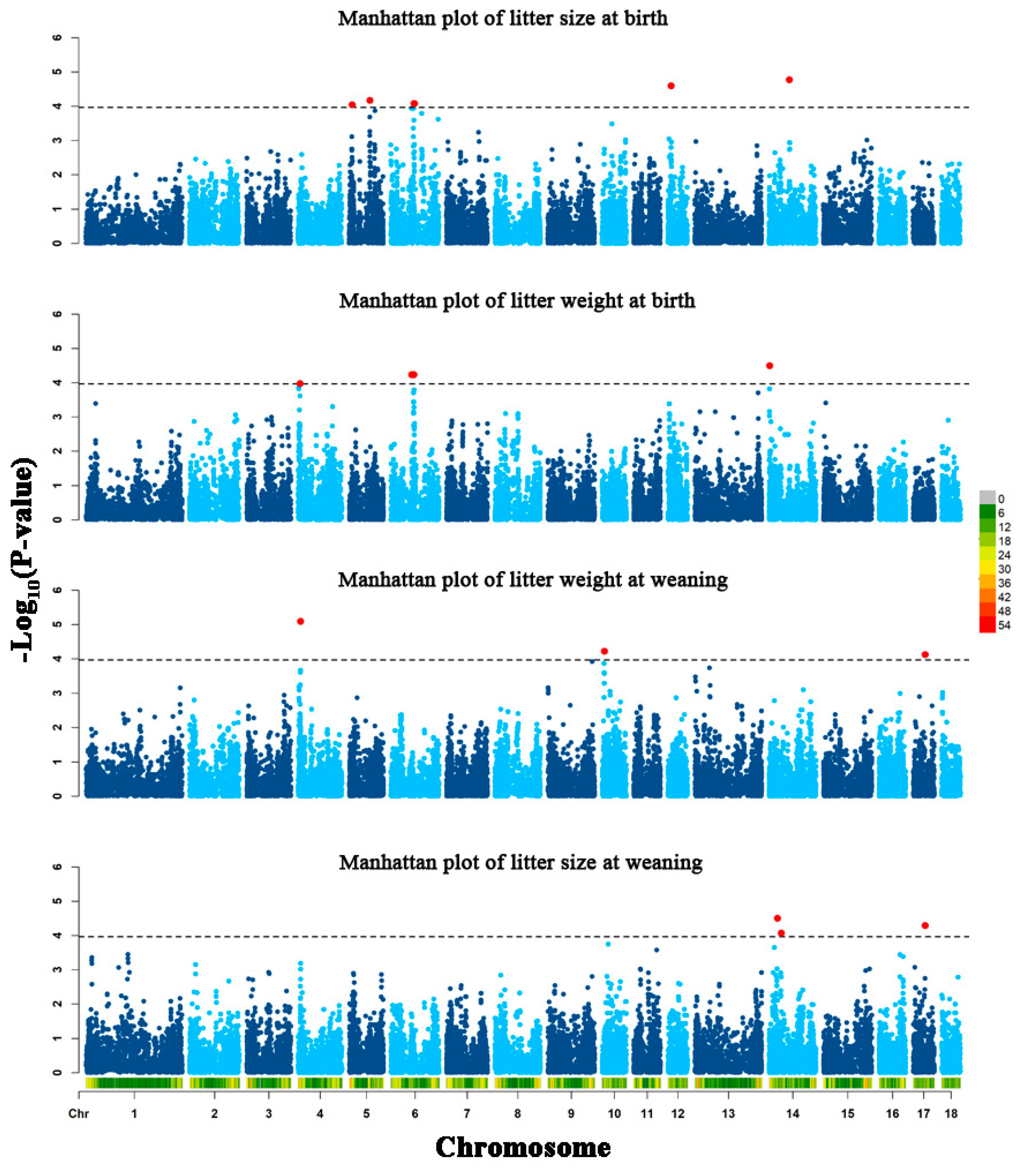
3.2. Genome-Wide Association Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Meuwissen, T.H.; Hayes, B.J.; Goddard, M.E. Prediction of total genetic value using genome-wide dense marker maps. Genetics 2001, 157, 1819–1829. [Google Scholar]
- Zhang, Z.; Ober, U.; Erbe, M.; Zhang, H.; Gao, N.; He, J.; Li, J.; Simianer, H. Improving the Accuracy of Whole Genome Prediction for Complex Traits Using the Results of Genome Wide Association Studies. PLoS ONE 2014, 9, e93017. [Google Scholar] [CrossRef] [PubMed]
- Edwards, A.O. Complement Factor H Polymorphism and Age-Related Macular Degeneration. Science 2005, 308, 421–424. [Google Scholar] [CrossRef] [Green Version]
- Sahana, G.; Kadlecová, V.; Hornshøj, H.; Nielsen, B.; Christensen, O.F. A genome-wide association scan in pig identifies novel regions associated with feed efficiency trait1. J. Anim. Sci. 2013, 91, 1041–1050. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Huang, Y.; Hou, L.; Ma, J.; Chen, C.; Ai, H.; Huang, L.; Ren, J. Genome-wide detection of genetic markers associated with growth and fatness in four pig populations using four approaches. Genet. Sel. Evol. 2017, 49, 21. [Google Scholar] [CrossRef] [Green Version]
- Uimari, P.; Sironen, A.; Sevón-Aimonen, M.-L. Whole-genome SNP association analysis of reproduction traits in the Finnish Landrace pig breed. Genet. Sel. Evol. 2011, 43, 42. [Google Scholar] [CrossRef]
- Tan, C.; Wu, Z.; Ren, J.; Huang, Z.; Liu, D.; He, X.; Prakapenka, D.; Zhang, R.; Li, N.; Da, Y.; et al. Genome-wide association study and accuracy of genomic prediction for teat number in Duroc pigs using genotyping-by-sequencing. Genet. Sel. Evol. 2017, 49, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, W.; Cheng, D.; Chen, S.; Wang, L.; Li, Y.; Ma, X.; Song, X.; Liu, X.; Li, W.; Liang, J.; et al. Genome-Wide Association Analysis of Meat Quality Traits in a Porcine Large White × Minzhu Intercross Population. Int. J. Biol. Sci. 2012, 8, 580–595. [Google Scholar] [CrossRef]
- Sato, S.; Uemoto, Y.; Kikuchi, T.; Egawa, S.; Kohira, K.; Saito, T.; Sakuma, H.; Miyashita, S.; Arata, S.; Kojima, T.; et al. SNP- and haplotype-based genome-wide association studies for growth, carcass, and meat quality traits in a Duroc multigenerational population. BMC Genet. 2016, 17, 60. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.-L.; Park, C.A.; Reecy, J.M. Building a livestock genetic and genomic information knowledgebase through integrative developments of Animal QTLdb and CorrDB. Nucleic Acids Res. 2019, 47, D701–D710. [Google Scholar] [CrossRef]
