

Prevalence and Molecular Characterization of Bovine Parainfluenza Virus Type 3 in Cattle Herds in China

Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. RNA Extraction and cDNA Synthesis
2.3. Detection of BPIV3
2.4. Amplification of HN and Genome Sequences
2.5. Sequence, Phylogenetic, and Recombination Analysis
3. Results
3.1. Detection of BPIV3
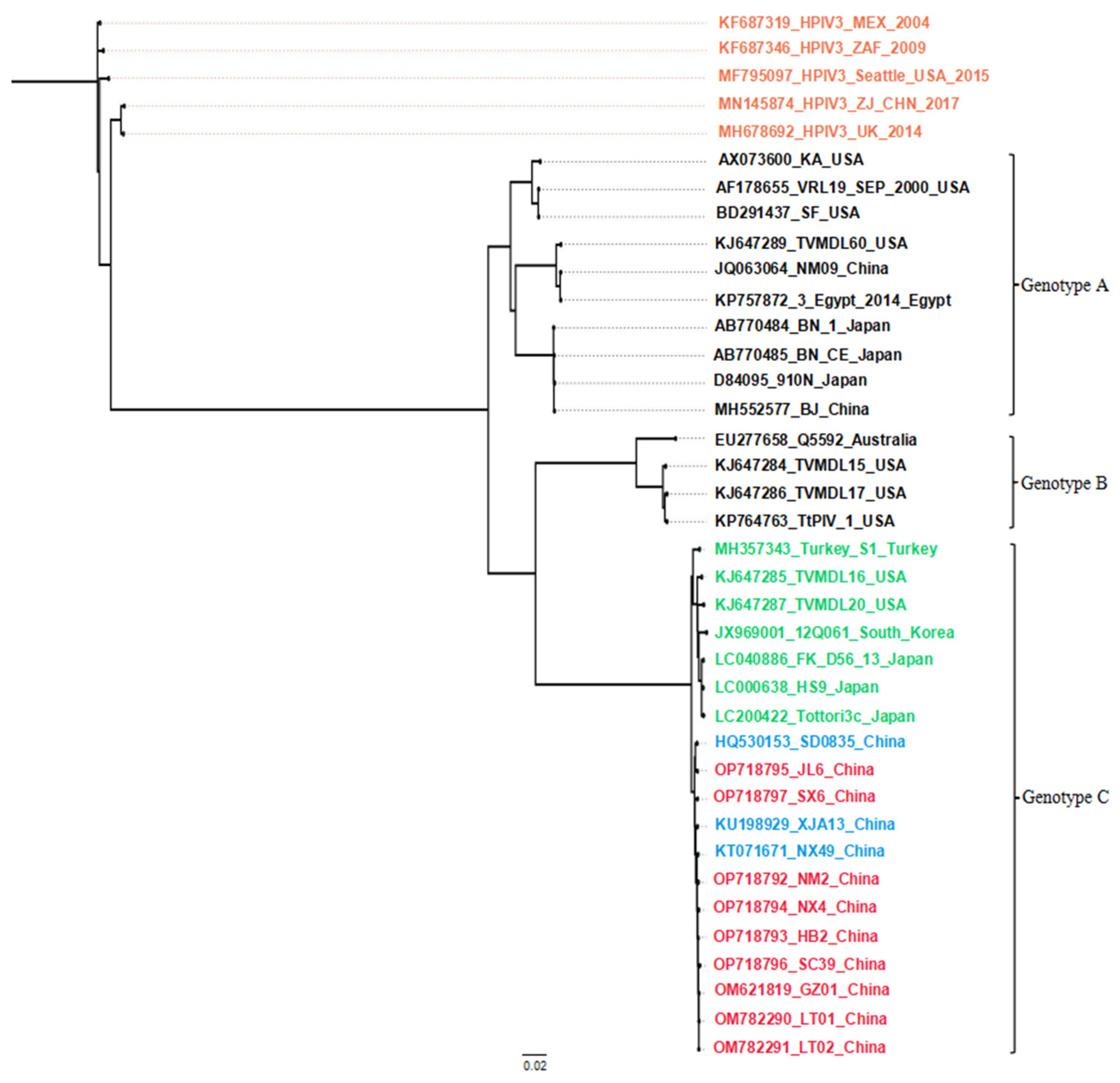
3.2. Bioinformatics Analysis of HN Gene Sequences
3.3. Bioinformatics Analysis of the Complete Genomes
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Buchmeier, M.; Clegg, J.; Franze-Fernandez, M. Virus taxonomy: Sixth Report of the International Committee on Taxonomy of Viruses. Arch. Virol. 1995, 10, 350–354. [Google Scholar]
- Miles, D.G. Overview of the North American beef cattle industry and the incidence of bovine respiratory disease (BRD). Anim. Health Res. Rev. 2009, 10, 101–103. [Google Scholar] [CrossRef] [PubMed]
- Ellis, J.A. Bovine parainfluenza-3 virus. Vet. Clin. N. Am. Food Anim. Pract. 2010, 26, 575–593. [Google Scholar] [CrossRef]
- Zhu, Y.M.; Shi, H.F.; Gao, Y.R.; Xin, J.Q.; Liu, N.H.; Xiang, W.H.; Ren, X.G.; Feng, J.K.; Zhao, L.P.; Xue, F. Isolation and genetic characterization of bovine parainfluenza virus type 3 from cattle in China. Vet. Microbiol. 2011, 149, 446–451. [Google Scholar] [CrossRef]
- Suzu, S.; Sakai, Y.; Shioda, T.; Shibuta, H. Nucleotide sequence of the bovine parainfluenza 3 virus genome: The genes of the F and HN glycoproteins. Nucleic Acids Res. 1987, 15, 2945–2958. [Google Scholar] [CrossRef] [Green Version]
