
Linkage Disequilibrium, Haplotype Block Structures, Effective Population Size and Genome-Wide Signatures of Selection of Two Conservation Herds of the South African Nguni Cattle


 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Populations and Samples
2.2. Genotyping and Quality Control
2.3. Minor Allele Frequency
2.4. PCA Analysis
2.5. Linkage Disequilibrium Estimation
2.6. Haplotype Block Structure
2.7. Effective Population Size
2.8. Detection of Selection Signatures
2.8.1. Integrated Haplotype Score (iHS)
2.8.2. Cross-Population Extended Haplotype Homozygosity (XP-EHH)
2.9. Annotation of Signatures of Selection Genomic Regions
3. Results
3.1. SNP Quality Control, MAF and FIS per Population
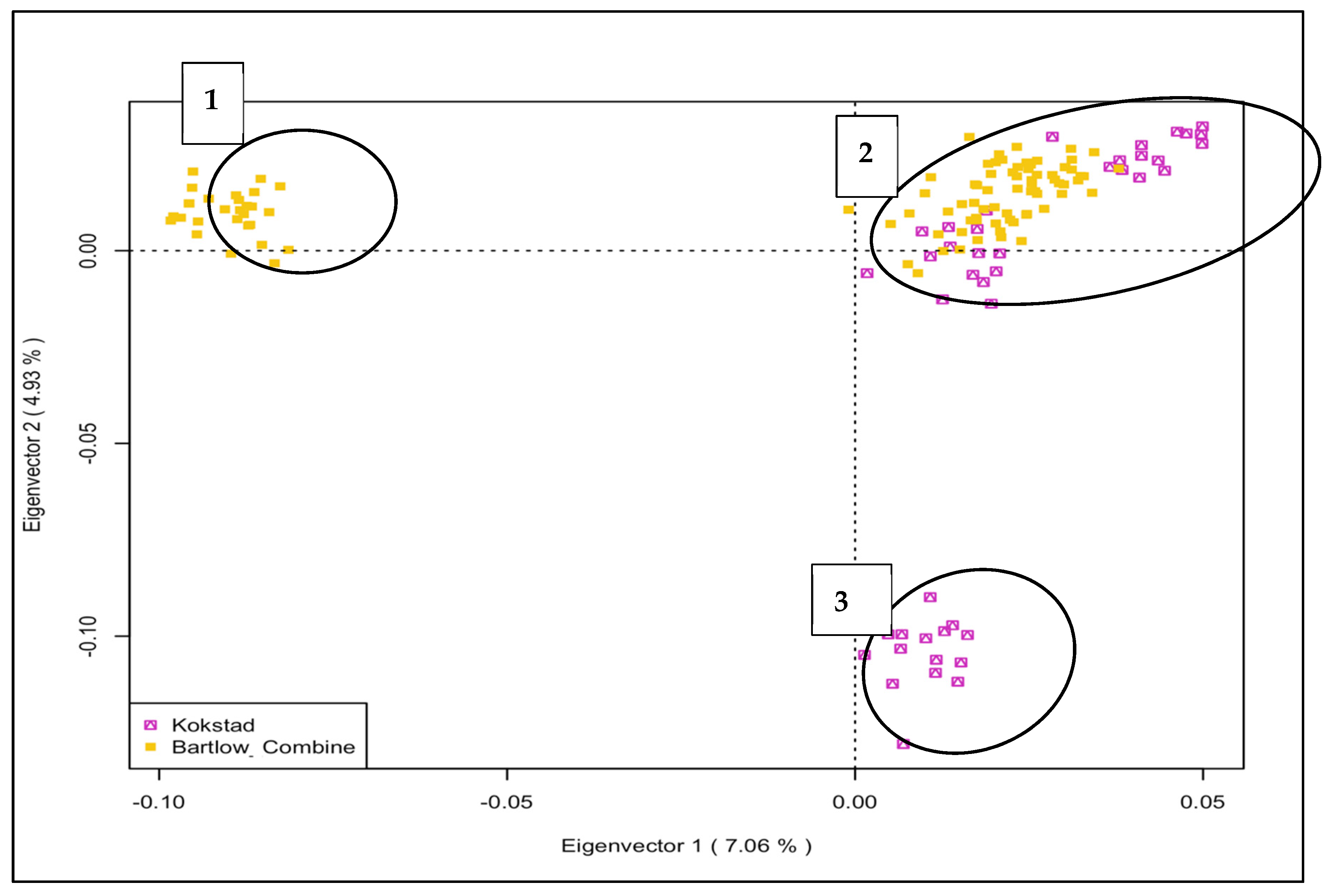
3.2. PCA Genetic Clustering
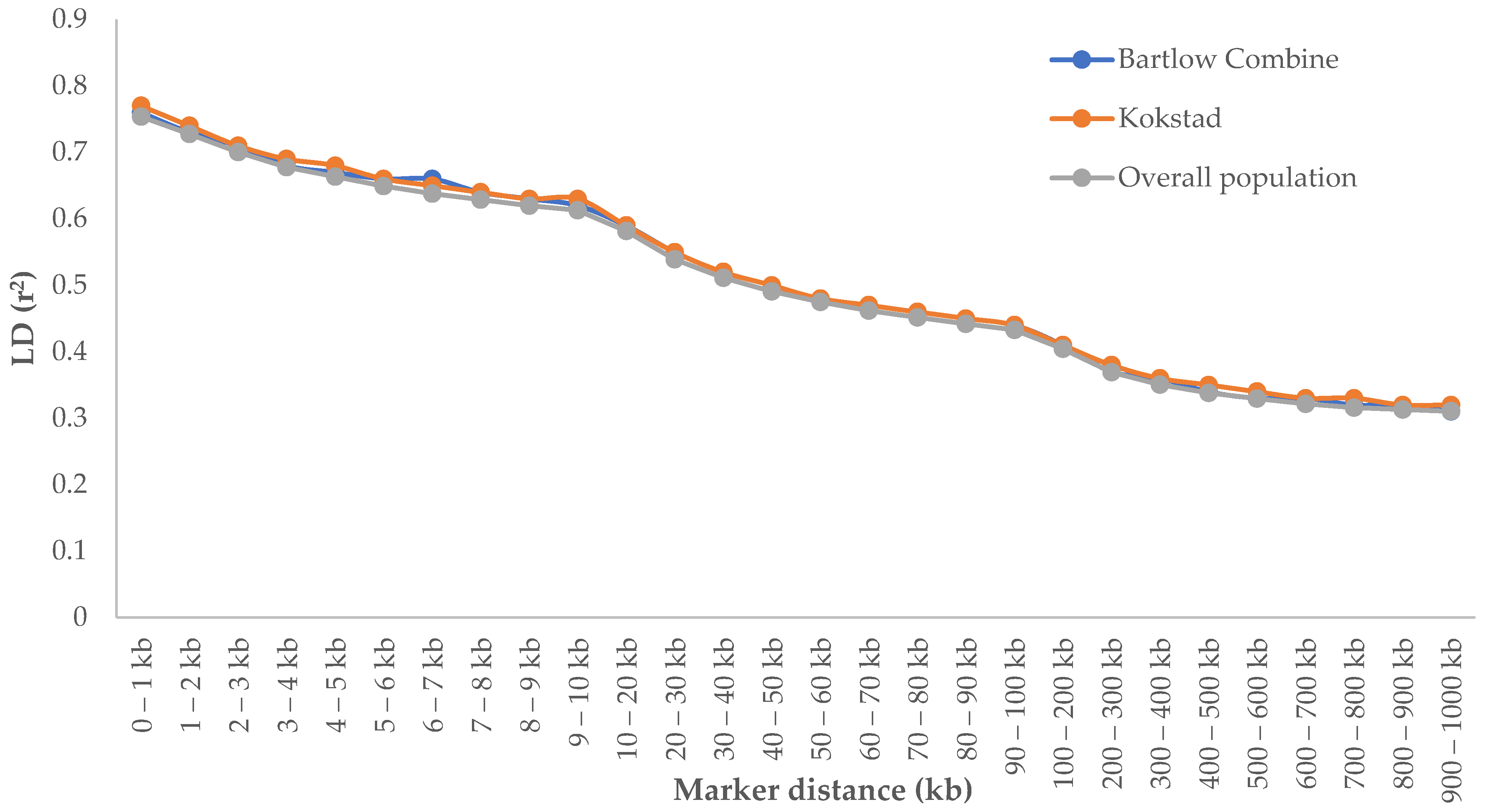
