
Integrative Analyses of Antler Cartilage Transcriptome and Proteome of Gansu Red Deer (Cervus elaphus kansuensis) at Different Growth Stages

 ,
,  , , , , and
, , , , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals, Sample Collection, and Preparation
2.2. RNA Extraction and Sequencing
2.3. Protein Extraction, iTRAQ Labeling, and Liquid Chromatography-Electrospray Ionization-Tandem Mass Spectrometry Analysis
2.4. Transcriptome and Proteome Data Analysis
2.5. Gene Functional Annotation of DEGs and DEPs
2.6. Integrated Analysis of Transcriptomic and Proteomic Data
3. Results
3.1. Data Availability
3.2. RNA Sequencing and Assembly
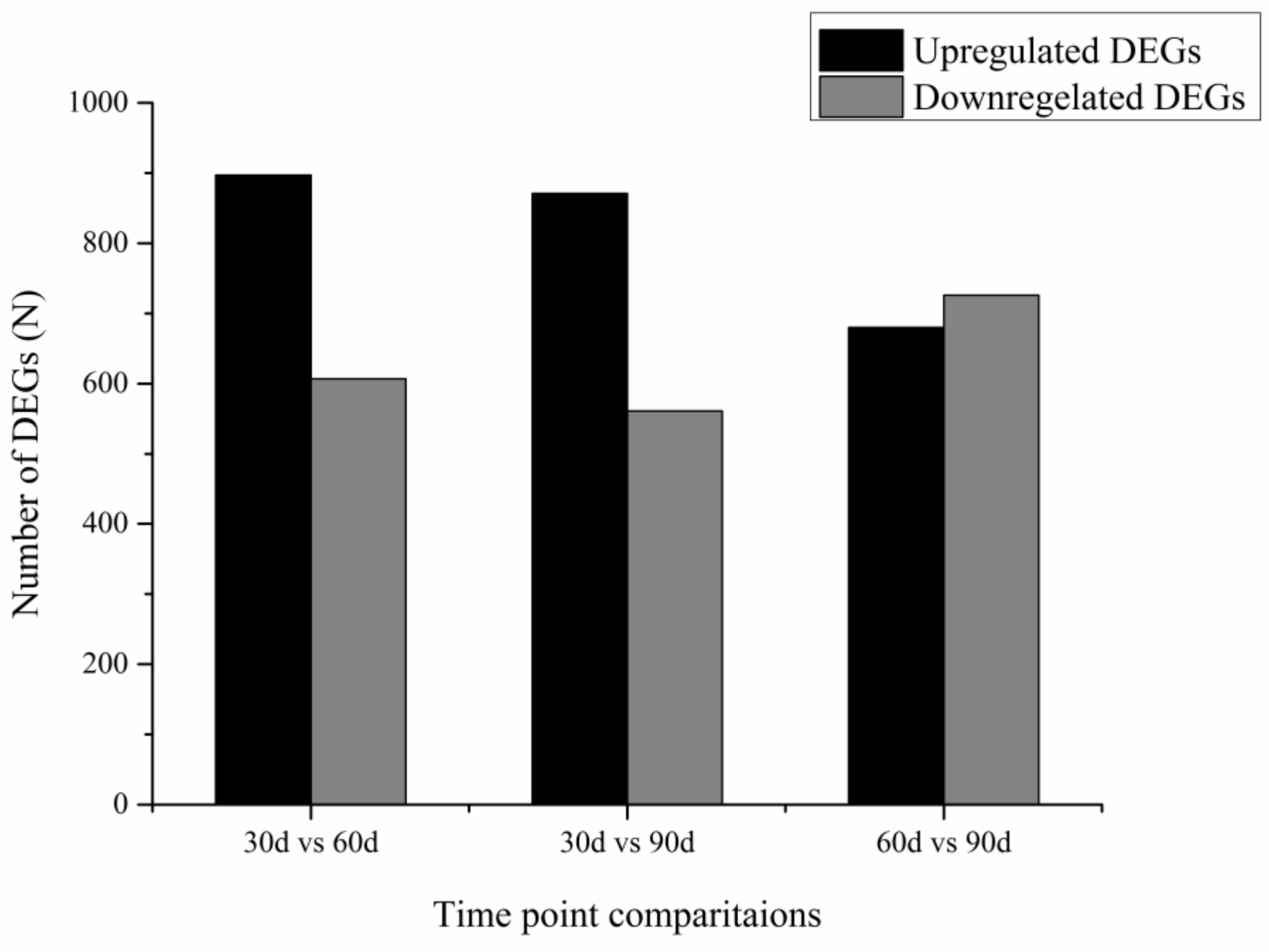
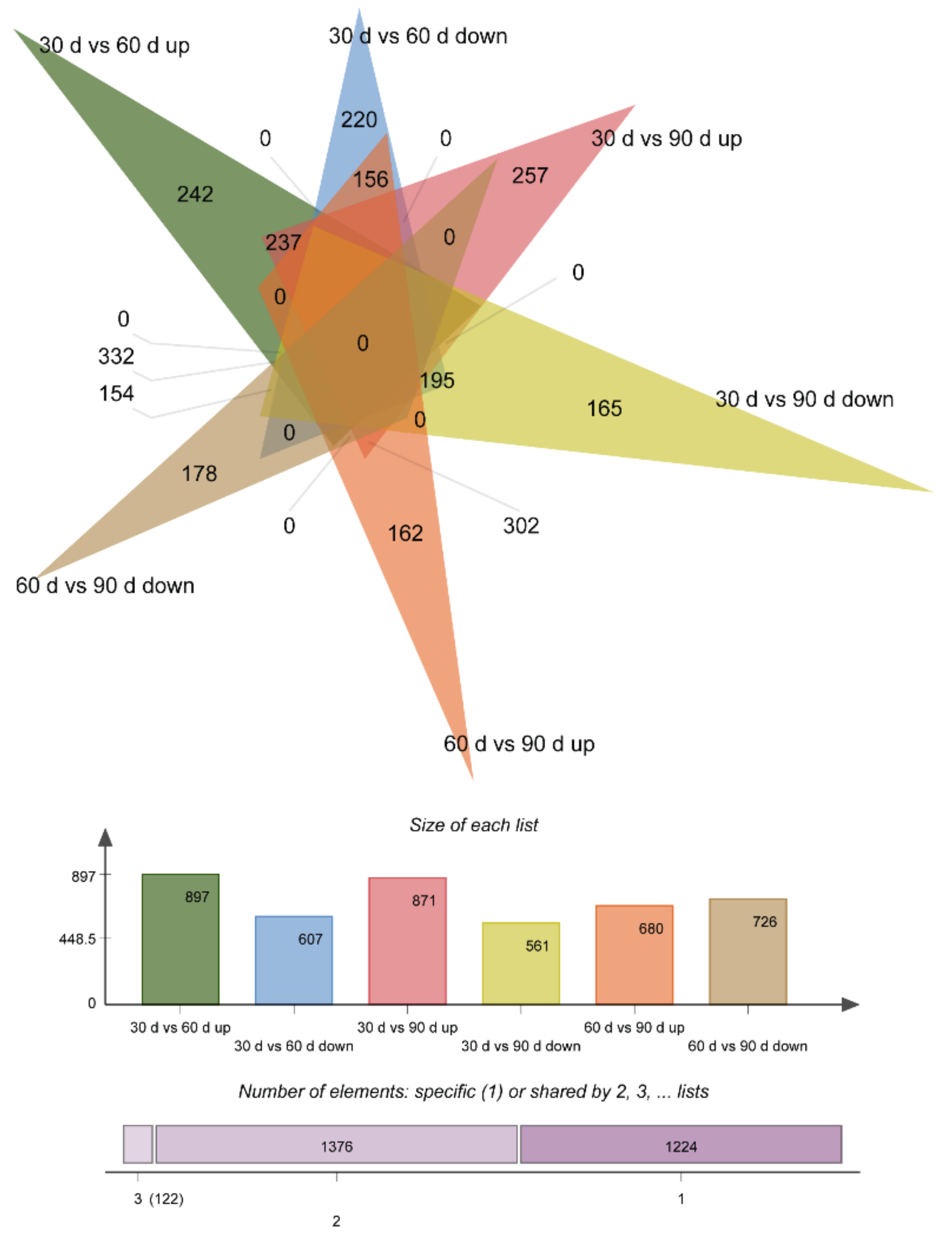
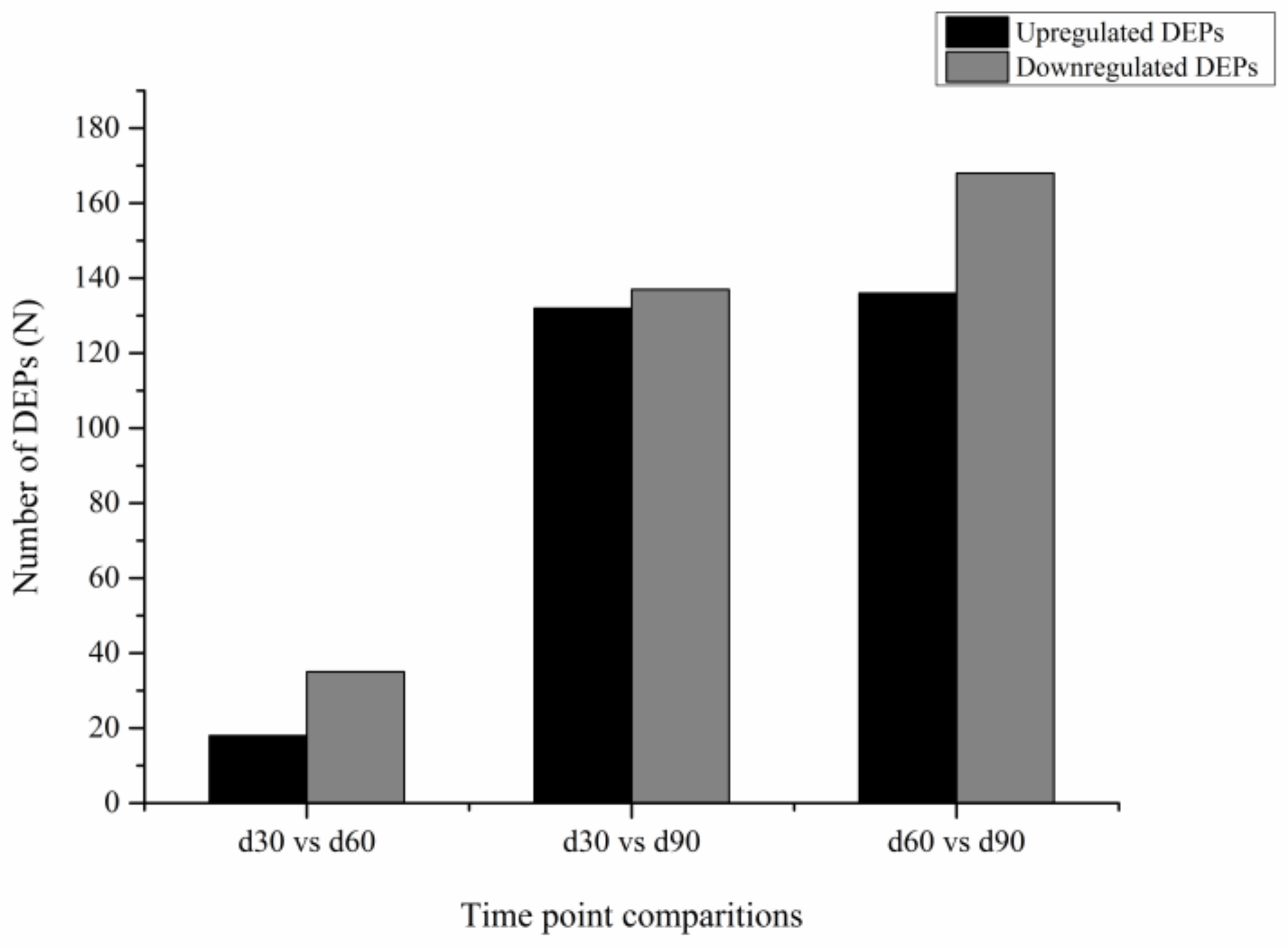
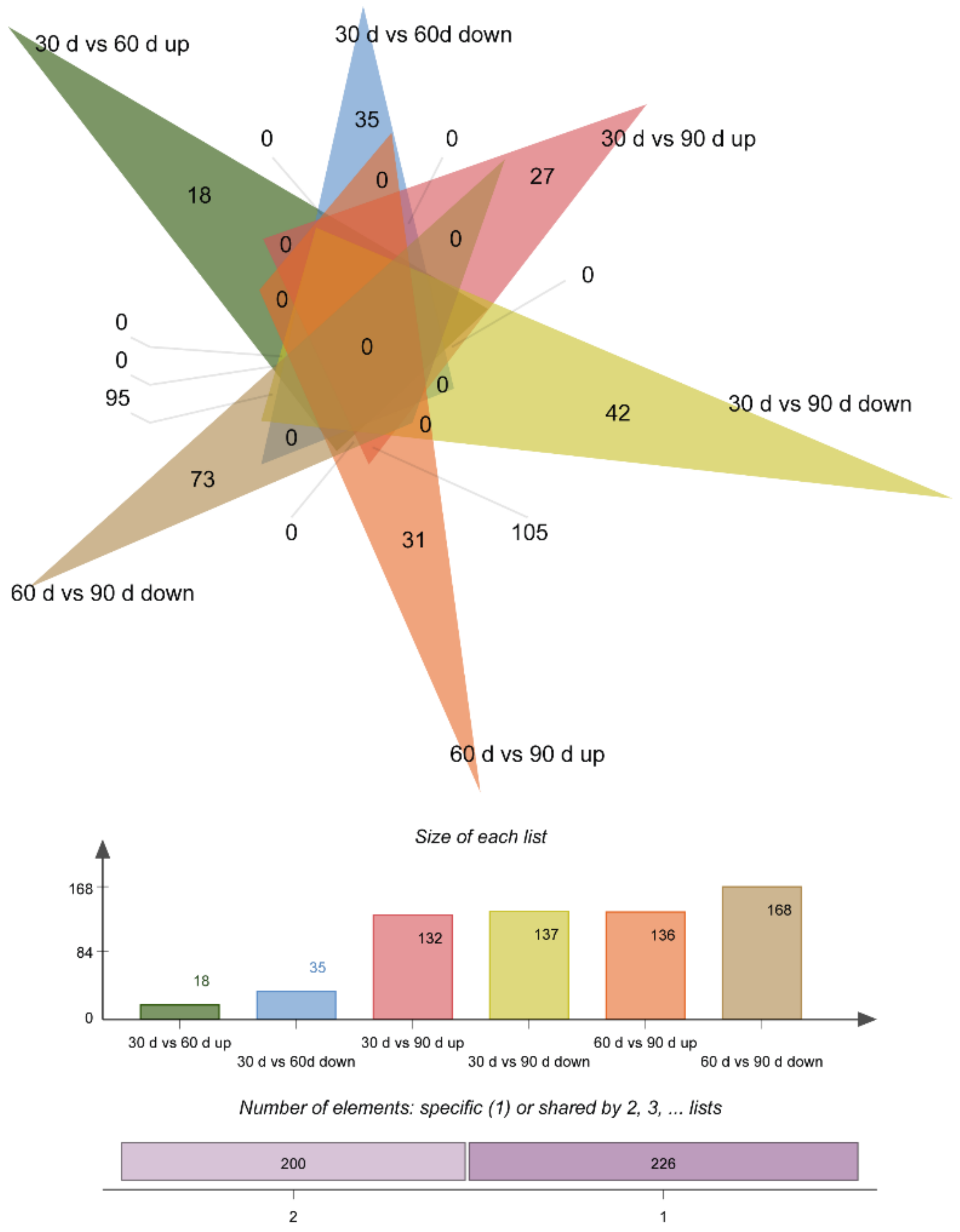
3.3. Differentially Expressed Genes
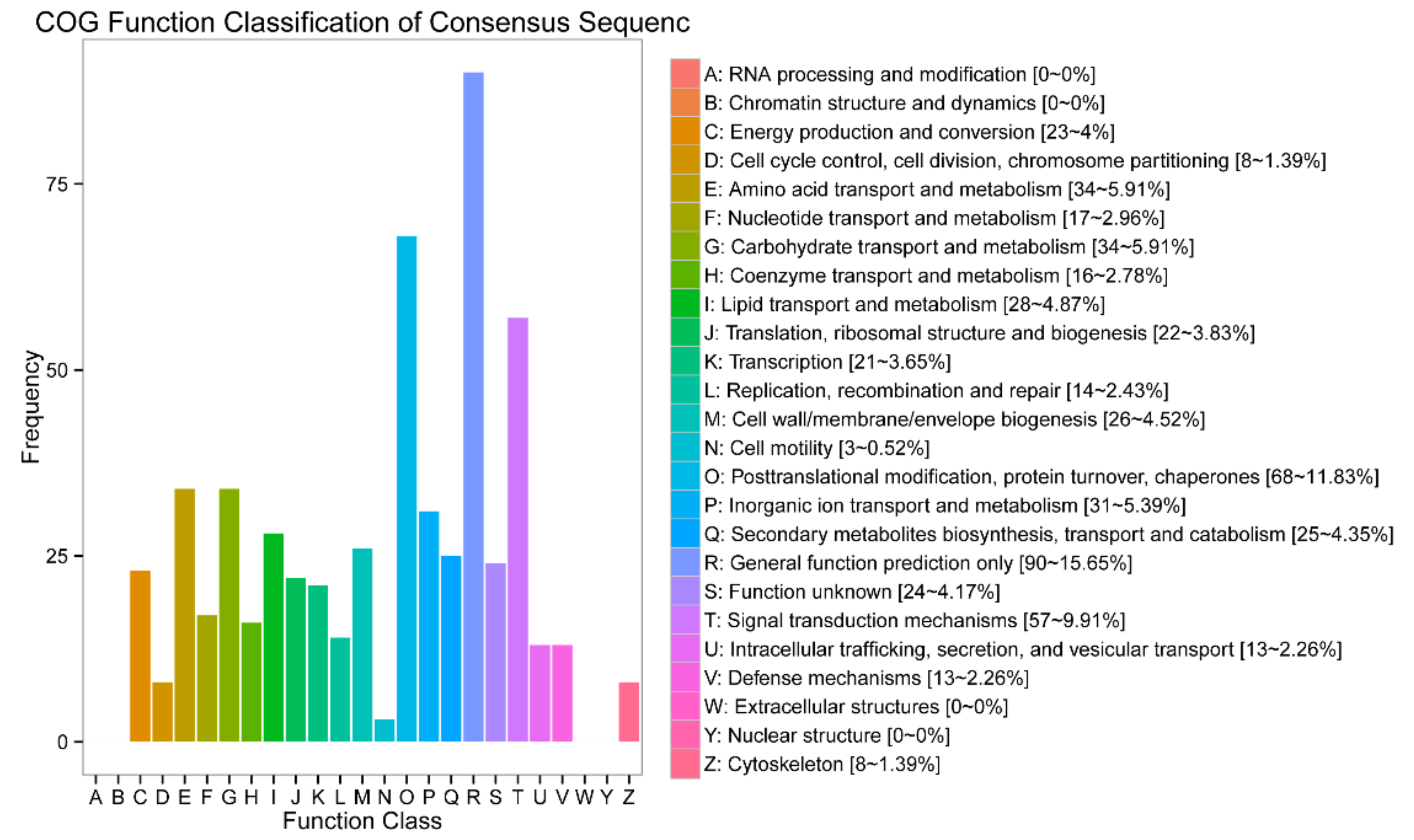
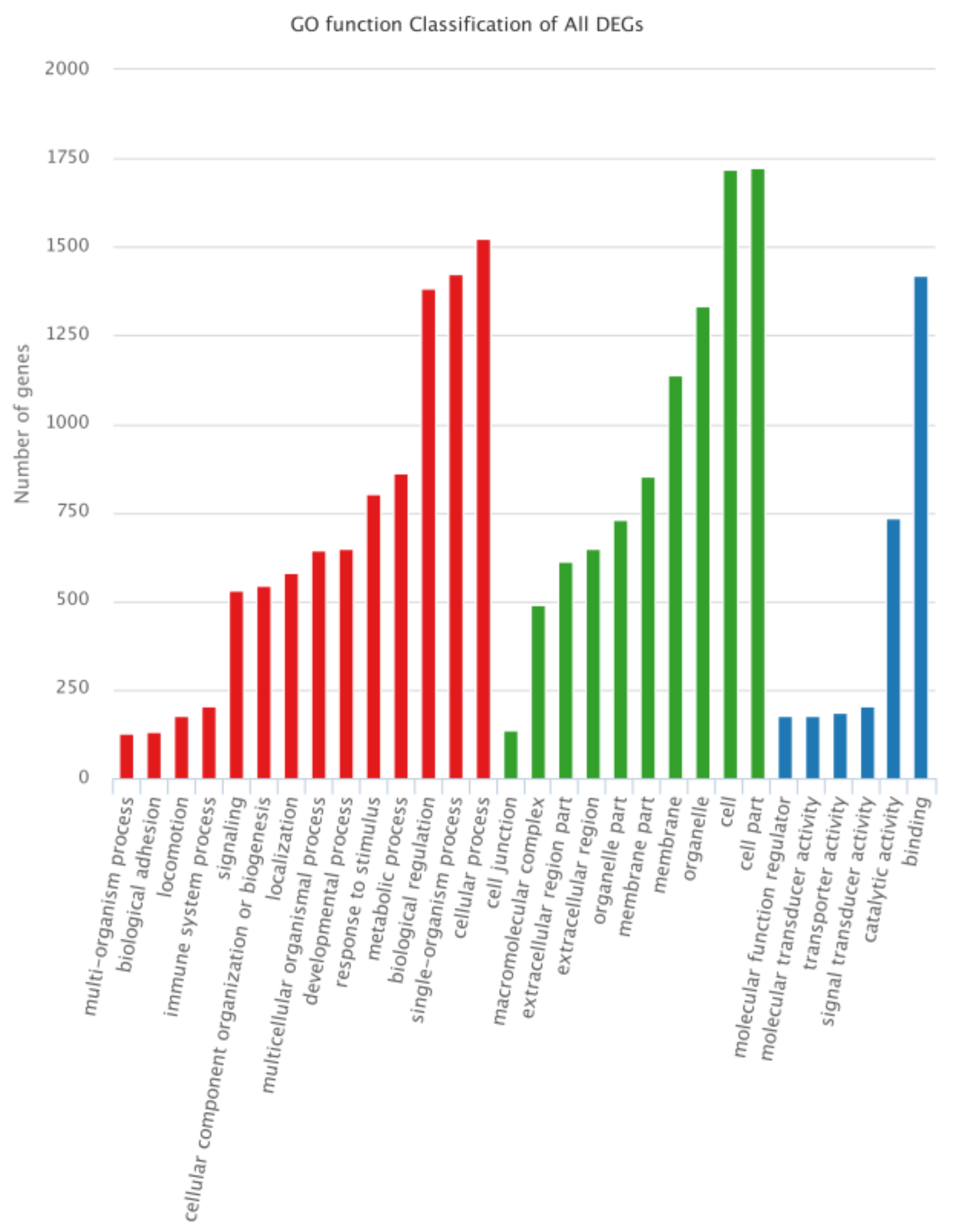
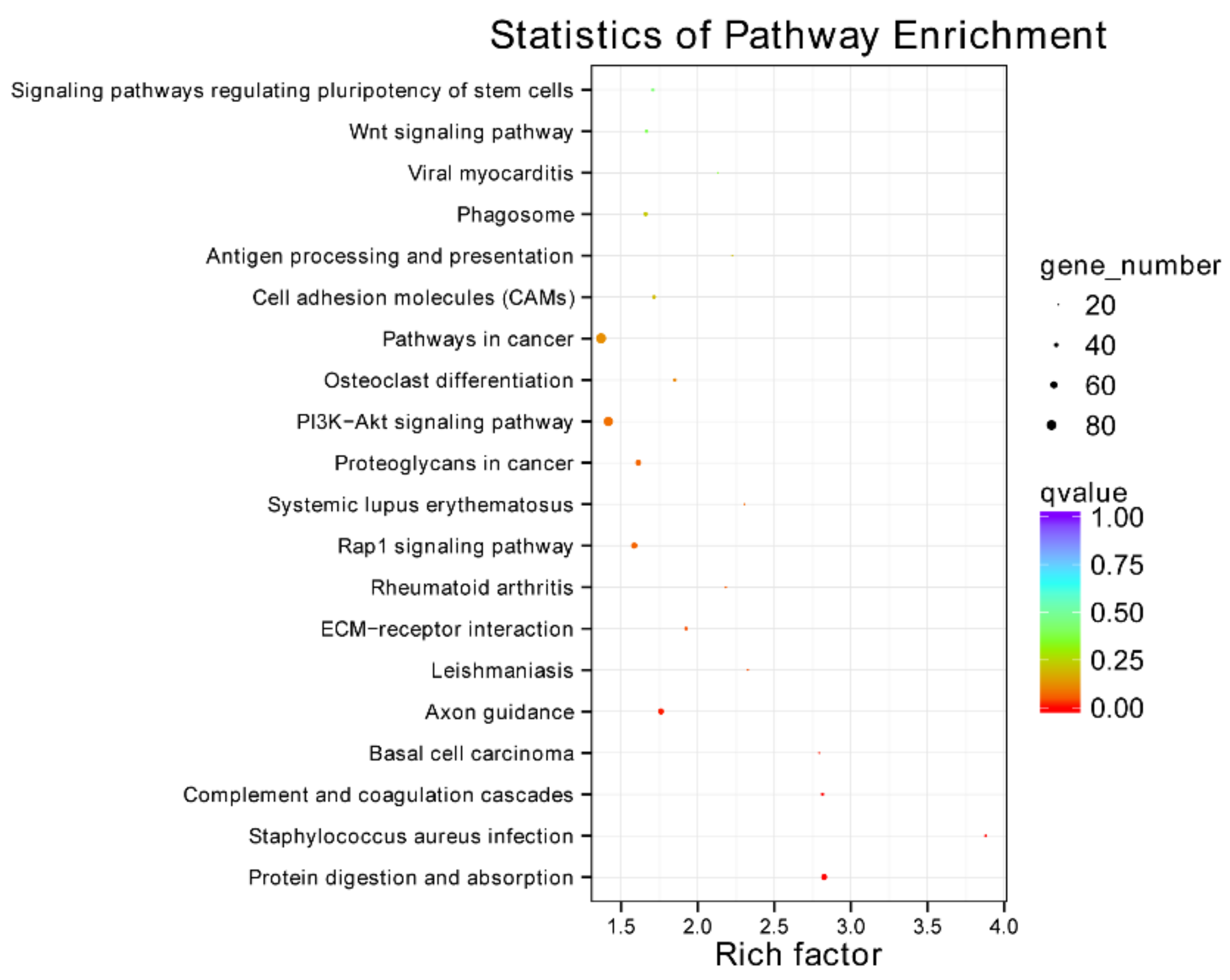
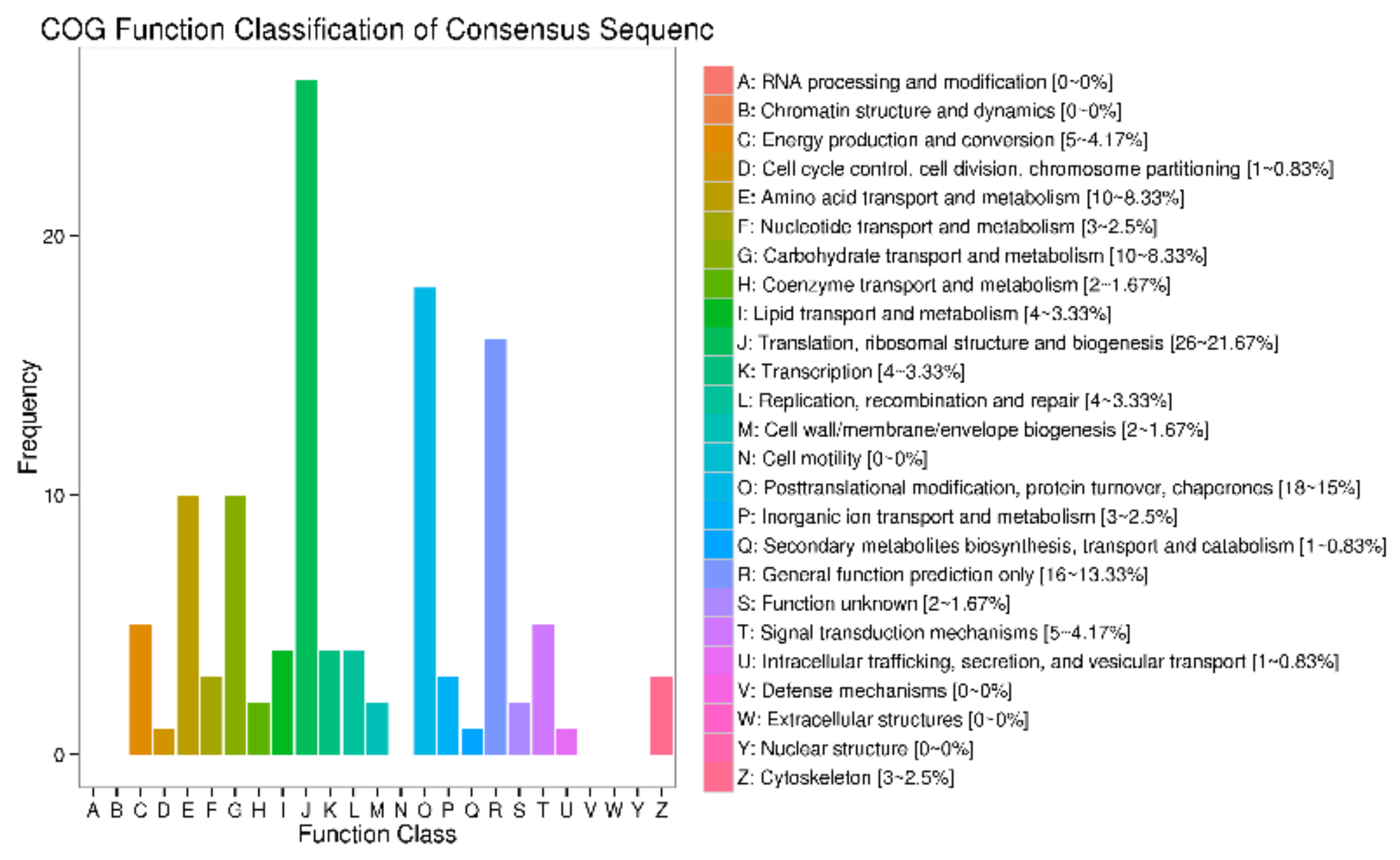
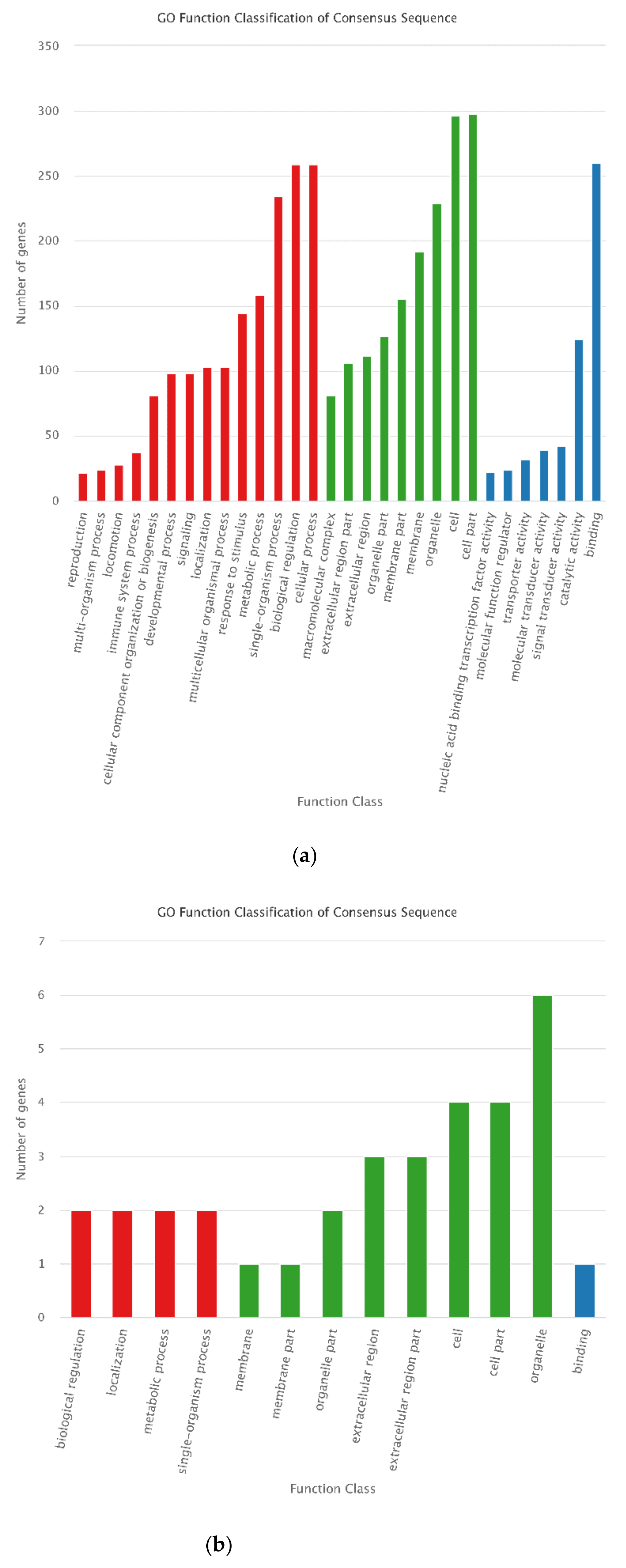
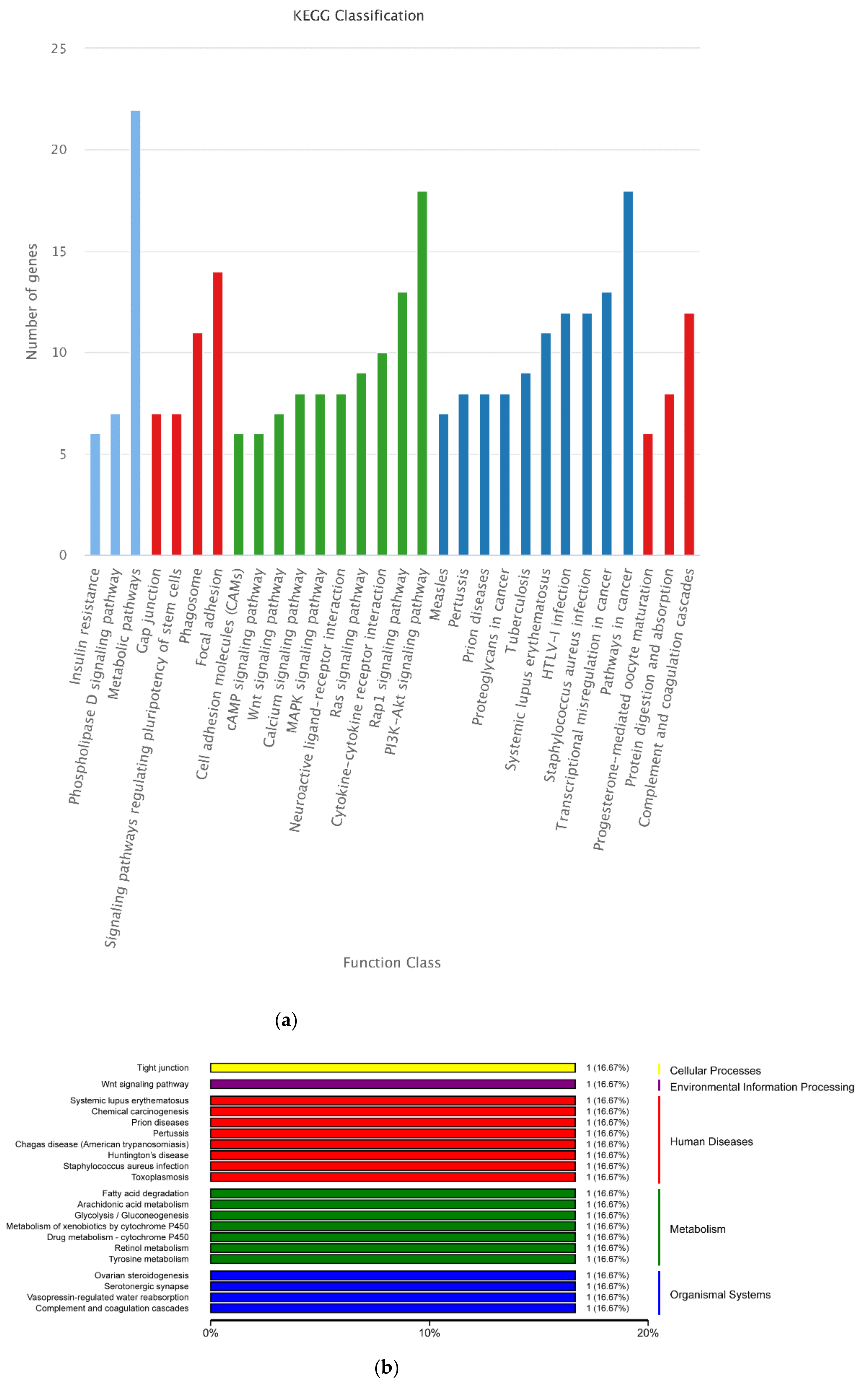
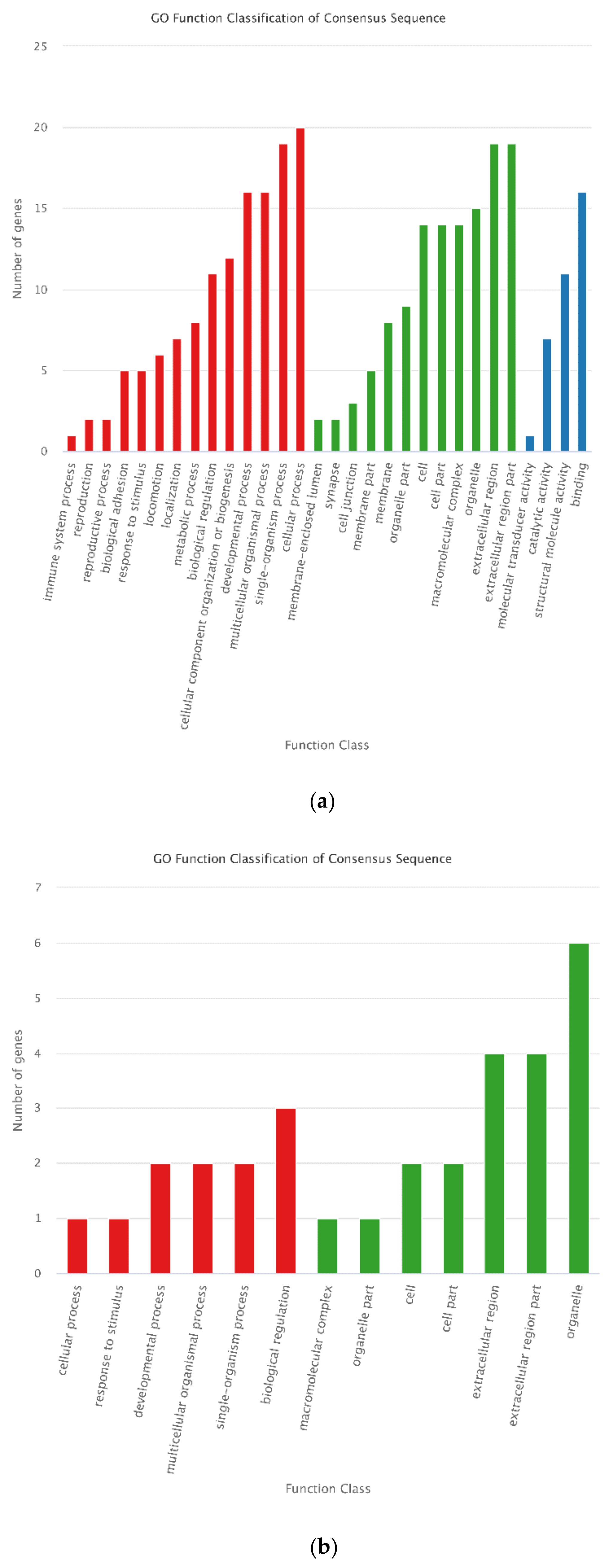
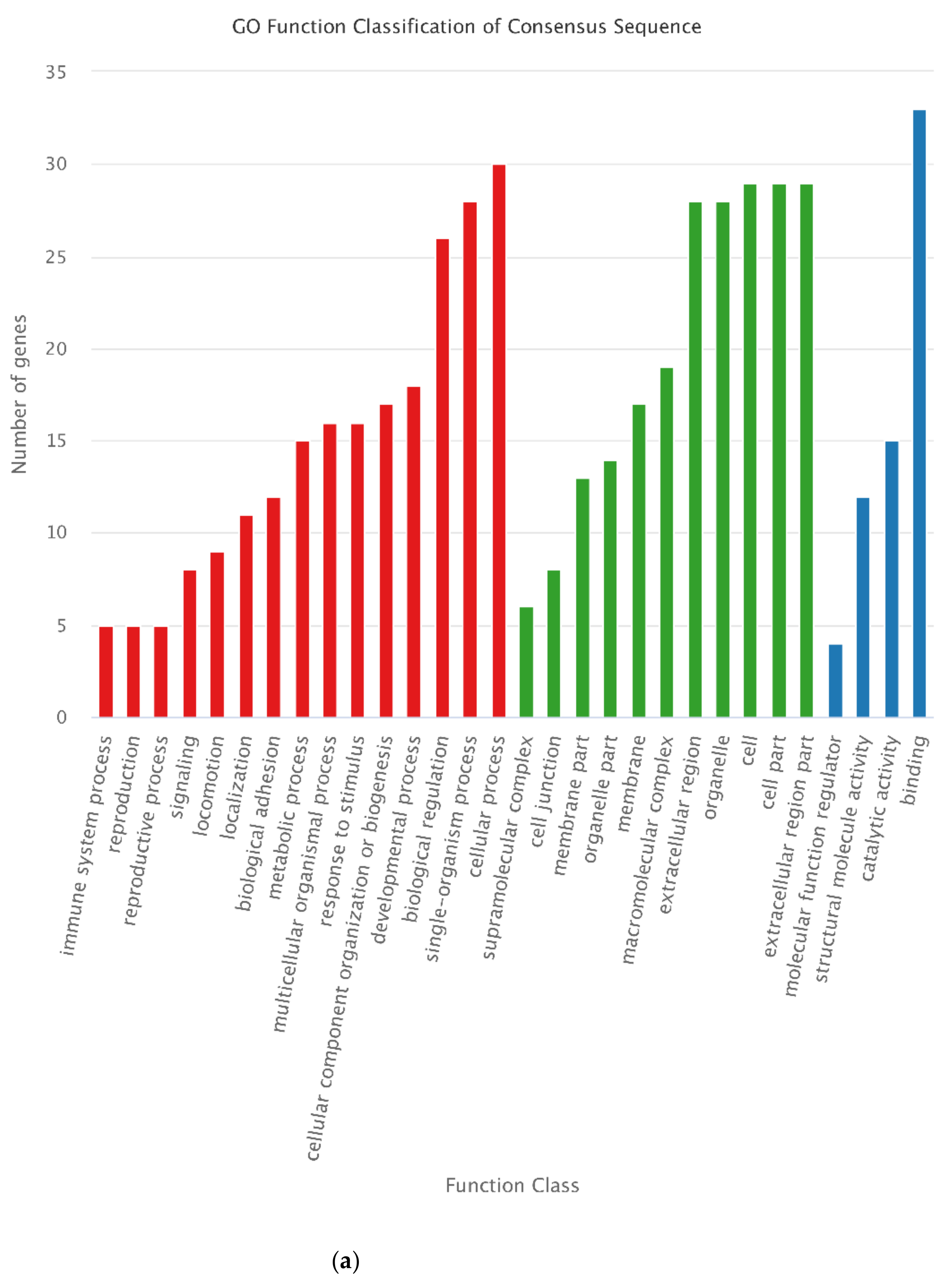
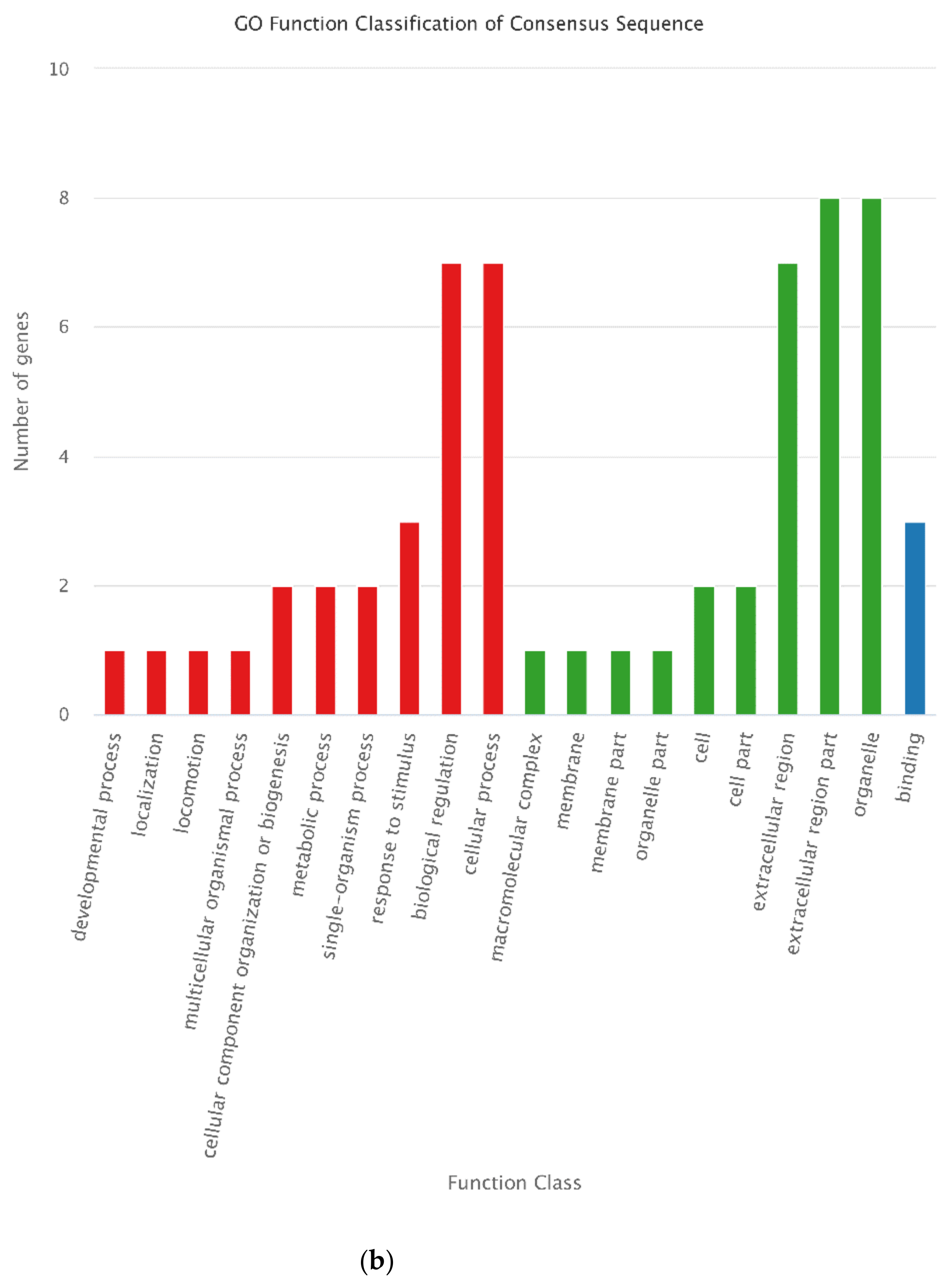
3.4. Functional Annotation of DEGs
3.5. Antler Proteins Revealed by iTRAQ Analysis
3.6. Differential Expression of Proteins
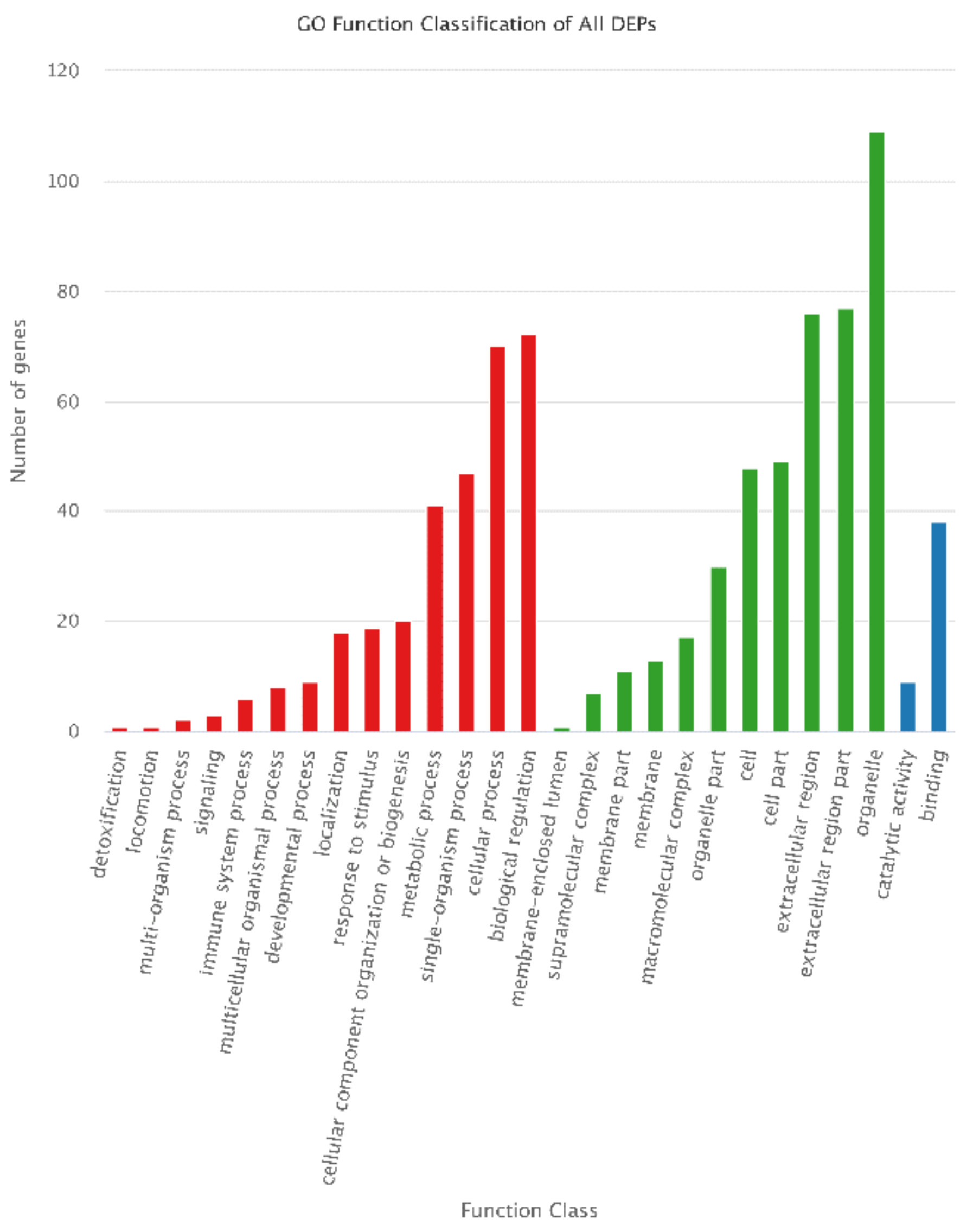
