
Molecular Analysis of the Heterakis dispar Population in Domestic Geese Based on the ITS1-5.8rRNA-ITS2 Fragment

Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Parasites Collection
2.2. DNA Extraction and PCR Reactions
2.3. Sequencing and Phylogenetic Analysis
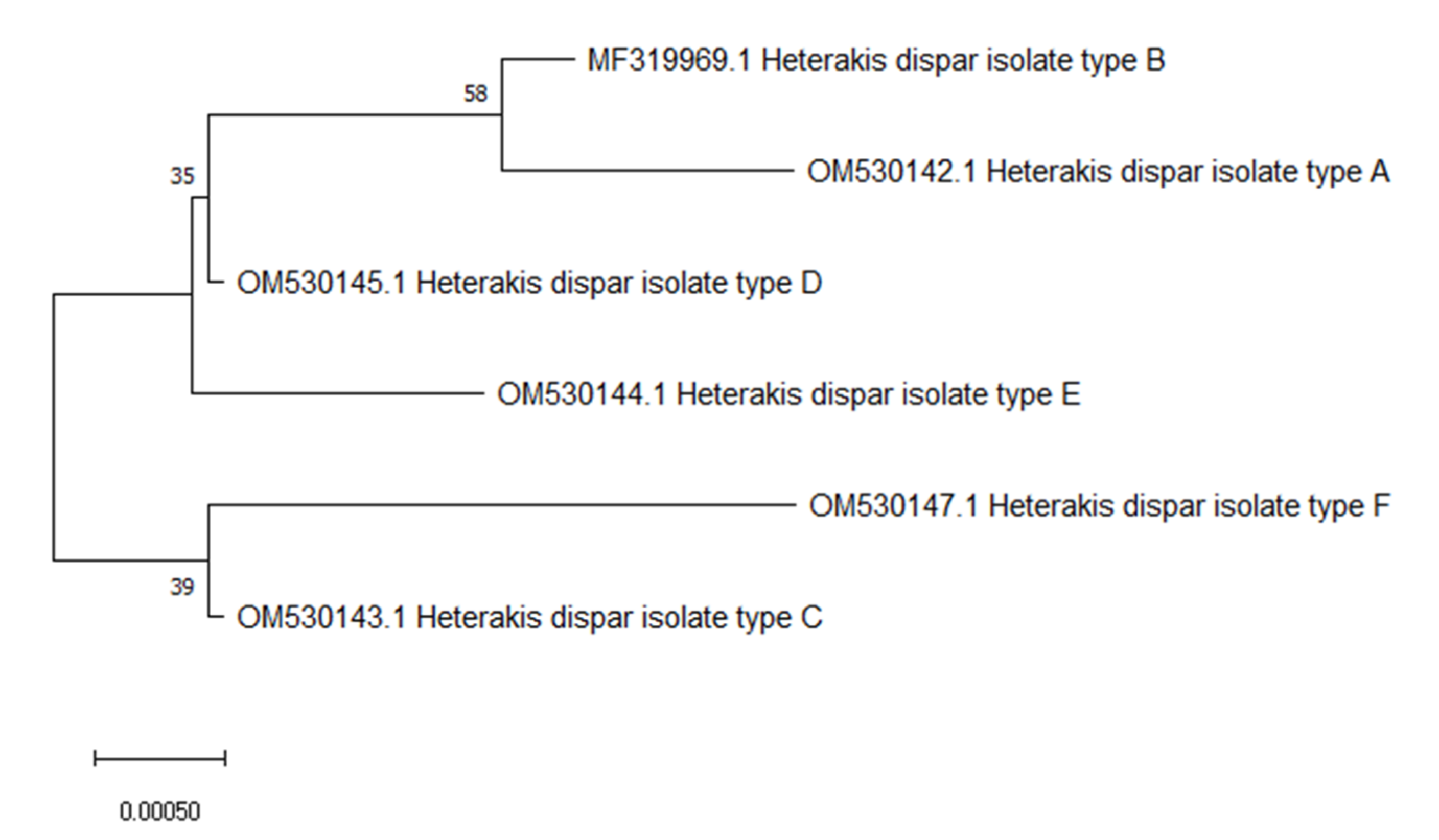
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bobrek, K.; Gaweł, A. Prevalence of Heterakis infection in parental flocks of geese. Avian Dis. 2020, 64, 552–555. [Google Scholar] [CrossRef] [PubMed]
- Yevstafyeva, V.A.; Melnychuk, V.V.; Nikiforova, O.V.; Suprunenko, K.V.; Korchan, L.N.; Lokes-Krupka, T.P.; Nehrebetskyi, I.S.; Korchan, N.I. Comparative morphology and biology of nematodes of genus Heterakis (Nematoda, Heterakidae), parasites of the domestic goose (Anser anser) in Ukraine. Regul. Mech. Biosyst. 2018, 9, 229–236. [Google Scholar] [CrossRef]
- Bobrek, K.; Hildebrand, J.; Urbanowicz, J.; Gaweł, A. Molecular identification and phylogenetic analysis of Heterakis dispar isolated from geese. Acta Parasitol. 2019, 64, 753–760. [Google Scholar] [CrossRef] [PubMed]
- Bazh, E.K. Molecular characterization of Ascaridia galli infecting native chickens in Egypt. Parasitol. Res. 2013, 112, 3223–3227. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Shi, L.; Li, D.; Sun, Y.; Niu, Y.; Chen, Z.; Luo, H.; Pang, X.; Sun, Z.; Liu, C.; et al. Extensive pyrosequencing reveals frequent intra-genomic variations of internal transcribed spacer regions of nuclear ribosomal DNA. PLoS ONE 2012, 7, e43971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amor, S.; Farjallah, S.; Mohammed, O.; Alagaili, A.; Bahri-Sfar, L. Molecular characterization of the nematode Heterakis gallinarum (Ascaridida:Heterakidae) infecting domestic chickens (Gallus gallus domesticus) in Tunisia. Turk. J. Vet. Anim. Sci. 2018, 42, 388–394. [Google Scholar] [CrossRef]
- Holguin, C.; Baeza, J.; Mueller, J.; Agudelo, P. High genetic diversity and geographical subdivision of three lance namatode species (Hoplolaimus spp) in the United States. Ecol. Evol. 2015, 5, 2929–2944. [Google Scholar] [CrossRef] [PubMed]