- Manolio, T.A.; Collins, F.S.; Cox, N.J.; Goldstein, D.B.; Hindorff, L.A.; Hunter, D.J.; McCarthy, M.I.; Ramos, E.M.; Cardon, L.R.; Chakravarti, A.; et al. Finding the missing heritability of complex diseases. Nature 2009, 461, 747–753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henderson, C.R. Best Linear Unbiased Estimation and Prediction under a Selection Model. Biometrics 1975, 31, 423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- VanRaden, P.M.; Wiggans, G.R. Derivation, Calculation, and Use of National Animal Model Information. J. Dairy Sci. 1991, 74, 2737–2746. [Google Scholar] [CrossRef]
- R Foundation for Statistical Computing. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2018; ISBN 3-900051-07-0. [Google Scholar]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Stephens, M. Genome-wide efficient mixed-model analysis for association studies. Nat. Genet. 2012, 44, 821–824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Onteru, S.K.; Fan, B.; Du, Z.-Q.; Garrick, D.J.; Stalder, K.J.; Rothschild, M.F. A whole-genome association study for pig reproductive traits. Anim. Genet. 2012, 43, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Tan, Q.; Kir, J.; Liu, D.; Bryant, D.; Guo, Y.; Stephens, R.; Baseler, M.W.; Lane, H.C.; et al. DAVID Bioinformatics Resources: Expanded annotation database and novel algorithms to better extract biology from large gene lists. Nucleic Acids Res. 2007, 35, W169–W175. [Google Scholar] [CrossRef]
- Chiang, K.-M.; Yang, H.-C.; Pan, W.-H. A Two-Stage Whole-Genome Gene Expression Association Study of Young-Onset Hypertension in Han Chinese Population of Taiwan. Sci. Rep. 2018, 8, 1800. [Google Scholar] [CrossRef] [PubMed]
- Davidson, S.; Shanley, L.; Cowie, P.; Lear, M.; McGuffin, P.; Quinn, J.P.; Barrett, P.; MacKenzie, A. Analysis of the effects of depression associated polymorphisms on the activity of the BICC1 promoter in amygdala neurones. Pharm. J. 2016, 16, 366–374. [Google Scholar] [CrossRef]
- Rothé, B.; Leal-Esteban, L.; Bernet, F.; Urfer, S.; Doerr, N.; Weimbs, T.; Iwaszkiewicz, J.; Constam, D.B. Bicc1 Polymerization Regulates the Localization and Silencing of Bound mRNA. Mol. Cell. Biol. 2015, 35, 3339–3353. [Google Scholar] [CrossRef] [Green Version]