3.7. Annotation of DEPs
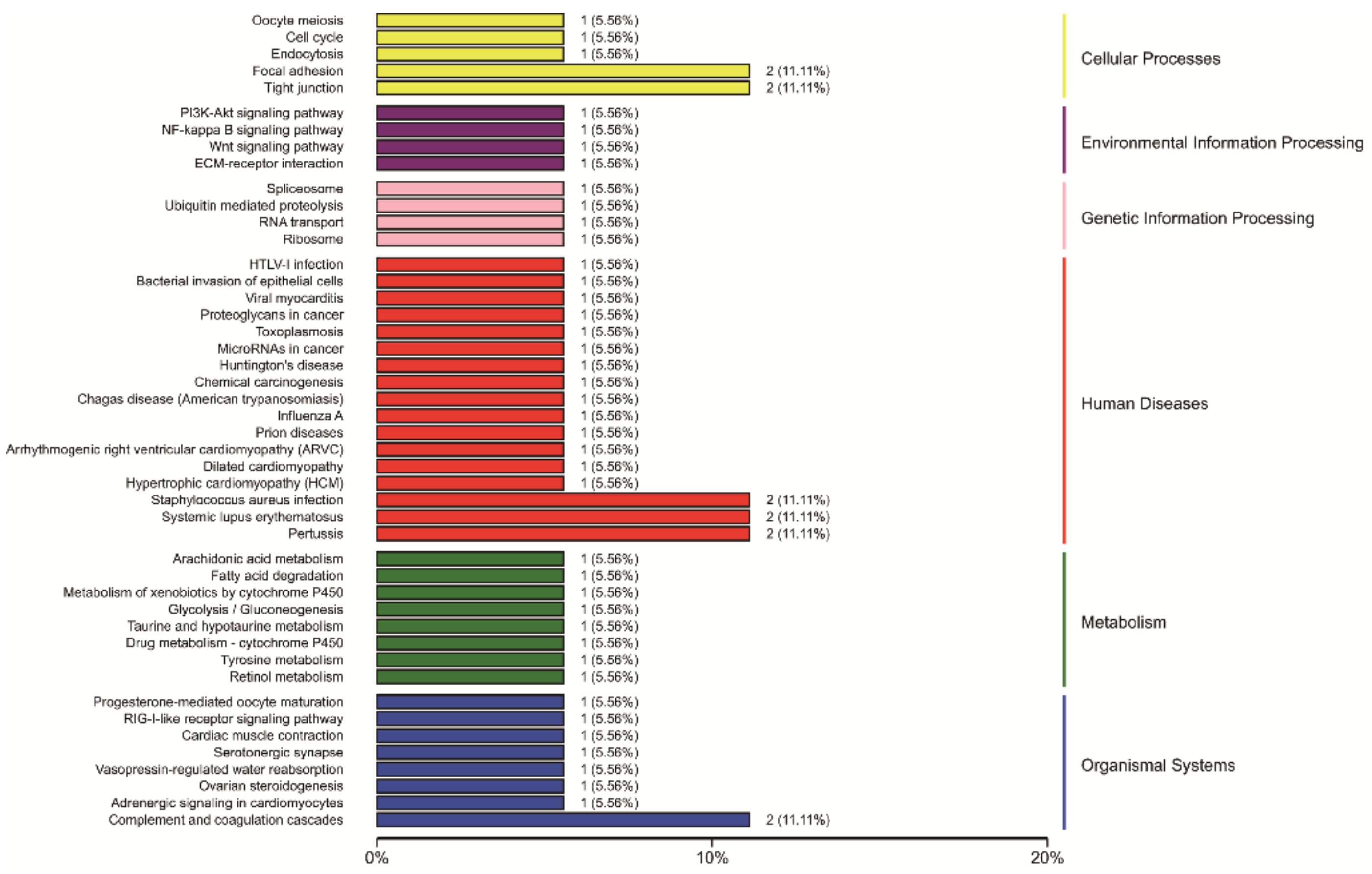
3.8. Integrated Analysis of Transcriptome and Proteome Data and Integrated Analysis of DEPs and DEG during Antler Growth
4. Discussion
4.1. General Features of the Transcriptomes and Proteomes of Antlers at Different Growth Stages
4.2. Analysis of Expression of Key DEGs and DEPs Involved in Rapid Antler Growth
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hu, P.; Shao, Y.; Xu, J.; Wang, T.; Li, Y.; Liu, H.; Rong, M.; Su, W.; Chen, B.; Cui, S.; et al. Genome-wide study on genetic diversity and phylogeny of five species in the genus Cervus. BMC Genom. 2019, 20, 384. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Liu, M.; Bai, X.; Li, Y.; Zhao, Y.; Wang, S.; Wang, J. Molecular cloning, recombinant expression, and purification of osteocalcin in sika deer (Cervus nippon) antler. Pol. J. Vet. Sci. 2019, 22, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Sui, Z.; Weng, Y.; Zhao, Q.; Deng, N.; Fang, F.; Zhu, X.; Shan, Y.; Zhang, L.; Zhang, Y. Ionic liquid-based method for direct proteome characterization of velvet antler cartilage. Talanta 2016, 161, 541–546. [Google Scholar] [CrossRef] [PubMed]
- Han, R.; Han, L.; Wang, S.; Li, H. Whole transcriptome analysis of mesenchyme tissue in sika deer antler revealed the CeRNAs regulatory network associated with antler development. Front. Genet. 2019, 10, 1403. [Google Scholar] [CrossRef] [Green Version]