- Pereira, F.; Moreira, C.; Fonseca, L.; van Asch, B.; Mota, M.; Abrantes, I.; Amorim, A. New insights into the phylogeny and worldwide dispersion of two closely related nematode species, Bursaphelenchus xylophilus and Bursaphelenchus mucronatus. PLoS ONE 2013, 8, e56288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, D.D.; Wang, J.F.; Zhang, D.Y.; Peng, Z.W.; Yang, T.Y.; Wang, Z.D.; Bowman, D.D.; Hou, Z.J.; Liu, Z.S. Genetic diversity of Haemonchus contortus isolated from sympatric wild blue sheep (Pseudois nayaur) and sheep in Helan mountains, China. Parasites Vectors 2017, 10, 473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, G.; Li, H.; Ryan, U.; Cong, M.; Hu, B.; Gao, M.; Ren, W.; Wang, X.; Zhang, S.; Lin, Q.; et al. Phylogenetic study of Baylisascaris schroederi isolated from Qinling subspecies of giant panda in China based on combined nuclear 5.8S and the second internal transcriber spacer (ITS-2) ribosomal DNA sequence. Parasitol. Int. 2012, 61, 497–500. [Google Scholar] [CrossRef] [PubMed]
- Johnson, A. Single- nucleotide polymorphism bioinformatics: A comprehensive review of resources. Circ. Cardiovasc. Genet. 2009, 2, 530–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aksenov, A.; Spiridonov, S. Diversity of the rDNA ITS haplotypes of the nematodes Haemonchus contortus (Trichostrongylidea, Rhabditida) of the same host. Izv. Akad. Nauk. Ser. Biol. 2013, 1, 43–52. [Google Scholar]



{kind=link}
{kind=link}
| Heterakis dispar Type | GenBank Accession Number | Nucleotide Position in the Obtained Sequences | Number of Parasites (n = 71) | |||
|---|---|---|---|---|---|---|
| Pos 47 | Pos 176 | Pos 728 | Pos 780 | |||
| B | MF319969 | A | C | G | C | 13 |
| A | OM530142 | G | 32 | |||
| C | OM530143 | A | T | 8 | ||
| D | OM530144 | T | 8 | |||
| E | OM530145 | T | T | 6 | ||
| F | OM530147 | G | T | A | T | 4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bobrek, K.; Gaweł, A.; Urbanowicz, J. Molecular Analysis of the Heterakis dispar Population in Domestic Geese Based on the ITS1-5.8rRNA-ITS2 Fragment. Animals 2022, 12, 926. https://doi.org/10.3390/ani12070926
Bobrek K, Gaweł A, Urbanowicz J. Molecular Analysis of the Heterakis dispar Population in Domestic Geese Based on the ITS1-5.8rRNA-ITS2 Fragment. Animals. 2022; 12(7):926. https://doi.org/10.3390/ani12070926
Chicago/Turabian StyleBobrek, Kamila, Andrzej Gaweł, and Joanna Urbanowicz. 2022. "Molecular Analysis of the Heterakis dispar Population in Domestic Geese Based on the ITS1-5.8rRNA-ITS2 Fragment" Animals 12, no. 7: 926. https://doi.org/10.3390/ani12070926