3.3. Linkage Disequilibrium Patterns and LD Decay
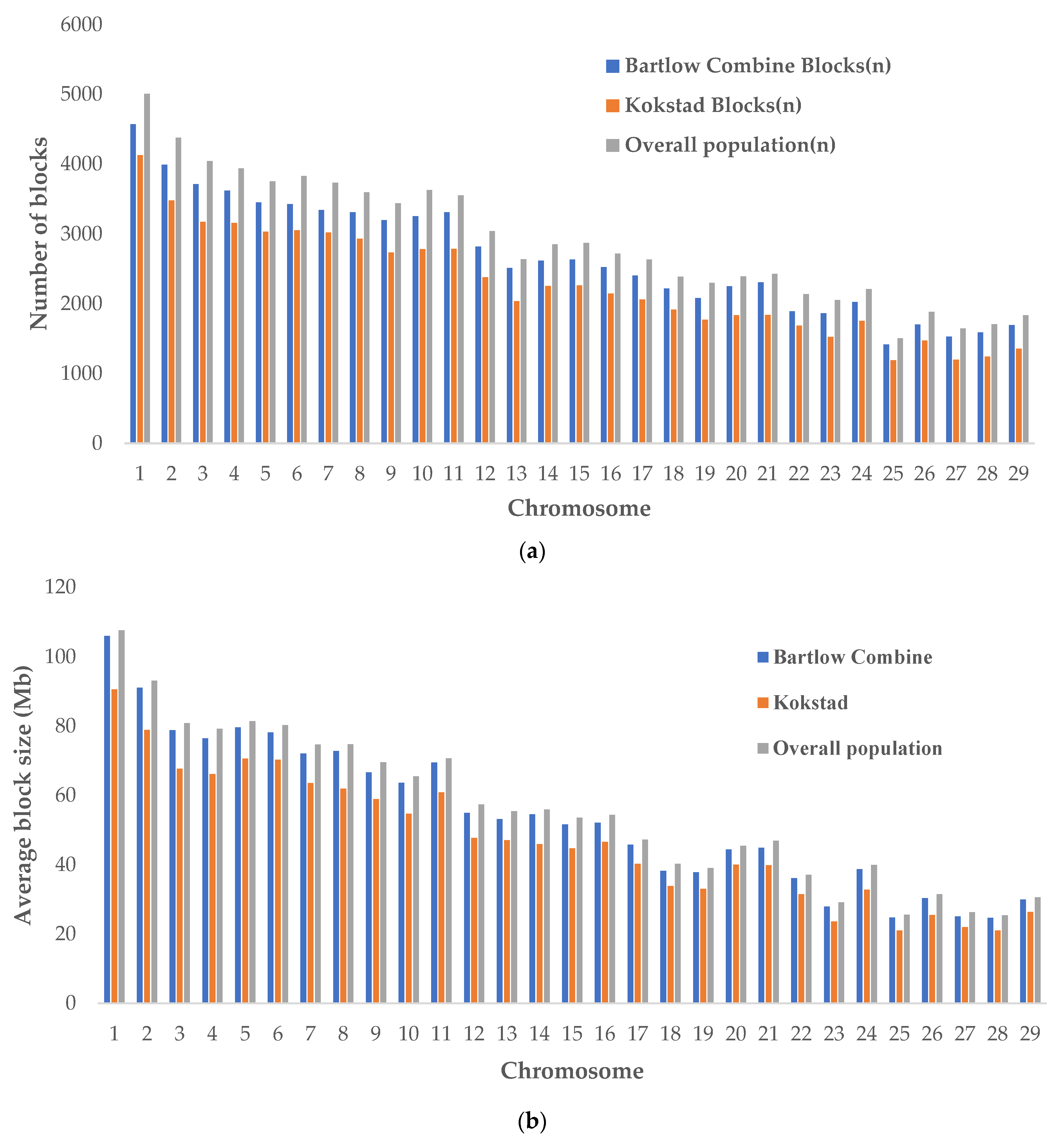
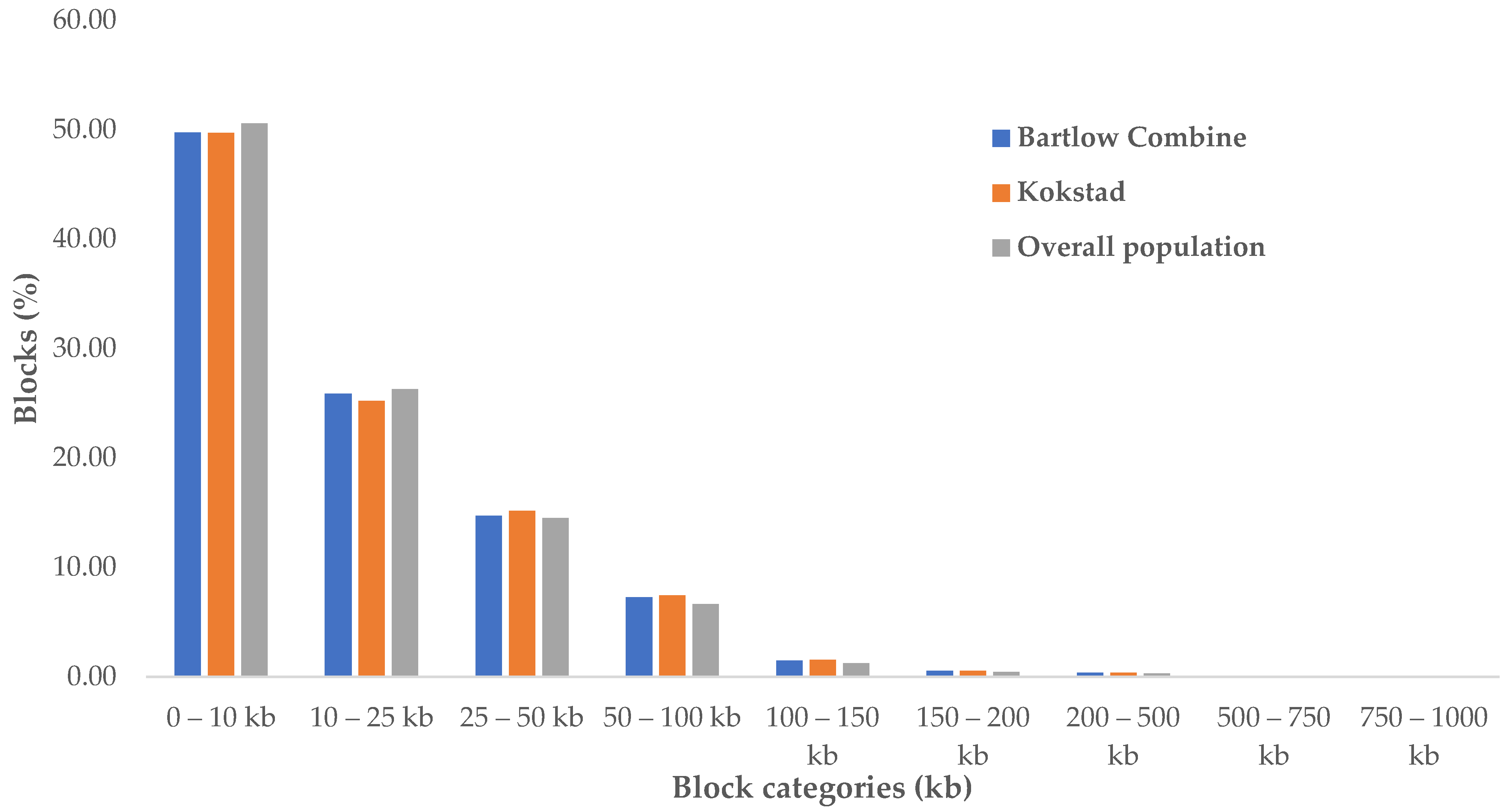
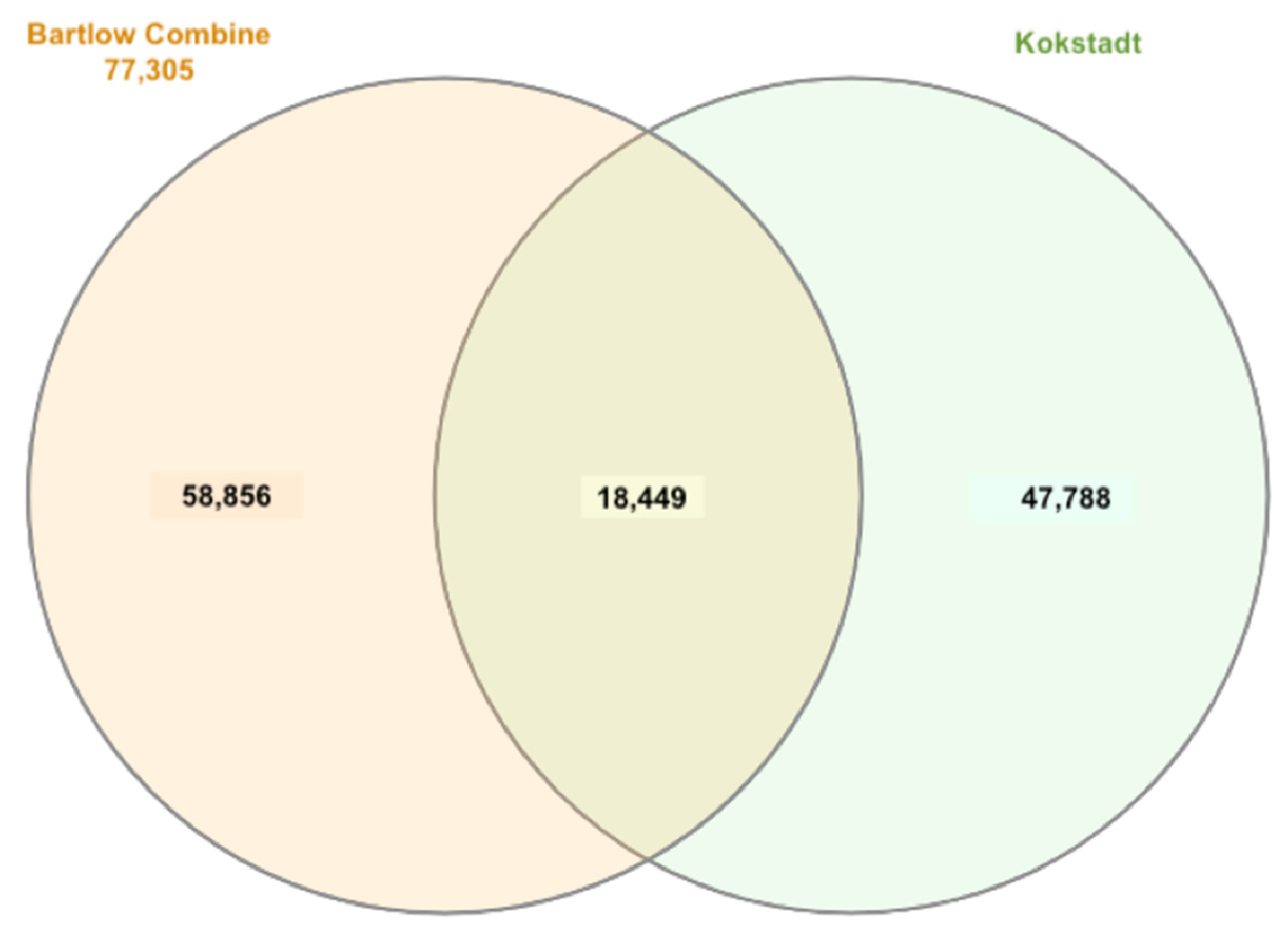