- Yao, B.; Zhang, M.; Liu, M.; Liu, Y.; Hu, Y.; Zhao, Y. Transcriptomic characterization elucidates a signaling network that controls antler growth. Genome 2018, 61, 829–841. [Google Scholar] [CrossRef]
- Park, H.J.; Lee, D.H.; Park, S.G.; Lee, S.C.; Cho, S.; Kim, H.K.; Kim, J.J.; Bae, H.; Park, B.C. Proteome analysis of red deer antlers. Proteomics 2004, 4, 3642–3653. [Google Scholar] [CrossRef]
- Hu, P.; Wang, T.; Liu, H.; Xu, J.; Wang, L.; Zhao, P.; Xing, X. Full-length transcriptome and microRNA sequencing reveal the specific gene-regulation network of velvet antler in sika deer with extremely different velvet antler weight. Mol. Genet. Genom. MGG 2019, 294, 431–443. [Google Scholar] [CrossRef]
- Akhtar, R.W.; Liu, Z.; Wang, D.; Ba, H.; Shah, S.A.H.; Li, C. Identification of proteins that mediate the role of androgens in antler regeneration using label free proteomics in sika deer (Cervus nippon). Gen. Comp. Endocrinol. 2019, 283, 113235. [Google Scholar] [CrossRef]
- Ba, H.; Wang, D.; Yau, T.O.; Shang, Y.; Li, C. Transcriptomic analysis of different tissue layers in antler growth center in Sika Deer (Cervus nippon). BMC Genom. 2019, 20, 173. [Google Scholar] [CrossRef] [Green Version]