- Sakai, Y.; Suzu, S.; Shioda, T.; Shibuta, H. Nucleotide sequence of the bovine parainfluenza 3 virus genome: Its 3’ end and the genes of NP, P, C and M proteins. Nucleic Acids Res. 1987, 15, 2927–2944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oem, J.K.; Lee, E.Y.; Lee, K.K.; Kim, S.H.; Lee, M.H.; Hyun, B.H. Molecular characterization of a Korean bovine parainfluenza virus type 3 isolate. Vet. Microbiol. 2013, 162, 224–227. [Google Scholar] [CrossRef]
- Konishi, M.; Ohkura, T.; Shimizu, M.; Akiyama, M.; Kameyama, K.I.; Takeuchi, K. Complete Genome Sequence of the First Isolate of Genotype C Bovine Parainfluenza Virus Type 3 in Japan. Genome Announc. 2014, 2, e01215-01214. [Google Scholar] [CrossRef] [Green Version]
- Neill, J.D.; Ridpath, J.F.; Valayudhan, B.T. Identification and genome characterization of genotype B and genotype C bovine parainfluenza type 3 viruses isolated in the United States. BMC Vet. Res. 2015, 11, 112. [Google Scholar] [CrossRef] [Green Version]
- Albayrak, H.; Yazici, Z.; Ozan, E.; Tamer, C.; Wahed, A.; Wehner, S.; Ulrich, K.; Weidmann, M. Characterisation of the First Bovine Parainfluenza Virus 3 Isolate Detected in Cattle in Turkey. Vet. Sci. 2019, 6, 56. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Hu, J.; Meng, F.; Cao, Y.; Wang, Z.; Zhang, Q.; Zhang, Q.; Zhang, X.; Han, M.; Wu, T.; et al. Isolation, Identification, and Genetic Phylogenetic Analysis of Two Different Genotypes of Bovine Parainfluenza 3 Virus in China. Viruses 2022, 14, 2221. [Google Scholar] [CrossRef]
- Guo, T.; Zhang, J.; Chen, X.; Wei, X.; Wu, C.; Cui, Q.; Hao, Y. Investigation of viral pathogens in cattle with bovine respiratory disease complex in Inner Mongolia, China. Microb. Pathog. 2021, 153, 104594. [Google Scholar] [CrossRef]