- Muráni, E.; Murániová, M.; Ponsuksili, S.; Schellander, K.; Wimmers, K. Identification of genes differentially expressed during prenatal development of skeletal muscle in two pig breeds differing in muscularity. BMC Dev. Biol. 2007, 7, 109. [Google Scholar] [CrossRef] [PubMed]
- Hirai, H.; Maru, Y.; Hagiwara, K.; Nishida, J.; Takaku, F. A novel putative tyrosine kinase receptor encoded by the eph gene. Science 1987, 238, 1717–1720. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.U.; Chen, Z.-F.; Anderson, D.J. Molecular Distinction and Angiogenic Interaction between Embryonic Arteries and Veins Revealed by ephrin-B2 and Its Receptor Eph-B4. Cell 1998, 93, 741–753. [Google Scholar] [CrossRef] [Green Version]
- Himanen, J.-P.; Henkemeyer, M.; Nikolov, D.B. Crystal structure of the ligand-binding domain of the receptor tyrosine kinase EphB2. Nature 1998, 396, 486–491. [Google Scholar] [CrossRef] [PubMed]
- Alonso-Martin, S.; Rochat, A.; Mademtzoglou, D.; Morais, J.; de Reyniès, A.; Auradé, F.; Chang, T.H.-T.; Zammit, P.S.; Relaix, F. Gene Expression Profiling of Muscle Stem Cells Identifies Novel Regulators of Postnatal Myogenesis. Front. Cell Dev. Biol. 2016, 4, 58. [Google Scholar] [CrossRef] [PubMed]
- Hackett, T.A.; Guo, Y.; Clause, A.; Hackett, N.J.; Garbett, K.; Zhang, P.; Polley, D.B.; Mirnics, K. Transcriptional maturation of the mouse auditory forebrain. BMC Genom. 2015, 16, 606. [Google Scholar] [CrossRef] [PubMed]
- Padilla, J.; Jenkins, N.T.; Thorne, P.K.; Martin, J.S.; Rector, R.S.; Davis, J.W.; Laughlin, M.H. Transcriptome-wide RNA sequencing analysis of rat skeletal muscle feed arteries. II. Impact of exercise training in obesity. J. Appl. Physiol. 2014, 116, 1033–1047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seabury, C.M.; Oldeschulte, D.L.; Saatchi, M.; Beever, J.E.; Decker, J.E.; Halley, Y.A.; Bhattarai, E.K.; Molaei, M.; Freetly, H.C.; Hansen, S.L.; et al. Genome-wide association study for feed efficiency and growth traits in U.S. beef cattle. BMC Genom. 2017, 18, 386. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, T.F.; Quintanilla, R.; Castelló, A.; González-Prendes, R.; Amills, M.; Cánovas, Á. Differential expression of mRNA isoforms in the skeletal muscle of pigs with distinct growth and fatness profiles. BMC Genom. 2018, 19, 145. [Google Scholar] [CrossRef]
- Chen, Z.; Ye, S.; Teng, J.; Diao, S.; Yuan, X.; Chen, Z.; Zhang, H.; Li, J.; Zhang, Z. Genome-wide association studies for the number of animals born alive and dead in duroc pigs. Theriogenology 2019, 139, 36–42. [Google Scholar] [CrossRef]