- Dong, Z.; Haines, S.; Coates, D. Proteomic profiling of stem cell tissues during regeneration of deer antler: A model of mammalian organ regeneration. J. Proteome Res. 2020, 19, 1760–1775. [Google Scholar] [CrossRef]
- Sui, Z.; Sun, H.; Weng, Y.; Zhang, X.; Sun, M.; Sun, R.; Zhao, B.; Liang, Z.; Zhang, Y.; Li, C.; et al. Quantitative proteomics analysis of deer antlerogenic periosteal cells reveals potential bioactive factors in velvet antlers. J. Chromatogr. A 2020, 1609, 460496. [Google Scholar] [CrossRef] [PubMed]
- Félix, C.; Meneses, R.; Gonçalves, M.F.M.; Tilleman, L.; Duarte, A.S.; Jorrín-Novo, J.V.; Van de Peer, Y.; Deforce, D.; Van Nieuwerburgh, F.; Esteves, A.C.; et al. A multi-omics analysis of the grapevine pathogen Lasiodiplodia theobromae reveals that temperature affects the expression of virulence- and pathogenicity-related genes. Sci. Rep. 2019, 9, 13144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Hua, J.; Yin, Y.; Gu, C.; Yu, C.; Shi, Q.; Guo, J.; Xuan, L.; Yu, F. An integrated transcriptome and proteome analysis reveals putative regulators of adventitious root formation in Taxodium ‘Zhongshanshan’. Int. J. Mol. Sci. 2019, 20, 1225. [Google Scholar] [CrossRef] [Green Version]
- Ye, X.; Wang, H.; Chen, P.; Fu, B.; Zhang, M.; Li, J.; Zheng, X.; Tan, B.; Feng, J. Combination of iTRAQ proteomics and RNA-seq transcriptomics reveals multiple levels of regulation in phytoplasma-infected Ziziphus jujuba Mill. Hortic. Res. 2017, 