- Kumar, S.; Tamura, K.; Nei, M. MEGA3: Integrated software for Molecular Evolutionary Genetics Analysis and sequence alignment. Brief Bioinform 2004, 5, 150–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, vev003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Wang, H.; Hou, P.; Xia, X.; He, H. A lateral flow dipstick combined with reverse transcription recombinase polymerase amplification for rapid and visual detection of the bovine respirovirus 3. Mol. Cell. Probes 2018, 41, 22–26. [Google Scholar] [CrossRef]
- Ren, Y.; Chen, X.; Tang, C.; Yue, H. First Isolation and Characteristics of Bovine Parainfluenza Virus Type 3 from Yaks. Pathogens 2022, 11, 962. [Google Scholar] [CrossRef]
- Cui, P.; Feng, L.; Zhang, L.; He, J.; An, T.; Fu, X.; Li, C.; Zhao, X.; Zhai, Y.; Li, H.; et al. Antimicrobial Resistance, Virulence Genes, and Biofilm Formation Capacity Among Enterococcus species From Yaks in Aba Tibetan Autonomous Prefecture, China. Front. Microbiol. 2020, 11, 1250. [Google Scholar] [CrossRef]
- Mi, R.; Wang, X.; Li, C.; Huang, Y.; Zhou, P.; Li, Z.; Lei, M.; Cai, J.; Chen, Z. Prevalence and genetic characterization of Cryptosporidium in yaks in Qinghai Province of China. PLoS ONE 2013, 8, e74985. [Google Scholar] [CrossRef] [Green Version]
- He, Q.; Guo, Z.; Zhang, B.; Yue, H.; Tang, C. First detection of bovine coronavirus in Yak (Bos grunniens) and a bovine coronavirus genome with a recombinant HE gene. J. Gen. Virol. 2019, 100, 793–803. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, F.X.; Sun, N.; Cao, L.; Zhang, S.Q.; Zhu, H.W.; Guo, L.; Cheng, S.P.; Wen, Y.J. Development and evaluation of two truncated recombinant NP antigen-based indirect ELISAs for detection of bovine parainfluenza virus type 3 antibodies in cattle. J. Virol. Methods 2015, 222, 47–54. [Google Scholar] [CrossRef]
- Zvirbliene, A.; Sezaite, I.; Pleckaityte, M.; Kucinskaite-Kodze, I.; Juozapaitis, M.; Sasnauskas, K. Mapping of an antigenic site on the nucleocapsid protein of human parainfluenza virus type 3. Viral Immunol. 2009, 22, 181–188. [Google Scholar] [CrossRef]