- Wang, Y.; Ding, X.; Tan, Z.; Xing, K.; Yang, T.; Pan, Y.; Wang, Y.; Mi, S.; Sun, D.; Wang, C. Genome-wide association study for reproductive traits in a Large White pig population. Anim. Genet. 2018, 49, 127–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, J.; Jorjani, H.; Carlborg, Ö. A genome-wide association study using international breeding-evaluation data identifies major loci affecting production traits and stature in the Brown Swiss cattle breed. BMC Genet. 2012, 13, 82. [Google Scholar] [CrossRef] [PubMed]
- Nayeri, S.; Sargolzaei, M.; Abo-Ismail, M.K.; May, N.; Miller, S.P.; Schenkel, F.; Moore, S.S.; Stothard, P. Genome-wide association for milk production and female fertility traits in Canadian dairy Holstein cattle. BMC Genet. 2016, 17, 75. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Kang, J.H.; Kim, J.M. Bayes Factor-Based Regulatory Gene Network Analysis of Genome-Wide Association Study of Economic Traits in a Purebred Swine Population. Genes 2019, 10, 293. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Trait a | Mean | SD | N | Mean Reliability b | h2 |
|---|---|---|---|---|---|
| LSB | −0.015 | 0.526 | 1067 | 0.435 | 0.158 |
| LWB | −0.061 | 0.099 | 1067 | 0.421 | 0.161 |
| LWW | 0.062 | 1.147 | 1067 | 0.511 | 0.173 |
| LSW | 1.604 | 6.454 | 1067 | 0.389 | 0.140 |
| Trait | LSB | LWB | LWW | LSW |
|---|---|---|---|---|
| LSB | ||||
| LWB | 0.835 ± 0.037 | |||
| LWW | 0.302 ± 0.127 | 0.500 ± 0.110 | ||
| LSW | 0.509 ± 0.119 | 0.551 ± 0.115 | 0.880 ± 0.038 |
| Traits | SNP | Chromosome | Position | p-Value | Allele Frequency | Allele Substitution Effect | Candidate Gene b |
|---|---|---|---|---|---|---|---|
| LSB | rs336638152 | 5 | 9006723 | 9.09 × 10−5 | 0.398 | −0.117 | Thioredoxin 2 (TXN2) |
| rs80999110 | 5 | 67782650 | 6.73 × 10−5 | 0.438 | −0.118 | Potassium voltage-gated channel subfamily A member 1 (KCNA1) | |
| rs81318862 | 6 | 76074229 | 8.29 × 10−5 | 0.109 | −0.187 | ENSSSCG00000003546 | |
| rs329711941 | 6 | 77726418 | 8.28 × 10−5 | 0.107 | −0.187 | Zinc finger DHHC-type containing 18 (ZDHHC18) | |
| rs81439394 | 12 | 10118697 | 2.53 × 10−5 | 0.290 | −0.141 | Mitogen-activated protein kinase 6 (MAP2K6) | |
| rs80979042 | 14 | 66823174 | 1.69 × 10−5 | 0.479 | −0.140 | BICC1 (BICC1) | |
| rs80825112 | 14 | 66854605 | 1.69 × 10−5 | 0.479 | −0.140 | BICC1 | |
| LWB | rs325089329 | 4 | 5662747 | 1.06× 10−4 | 0.155 | −0.345 | Family with sequence similarity 135 member B (FAM135B) |