4, 17080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, N.; Yang, Z.; Zhang, Z.; Hu, L.; Chen, L. Comparative transcriptome combined with proteome analyses revealed key factors involved in Alfalfa (Medicago sativa) response to waterlogging stress. Int. J. Mol. Sci. 2019, 20, 1359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [Green Version]
- Leng, N.; Dawson, J.A.; Thomson, J.A.; Ruotti, V.; Rissman, A.I.; Smits, B.M.; Haag, J.D.; Gould, M.N.; Stewart, R.M.; Kendziorski, C. EBSeq: An empirical Bayes hierarchical model for inference in RNA-seq experiments. Bioinformatics 2013, 29, 1035–1043. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Cai, R.; Jin, X.; Shafer, A.B.A.; Hu, X.; Yang, S.; Li, Y.; Qi, L.; Liu, S.; Hu, D. Blood transcriptomics of captive forest musk deer (Moschus berezovskii) and possible associations with the immune response to abscesses. Sci. Rep. 2018, 8, 599. [Google Scholar] [CrossRef] [Green Version]
- Li, J.N.; Zhao, Y.T.; Cao, S.L.; Wang, H.; Zhang, J.J. Integrated transcriptomic and proteomic analyses of grass carp intestines after vaccination with a double-targeted DNA vaccine of Vibrio mimicus. Fish Shellfish Immunol. 2020, 98, 641–652. [Google Scholar] [CrossRef]