- Heminway, B.; Yu, Y.; Galinski, M. Paramyxovirus mediated cell fusion requires co-expression of both the fusion and hemagglutinin-neuraminidase glycoproteins. Virus Res. 1994, 31, 1–16. [Google Scholar] [CrossRef]
- Dutch, R.E.; Hagglund, R.N.; Nagel, M.A.; Paterson, R.G.; Lamb, R.A. Paramyxovirus fusion (F) protein: A conformational change on cleavage activation. Virology 2001, 281, 138–150. [Google Scholar] [CrossRef]
- Liu, Y.; Xie, W.; Chi, M.; Wen, H.; Zhao, L.; Song, Y.; Liu, N.; Chi, L.; Wang, Z. Mutations in the HRB linker of human parainfluenza virus type 3 fusion protein reveal its importance for fusion activity. Virus Res. 2020, 275, 197791. [Google Scholar] [CrossRef]
- Yin, H.; Paterson, R.; Wen, X.; Lamb, R.; Jardetzky, T. Structure of the uncleaved ectodomain of the paramyxovirus (hPIV3) fusion protein. Proc. Natl. Acad. Sci. USA 2005, 102, 9288–9293. [Google Scholar] [CrossRef] [Green Version]
- Ji, Y.; Liu, T.; Jia, Y.; Liu, B.; Yu, Q.; Cui, X.; Guo, F.; Chang, H.; Zhu, Q. Two single mutations in the fusion protein of Newcastle disease virus confer hemagglutinin-neuraminidase independent fusion promotion and attenuate the pathogenicity in chickens. Virology 2017, 509, 146–151. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, Y.; Huang, Y.; Wen, H.; Zhao, L.; Song, Y.; Wang, Z. The effect of the HRB linker of Newcastle disease virus fusion protein on the fusogenic activity. J. Microbiol. 2021, 59, 513–521. [Google Scholar] [CrossRef]
- Bousse, T.L.; Taylor, G.; Krishnamurthy, S.; Portner, A.; Samal, S.K.; Takimoto, T. Biological significance of the second receptor binding site of Newcastle disease virus hemagglutinin-neuraminidase protein. J. Virol. 2004, 78, 13351–13355. [Google Scholar] [CrossRef] [Green Version]