| rs329734169 | 4 | 5677434 | 1.06× 10−4 | 0.155 | −0.345 | FAM135B | |
| rs81476258 | 6 | 68258924 | 5.85 × 10−5 | 0.407 | 0.276 | EPH receptor B2 (EPHB2) | |
| rs81326131 | 6 | 74780466 | 5.85 × 10−5 | 0.407 | 0.276 | EPHB2 | |
| rs332491771 | 14 | 1176591 | 3.18 × 10−5 | 0.413 | 0.251 | Semaphorin 4D (SEMA4D) | |
| LWW | rs80808642 | 4 | 7289023 | 8.01 × 10−6 | 0.480 | −1.629 | ST3 beta-galactoside alpha-2,3-sialyltransferase 1 (ST3GAL1) |
| rs81427863 | 10 | 7002238 | 5.98 × 10−5 | 0.215 | −1.718 | Potassium channel tetramerization domain containing 3 (KCTD3) | |
| rs322567083 | 10 | 7029718 | 5.98 × 10−5 | 0.215 | −1.718 | KCTD3 | |
| rs81428034 | 10 | 7059506 | 5.98 × 10−5 | 0.215 | −1.718 | KCTD3 | |
| rs328230332 | 17 | 39032680 | 7.50 × 10−5 | 0.100 | 2.338 | Family with sequence similarity 110 member A (FAM110A) | |
| LSW | rs339777110 | 14 | 27027253 | 3.12 × 10−5 | 0.168 | 0.032 | Transmembrane protein 132D (TMEM132D) |
| rs80947288 | 14 | 39275817 | 8.41 × 10−5 | 0.259 | 0.025 | T-box 3 (TBX3) | |
| rs328230332 | 17 | 39032680 | 5.12 × 10−5 | 0.100 | 0.038 | FAM110A |
| Trait | Biological Process GO Terms |
|---|---|
| LSB | GO: 0061436, establishment of skin barrier; GO: 0045606, positive regulation of epidermal cell differentiation; GO: 0010482, regulation of epidermal cell division |
| LWB | GO: 0042742, defense response to bacterium; GO: 0045087, innate immune response |
| LWW | GO: 0061436, establishment of skin barrier; GO: 0043552, positive regulation of phosphatidylinositol 3-kinase activity; GO: 0034644, cellular response to UV |
| LSW | GO: 0042742, defense response to bacterium; GO: 0045087, innate immune response; GO: 0035115, embryonic forelimb morphogenesis |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Z.; Chen, Z.; Ye, S.; He, Y.; Huang, S.; Yuan, X.; Chen, Z.; Zhang, H.; Li, J. Genome-Wide Association Study for Reproductive Traits in a Duroc Pig Population. Animals 2019, 9, 732. https://doi.org/10.3390/ani9100732
Zhang Z, Chen Z, Ye S, He Y, Huang S, Yuan X, Chen Z, Zhang H, Li J. Genome-Wide Association Study for Reproductive Traits in a Duroc Pig Population. Animals. 2019; 9(10):732. https://doi.org/10.3390/ani9100732
Chicago/Turabian StyleZhang, Zhe, Zitao Chen, Shaopan Ye, Yingting He, Shuwen Huang, Xiaolong Yuan, Zanmou Chen, Hao Zhang, and Jiaqi Li. 2019. "Genome-Wide Association Study for Reproductive Traits in a Duroc Pig Population" Animals 9, no. 10: 732. https://doi.org/10.3390/ani9100732