- Medina, I.; Montaner, D.; Tárraga, J.; Dopazo, J. Prophet, a web-based tool for class prediction using microarray data. Bioinformatics 2007, 23, 390–391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez-Riverol, Y.; Bai, J.; Bandla, C.; Garcia-Seisdedos, D.; Hewapathirana, S.; Kamatchinathan, S.; Kundu, D.J.; Prakash, A.; Frericks-Zipper, A.; Eisenacher, M.; et al. The PRIDE database resources in 2022: A hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 2022, 50, D543–D552. [Google Scholar] [CrossRef]
- Tatusov, R.L.; Galperin, M.Y.; Natale, D.A.; Koonin, E.V. The COG database: A tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000, 28, 33–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Li, Y.; Wang, M.; Li, Q.; Gao, Y.; Li, Q.; Li, J.; Cao, Y. Transcriptome profiling of longissimus lumborum in Holstein bulls and steers with different beef qualities. PloS ONE 2020, 15, e0235218. [Google Scholar] [CrossRef]
- Wu, J.; Mao, X.; Cai, T.; Luo, J.; Wei, L. KOBAS server: A web-based platform for automated annotation and pathway identification. Nucleic Acids Res. 2006, 34, W720–W724. [Google Scholar] [CrossRef]
- Xiao, C.; Ye, J.; Esteves, R.M.; Rong, C. Using Spearman’s correlation coefficients for exploratory data analysis on big dataset. Concurr. Comput. Pract. Exp. 2016, 28, 3866–3878. [Google Scholar] [CrossRef]
- Chen, Y. Transcriptome Profiles of Velvet Antler in Gansu Red Deer. 2021. Available online: https://www.ncbi.nlm.nih.gov/bioproject/772802 (accessed on 19 October 2021).