- Shibuta, H.; Suzu, S.; Shioda, T. Differences in bovine parainfluenza 3 virus variants studied by monoclonal antibodies against viral glycoproteins. Virology 1986, 155, 688–696. [Google Scholar] [CrossRef]
- Khattar, S.K.; Yan, Y.; Panda, A.; Collins, P.L.; Samal, S.K. A Y526Q mutation in the Newcastle disease virus HN protein reduces its functional activities and attenuates virus replication and pathogenicity. J. Virol. 2009, 83, 7779–7782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Jia, Y.; Wei, N.; Ye, C.; Hao, H.; Xiao, S.; Wang, X.; Liu, H.; Yang, Z. Identification of a new amino acid mutation in the HN protein of NDV involved in pathogenicity. Vet. Res. 2021, 52, 147. [Google Scholar] [CrossRef] [PubMed]
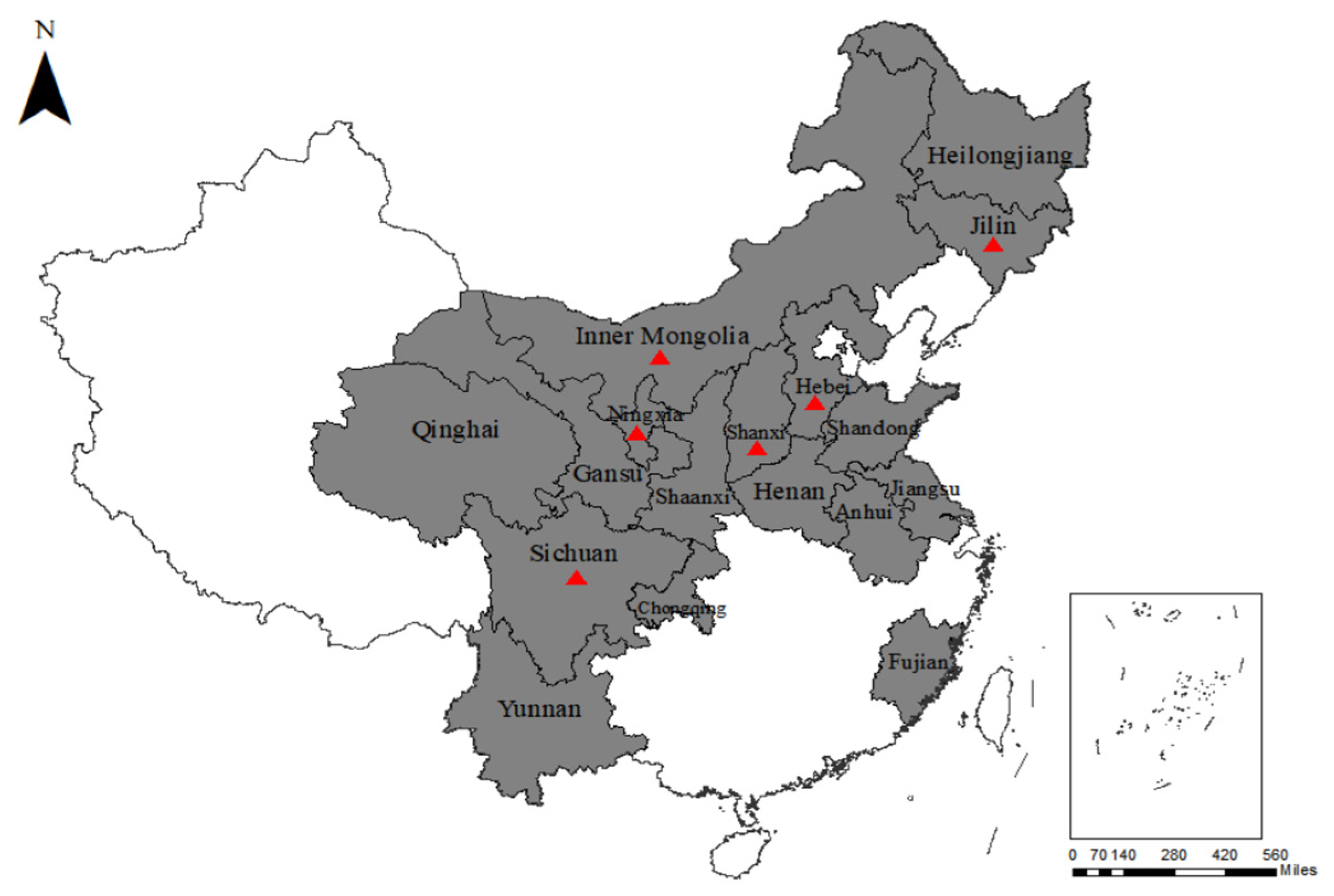
 ) represent the sampling areas that tested positive for BPIV3.
) represent the sampling areas that tested positive for BPIV3.
) represent the sampling areas that tested positive for BPIV3.
) represent the sampling areas that tested positive for BPIV3.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Region | Sample Source | Sample Type | Number of Farms | Number of Samples | Positive Rate (%) |
|---|---|---|---|---|---|
| Sichuan | Beef cattle | Nasal swab | 10 | 173 | 34.68% (60/173) |
| Fujian | Beef cattle | Nasal swab | 1 | 10 | 0.00% (0/10) |
| Ningxia | Beef cattle | Nasal swab | 2 | 30 | 13.33% (4/30) |
| Inner Mongolia | Beef cattle | Nasal swab | 5 | 58 | 27.59% (16/58) |
| Jiangsu | Beef cattle | Nasal swab | 1 | 15 | 0.00% (0/15) |
| Henan | Beef cattle | Nasal swab | 4 | 40 | 0.00% (0/40) |
| Hebei | Beef cattle | Nasal swab | 4 | 61 | 19.67% (12/61) |
| Shanxi | Beef cattle | Nasal swab | 4 | 52 | 23.08% (12/52) |
| Chongqing | Beef cattle | Nasal swab | 4 | 83 | 0.00% (0/83) |
| Shandong | Beef cattle | Nasal swab | 1 | 30 | 0.00% (0/30) |
| Jilin | Beef cattle | Nasal swab | 1 | 10 | 30.00% (3/10) |
| Qinghai | Beef cattle | Nasal swab | 2 | 16 | 0.00% (0/16) |
| Anhui | Beef cattle | Nasal swab | 1 | 15 | 0.00% (0/15) |
| Yunnan | Beef cattle | Nasal swab | 1 | 10 | 0.00% (0/10) |
| Heilongjiang | Beef cattle | Nasal swab | 1 | 12 | 0.00% (0/12) |
| Shaanxi | Beef cattle | Nasal swab | 1 | 10 | 0.00% (0/10) |
| Gansu | Beef cattle | Nasal swab | 2 | 20 | 0.00% (0/20) |
| Sichuan | Yak | Nasal swab | 7 | 71 | 16.9% (12/71) |
| Qinghai | Yak | Nasal swab | 1 | 10 | 0.00% (0/10) |
| Sichuan | Yak | Lung tissue | 5 | 50 | 44.00% (22/50) |
| Name | Primer Sequence (5′-3′) | Position |
|---|---|---|
| BPIV3-1F | ACCAAACAAGAGGAGAGACTTG | 1–1963 |
| BPIV3-1R | ATTGTTGTGCTGAGCCTTGT | |
| BPIV3-2F | AGACTCCATCCACAACCCA | 1750–3697 |
| BPIV3-2R | CTTGTGTCTGGGAACTACTGTG | |
| BPIV3-3F | TCAAAGGCAAAACAGTCATACAT | 3482–5728 |
| BPIV3-3R | TCCTACTGAGCTTTGAATTGACTGT | |
| BPIV3-4F | AACAGTACTAGTTCCAGGAAGAAGC | 5396–7884 |
| BPIV3-4R | GGACAGCCAGTTAAATTGCATATTAC | |
| BPIV3-5F | CAGTAGGACCGGGGATTTA | 7880–9902 |
| BPIV3-5R | TGTCCTCCGTGTCTTTCTCTA | |
| BPIV3-6F | CCTTTTTCCGAACTTTTGG | 9734–12,509 |
| BPIV3-6R | GTAGTCACTGGTGTCAGAATCTTTA | |
| BPIV3-7F | AGGAGGAAGAATGATAAATGG | 12,105–13,822 |
| BPIV3-7R | TTCTTGGATTATCGTCACAGTTA | |
| BPIV3-8F | GTGTGTTGTTTAGCAGAAATAGC | 13,501–15,474 |
| BPIV3-8R | ACCAAACAAGAGAAAAACTCTGT |
| Nucleotide Position | Nucleotide Mutations | Amino Acid Mutations | ||
|---|---|---|---|---|
| Chinese Strains | Overseas Strains | Chinese Strains | Overseas Strains | |
| 1088 | T | C | - | - |
| 1169 | A | G | - | - |
| 1355 | T | C | - | - |