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [Green Version]
- Mount, J.G.; Muzylak, M.; Allen, S.; Althnaian, T.; McGonnell, I.M.; Price, J.S. Evidence that the canonical Wnt signalling pathway regulates deer antler regeneration. Dev. Dyn. Off. Publ. Am. Assoc. Anat. 2006, 235, 1390–1399. [Google Scholar] [CrossRef]
- Zhao, Y.; Yao, B.; Zhang, M.; Wang, S.; Zhang, H.; Xiao, W. Comparative analysis of differentially expressed genes in Sika deer antler at different stages. Mol. Biol. Rep. 2013, 40, 1665–1676. [Google Scholar] [CrossRef] [PubMed]
- Bana, N.A.; Nyiri, A.; Nagy, J.; Frank, K.; Nagy, T.; Steger, V.; Schiller, M.; Lakatos, P.; Sugar, L.; Horn, P.; et al. The red deer Cervus elaphus genome CerEla1.0: Sequencing, annotating, genes, and chromosomes. Mol. Genet. Genom. MGG 2018, 293, 665–684. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chapman, D.I. Antlers–bones of contention. Mammal Rev. 1975, 5, 121–172. [Google Scholar] [CrossRef]
- Price, J.S.; Allen, S.; Faucheux, C.; Althnaian, T.; Mount, J.G. Deer antlers: A zoological curiosity or the key to understanding organ regeneration in mammals? J. Anat. 2005, 207, 603–618. [Google Scholar] [CrossRef]
- Li, C.; Clark, D.E.; Lord, E.A.; Stanton, J.A.; Suttie, J.M. Sampling technique to discriminate the different tissue layers of growing antler tips for gene discovery. Anat. Rec. 2002, 268, 125–130. [Google Scholar] [CrossRef]
- Yao, B.; Zhao, Y.; Zhang, H.; Zhang, M.; Liu, M.; Liu, H.; Li, J. Sequencing and de novo analysis of the Chinese Sika deer antler-tip transcriptome during the ossification stage using Illumina RNA-Seq technology. Biotechnol. Lett. 2012, 34, 813–822. [Google Scholar] [CrossRef]
- Yang, X.G.; Chen, Y.X.; Liu, X.D.; Liu, Q.Z.; Pi, X.M.; Liu, Y.H.; Dong, Z. De novo characterization of velvet skin transcriptome at the antlers tips of red deer (Cervus elaphus) and analysis of growth factors and their receptors related to regeneration. Pak. J. Zool. 2016, 48, 151–157. [Google Scholar]
- Jia, B.; Liu, Y.; Li, Q.; Zhang, J.; Ge, C.; Wang, G.; Chen, G.; Liu, D.; Yang, F. Altered miRNA and mRNA Expression in Sika Deer Skeletal Muscle with Age. Genes 2020, 11, 172. [Google Scholar] [CrossRef] [Green Version]
- Jia, B.; Zhang, L.; Zhang, Y.; Ge, C.; Yang, F.; Du, R.; Ba, H. Integrated analysis of miRNA and mRNA transcriptomic reveals antler growth regulatory network. Mol. Genet. Genom. MGG 2021, 296, 689–703. [Google Scholar] [CrossRef]
- Wu, B.; Li, Y.; Li, J.; Xie, Z.; Luan, M.; Gao, C.; Shi, Y.; Chen, S. Genome-wide analysis of alternative splicing and non-coding RNAs reveal complicated transcriptional regulation in Cannabis sativa L. Int. J. Mol. Sci. 2021, 22, 11989. [Google Scholar] [CrossRef] [PubMed]
- Venkatachalam, D.; Chambers, J.P.; Kongara, K.; Singh, P. Analgesic efficacy of articaine hydrochloride for velvet antler removal in red deer (Cervus elaphus) and analysis of drug residues in the harvested velvet antlers. N. Z. Vet. J. 2019, 67, 228–233. [Google Scholar] [CrossRef] [PubMed]
- Bi, X.; Zhai, J.; Xia, Y.; Li, H. Analysis of genetic information from the antlers of Rangifer tarandus (reindeer) at the rapid growth stage. PLoS ONE 2020, 15, e0230168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Waldrup, K.A.; Corson, I.D.; Littlejohn, R.P.; Suttie, J.M. Histogenesis of antlerogenic tissues cultivated in diffusion chambers in vivo in red deer (Cervus elaphus). J. Exp. Zool. 1995, 272, 345–355. [Google Scholar] [CrossRef] [PubMed]
- Goss, R.J. Tumor-like growth of antlers in castrated fallow deer: An electron microscopic study. Scanning Microsc. 1990, 4, 715–720; discussion 720–721. [Google Scholar] [PubMed]
- Rodon, J.; Dienstmann, R.; Serra, V.; Tabernero, J. Development of PI3K inhibitors: Lessons learned from early clinical trials. Nat. Rev. Clin. Oncol. 2013, 10, 143–153. [Google Scholar] [CrossRef]
- Xu, W.; Yang, Z.; Lu, N. A new role for the PI3K/Akt signaling pathway in the epithelial-mesenchymal transition. Cell Adhes. Migr. 2015, 9, 317–324. [Google Scholar] [CrossRef] [Green Version]
- Ediriweera, M.K.; Tennekoon, K.H.; Samarakoon, S.R. Role of the PI3K/AKT/mTOR signaling pathway in ovarian cancer: Biological and therapeutic significance. Semin. Cancer Biol. 2019, 59, 147–160. [Google Scholar] [CrossRef]
- Chang, L.; Graham, P.H.; Ni, J.; Hao, J.; Bucci, J.; Cozzi, P.J.; Li, Y. Targeting PI3K/Akt/mTOR signaling pathway in the treatment of prostate cancer radioresistance. Crit. Rev. Oncol./Hematol. 2015, 96, 507–517. [Google Scholar] [CrossRef]
- Dimitrova, V.; Arcaro, A. Targeting the PI3K/AKT/mTOR signaling pathway in medulloblastoma. Curr. Mol. Med. 2015, 15, 82–93. [Google Scholar] [CrossRef]
- Ye, B.; Jiang, L.L.; Xu, H.T.; Zhou, D.W.; Li, Z.S. Expression of PI3K/AKT pathway in gastric cancer and its blockade suppresses tumor growth and metastasis. Int. J. Immunopathol. Pharmacol. 2012, 25, 627–636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, F.B.; Qiu, H.Y. Effects of Artesunate on chondrocyte proliferation, apoptosis and autophagy through the PI3K/AKT/mTOR signaling pathway in rat models with rheumatoid arthritis. Biomed. Pharmacother. 2018, 102, 1209–1220. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Sha, Y.; Wang, S.; Chi, Q.; Liu, Y.; Wang, C.; Yang, L. Effects of Bakuchiol on chondrocyte proliferation via the PI3K-Akt and ERK1/2 pathways mediated by the estrogen receptor for promotion of the regeneration of knee articular cartilage defects. Cell Prolif. 2019, 52, e12666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, X.; Ni, B.; Mao, Z.; Xi, Y.; Chu, X.; Zhang, R.; Ma, X.; You, H. NOV/CCN3 induces cartilage protection by inhibiting PI3K/AKT/mTOR pathway. J. Cell. Mol. Med. 2019, 23, 7525–7534. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Lai, S.; Hou, X.; Cao, W.; Zhang, Y.; Zhang, Z. Protective effects of PI3K/Akt signal pathway induced cell autophagy in rat knee joint cartilage injury. Am. J. Transl. Res. 2018, 10, 762–770. [Google Scholar]
- Huang, X.; Xi, Y.; Mao, Z.; Chu, X.; Zhang, R.; Ma, X.; Ni, B.; Cheng, H.; You, H. Vanillic acid attenuates cartilage degeneration by regulating the MAPK and PI3K/AKT/NF-κB pathways. Eur. J. Pharmacol. 2019, 859, 172481. [Google Scholar] [CrossRef]
- Tao, H.; Cheng, L.; Yang, R. Downregulation of miR-34a promotes proliferation and inhibits apoptosis of rat osteoarthritic cartilage cells by activating PI3K/Akt pathway. Clin. Interv. Aging 2020, 15, 373–385. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Lu, H.; Yao, J.; Zhang, X.; Huang, Y.; Ma, S.; Zou, K.; Wei, Y.; Yang, Z.; Li, J.; et al. GABARAP promotes bone marrow mesenchymal stem cells-based the osteoarthritis cartilage regeneration through the inhibition of PI3K/AKT/mTOR signaling pathway. J. Cell. Physiol. 2019, 234, 21014–21026. [Google Scholar] [CrossRef]
- Liu, Z.; Zhao, H.; Wang, D.; McMahon, C.; Li, C. Differential effects of the PI3K/AKT pathway on antler stem cells for generation and regeneration of antlers in vitro. Front Biosci Landmark Ed. 2018, 23, 1848–1863. [Google Scholar] [CrossRef]
- Dong, Z.; Ba, H.; Zhang, W.; Coates, D.; Li, C. iTRAQ-based quantitative proteomic analysis of the potentiated and dormant antler stem cells. Int. J. Mol. Sci. 2016, 17, 1778. [Google Scholar] [CrossRef] [Green Version]