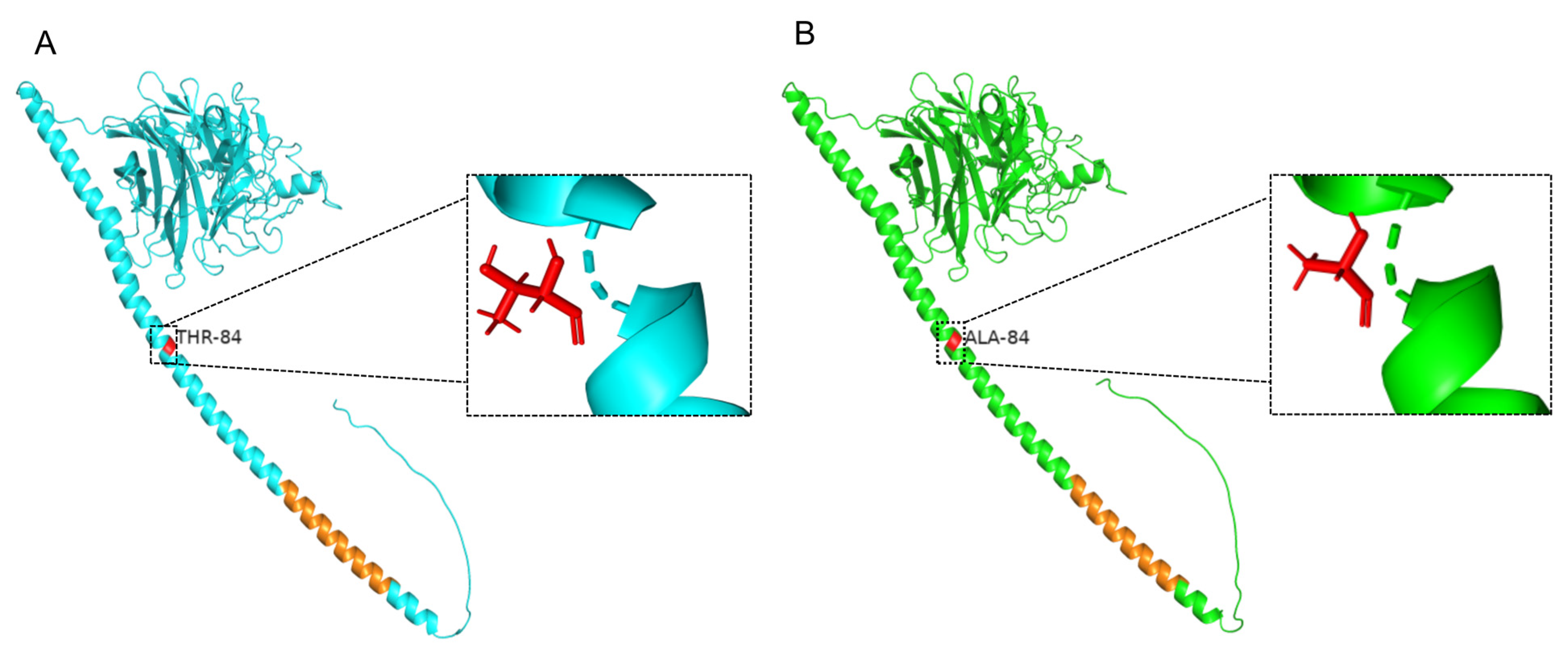
| 1426 | C | T | P | L |
| 1436 | C | T | - | - |
| 1590 | A | G | T | A |
| 1666 | A | G | / | / |
| 3659 | A | C | / | / |
| 4096 | T | C | - | - |
| 4234 | C | T | - | - |
| 4357 | C | T | - | - |
| 4603 | T | C | - | - |
| 4795 | A | G | - | - |
| 4938 | A | G | / | / |
| 4968 | A | C | / | / |
| 5654 | T | C | - | - |
| 6474 | A | G | N | D |
| 6611 | G | A | M | I |
| 6767 | A | G | / | / |
| 7082 | C | T | - | - |
| 7104 | A | G | T | A |
| 7310 | T | C | - | - |
| 7847 | A | G | - | - |
| 7916 | C | T | - | - |
| 7943 | C | T | - | - |
| 7946 | C | T | - | - |
| 7952 | C | T | - | - |
| 7991 | T | C | - | - |
| 8180 | T | C | - | - |
| 8282 | A | G | - | - |
| 8453 | G | A | - | - |
| 8701 | A | C | / | / |
| 9424 | C | T | - | - |
| 9784 | C | T | - | - |
| 10,342 | A | G | - | - |
| 11,815 | T | C | - | - |
| 12,082 | A | G | - | - |
| 12,853 | T | C | - | - |
| 13,853 | C | T | - | - |
| 14,261 | C | T | - | - |
| 14,367 | T | C | - | - |
| 14,607 | C | A | - | - |
| 14,637 | A | G | - | - |
| 14,904 | G | A | - | - |
| 14,961 | A | C | - | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ren, Y.; Tang, C.; Yue, H. Prevalence and Molecular Characterization of Bovine Parainfluenza Virus Type 3 in Cattle Herds in China. Animals 2023, 13, 793. https://doi.org/10.3390/ani13050793
Ren Y, Tang C, Yue H. Prevalence and Molecular Characterization of Bovine Parainfluenza Virus Type 3 in Cattle Herds in China. Animals. 2023; 13(5):793. https://doi.org/10.3390/ani13050793
Chicago/Turabian StyleRen, Yunxin, Cheng Tang, and Hua Yue. 2023. "Prevalence and Molecular Characterization of Bovine Parainfluenza Virus Type 3 in Cattle Herds in China" Animals 13, no. 5: 793. https://doi.org/10.3390/ani13050793