- Clevers, H. Wnt/beta-catenin signaling in development and disease. Cell 2006, 127, 469–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lerner, U.H.; Ohlsson, C. The WNT system: Background and its role in bone. J. Int. Med. 2015, 277, 630–649. [Google Scholar] [CrossRef]
- Zhang, H.L.; Yang, Z.Q.; Duan, C.C.; Geng, S.; Wang, K.; Yu, H.F.; Yue, Z.P.; Guo, B. WNT4 acts downstream of BMP2 to mediate the regulation of ATRA signaling on RUNX1 expression: Implications for terminal differentiation of antler chondrocytes. J. Cell. Physiol. 2018, 233, 1129–1145. [Google Scholar] [CrossRef] [PubMed]
- Yin, P.; Wang, W.; Zhang, Z.; Bai, Y.; Gao, J.; Zhao, C. Wnt signaling in human and mouse breast cancer: Focusing on Wnt ligands, receptors and antagonists. Cancer Sci. 2018, 109, 3368–3375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haraguchi, R.; Kitazawa, R.; Mori, K.; Tachibana, R.; Kiyonari, H.; Imai, Y.; Abe, T.; Kitazawa, S. sFRP4-dependent Wnt signal modulation is critical for bone remodeling during postnatal development and age-related bone loss. Sci. Rep. 2016, 6, 25198. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Gao, X.; Chen, S.; Li, D.; Chen, S.; Xie, M.; Xu, Z.; Yang, G. Genome-wide identification and expression analysis of ethylene responsive factor family transcription factors in Juglans regia. PeerJ 2021, 9, e12429. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Yan, C.; Liu, Y.; Wu, Z.; Zhang, J.; Sun, Y. Cloning, expression analysis and RNAi of farnesoic acid O-methylransferase gene from Neocaridina denticulata sinensis. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2022, 259, 110719. [Google Scholar] [CrossRef]
- Lu, S.; Wang, J.; Chitsaz, F.; Derbyshire, M.K.; Geer, R.C.; Gonzales, N.R.; Gwadz, M.; Hurwitz, D.I.; Marchler, G.H.; Song, J.S.; et al. CDD/SPARCLE: The conserved domain database in 2020. Nucleic Acids Res. 2020, 48, D265–D268. [Google Scholar] [CrossRef] [Green Version]
- National Research Council (US) Committee for the Update of the Guide for the Care and Use of Laboratory Animals. Guide for the Care and Use of Laboratory Animals, 8th ed.; The National Academies Press: Washington, DC, USA, 2011. [Google Scholar] [CrossRef]
- Percie du Sert, N.; Ahluwalia, A.; Alam, S.; Avey, M.T.; Baker, M.; Browne, W.J.; Clark, A.; Cuthill, I.C.; Dirnagl, U.; Emerson, M.; et al. Reporting animal research: Explanation and elaboration for the ARRIVE guidelines 2.0. PLoS Biol. 2020, 18, e3000411. [Google Scholar] [CrossRef]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| d30 | d60 | d90 | |||
|---|---|---|---|---|---|
| Gene ID | FPKM | Gene ID | FPKM | Gene ID | FPKM |
| gene4406 | 21762.164 | gene4406 | 22433.19609 | gene4406 | 18476.40401 |
| gene13363 | 9940.114655 | gene13363 | 17511.18606 | gene13363 | 13310.13206 |
| gene7568 | 6636.798024 | gene7568 | 7031.922373 | gene1629 | 9726.051422 |
| gene2851 | 3166.407348 | gene21360 | 3111.317734 | gene7568 | 8106.755774 |
| gene21360 | 3036.281532 | gene2851 | 3085.655884 | gene21360 | 3165.661509 |
| gene7842 | 2921.125488 | gene2852 | 2773.071777 | gene2851 | 2506.976623 |
| gene7323 | 2801.431152 | gene7323 | 2708.365234 | gene17381 | 2460.772705 |
| gene2852 | 2557.674316 | gene17381 | 2611.812988 | gene7842 | 2424.758789 |
| gene7381 | 2540.665881 | gene10381 | 2538.190388 | gene2852 | 2174.088867 |
| gene10381 | 2413.023017 | gene7842 | 2481.294678 | gene7323 | 2160.519531 |
| gene19732 | 2199.416988 | gene1629 | 2477.907784 | gene3336 | 2003.781029 |
| gene3336 | 2107.003662 | gene9487 | 2229.203369 | gene10381 | 1976.832594 |
| gene16931 | 2031.898727 | gene3336 | 2209.905213 | gene19732 | 1887.025254 |
| gene17381 | 2001.166626 | gene15888 | 2127.262207 | newGene_15708 | 1807.805496 |
| gene9487 | 1994.406006 | gene18121 | 2125.092085 | newGene_29845 | 1741.988892 |
| gene22181 | 1956.759766 | gene19732 | 1942.814499 | newGene_2805 | 1664.798706 |
| gene18121 | 1939.410617 | gene3351 | 1925.796814 | gene3351 | 1636.892456 |
| gene723 | 1862.862183 | gene16931 | 1909.572655 | gene15888 | 1556.295532 |
| newGene_2805 | 1826.791504 | gene13127 | 1883.147461 | gene16931 | 1527.461914 |
| gene15888 | 1821.269531 | gene18131 | 1821.30127 | gene723 | 1490.671143 |
| d30 | d60 | d90 | |||
|---|---|---|---|---|---|
| Protein ID | Expression | Protein ID | Expression | Protein ID | Expression |
| protein7361 | 1.686 | protein1174 | 1.719 | protein3419 | 3.398 |
| protein14704 | 1.666 | protein19035 | 1.691 | protein17593 | 3.243 |
| protein1519 | 1.664 | protein11670 | 1.6 | protein16793 | 3.082 |
| protein4597 | 1.66 | protein10984 | 1.596 | protein9356 | 2.86 |
| protein4576 | 1.648 | protein1517 | 1.5 | protein10194 | 2.786 |
| protein1521 | 1.635 | protein11698 | 1.496 | protein11790 | 2.785 |
| protein11686 | 1.592 | protein13127 | 1.464 | protein4470 | 2.669 |
| protein2427 | 1.543 | protein457 | 1.425 | protein6646 | 2.177 |
| protein1512 | 1.497 | protein10013 | 1.417 | protein8135 | 2.087 |
| protein15147 | 1.443 | protein14729 | 1.416 | protein10206 | 2.007 |
| protein4602 | 1.412 | protein16576 | 1.405 | newProtein_14627.3 | 1.908 |
| protein1875 | 1.389 | protein11169 | 1.403 | protein1493 | 1.899 |
| newProtein_6084.1 | 1.386 | protein2538 | 1.402 | protein3325 | 1.867 |
| protein3788 | 1.371 | protein13191 | 1.401 | protein8462 | 1.828 |
| protein4604 | 1.369 | protein10014 | 1.393 | protein2690 | 1.827 |
| protein1514 | 1.367 | protein1982 | 1.383 | protein16950 | 1.805 |
| protein629 | 1.353 | protein7765 | 1.362 | protein6800 | 1.775 |
| newProtein_16623.2 | 1.348 | protein14730 | 1.359 | protein13164 | 1.68 |
| newProtein_20523.1 | 1.348 | protein7142 | 1.354 | protein6020 | 1.66 |
| protein15292 | 1.345 | protein5871 | 1.352 | protein16569 | 1.66 |
| d30 vs. d60 | d30 vs. d90 | d60 vs. d90 | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| mRNA | Protein | mRNA | Protein | mRNA | Protein | ||||||||||
| Total | Up | Down | Up | Down | Total | Up | Down | Up | Down | Total | Up | Down | Up | Down | |
| NDEPs_DEGs | 76 | 44 | 32 | 0 | 0 | 76 | 36 | 40 | 0 | 0 | 80 | 18 | 52 | 0 | 0 |
| DEPs_NDEGs | 24 | 0 | 0 | 11 | 13 | 213 | 0 | 0 | 117 | 96 | 257 | 0 | 0 | 125 | 132 |
| DEPs_DEGs | 8 | 5 | 3 | 3 | 5 | 25 | 10 | 15 | 16 | 9 | 45 | 13 | 32 | 30 | 15 |
| DEPs_DEGs same tend | 0 | 0 | 0 | 0 | 0 | 9 | 5 | 4 | 5 | 4 | 16 | 7 | 9 | 7 | 9 |
| DEPs_DEGs reverse tend | 8 | 5 | 3 | 3 | 5 | 16 | 5 | 11 | 11 | 5 | 29 | 6 | 23 | 23 | 6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Y.; Zhang, Z.; Jin, W.; Li, Z.; Bao, C.; He, C.; Guo, Y.; Li, C. Integrative Analyses of Antler Cartilage Transcriptome and Proteome of Gansu Red Deer (Cervus elaphus kansuensis) at Different Growth Stages. Animals 2022, 12, 934. https://doi.org/10.3390/ani12070934
Chen Y, Zhang Z, Jin W, Li Z, Bao C, He C, Guo Y, Li C. Integrative Analyses of Antler Cartilage Transcriptome and Proteome of Gansu Red Deer (Cervus elaphus kansuensis) at Different Growth Stages. Animals. 2022; 12(7):934. https://doi.org/10.3390/ani12070934
Chicago/Turabian StyleChen, Yanxia, Zhenxiang Zhang, Wenjie Jin, Zhaonan Li, Changhong Bao, Caixia He, Yuqin Guo, and Changzhong Li. 2022. "Integrative Analyses of Antler Cartilage Transcriptome and Proteome of Gansu Red Deer (Cervus elaphus kansuensis) at Different Growth Stages" Animals 12, no. 7: 934. https://doi.org/10.3390/ani12070934