3.4. Haplotype Frequencies and Haplotype Block Structure
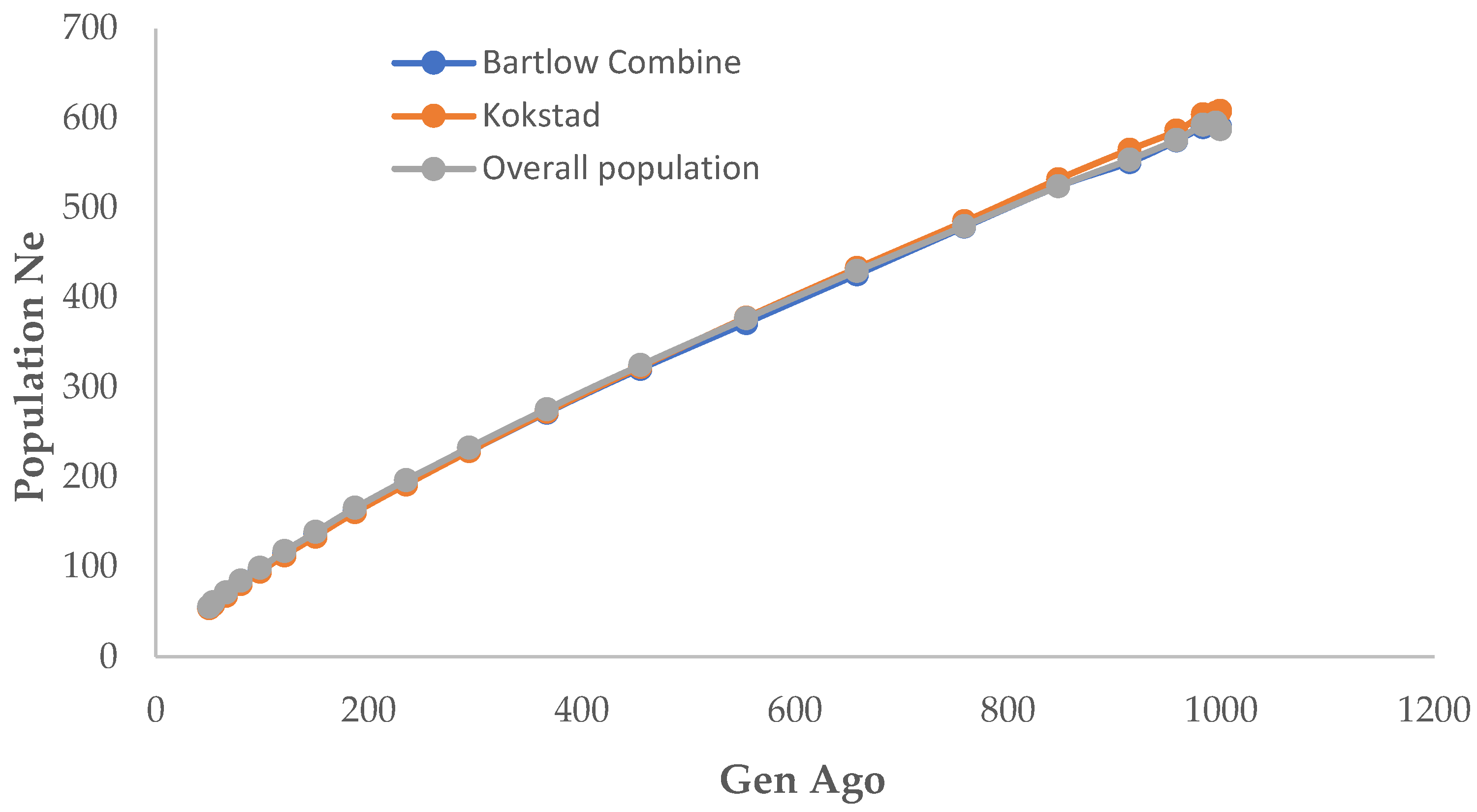
3.5. Effective Population Size
3.6. Selective Sweeps in Bartlow Combine and Kokstad Nguni Cattle Populations
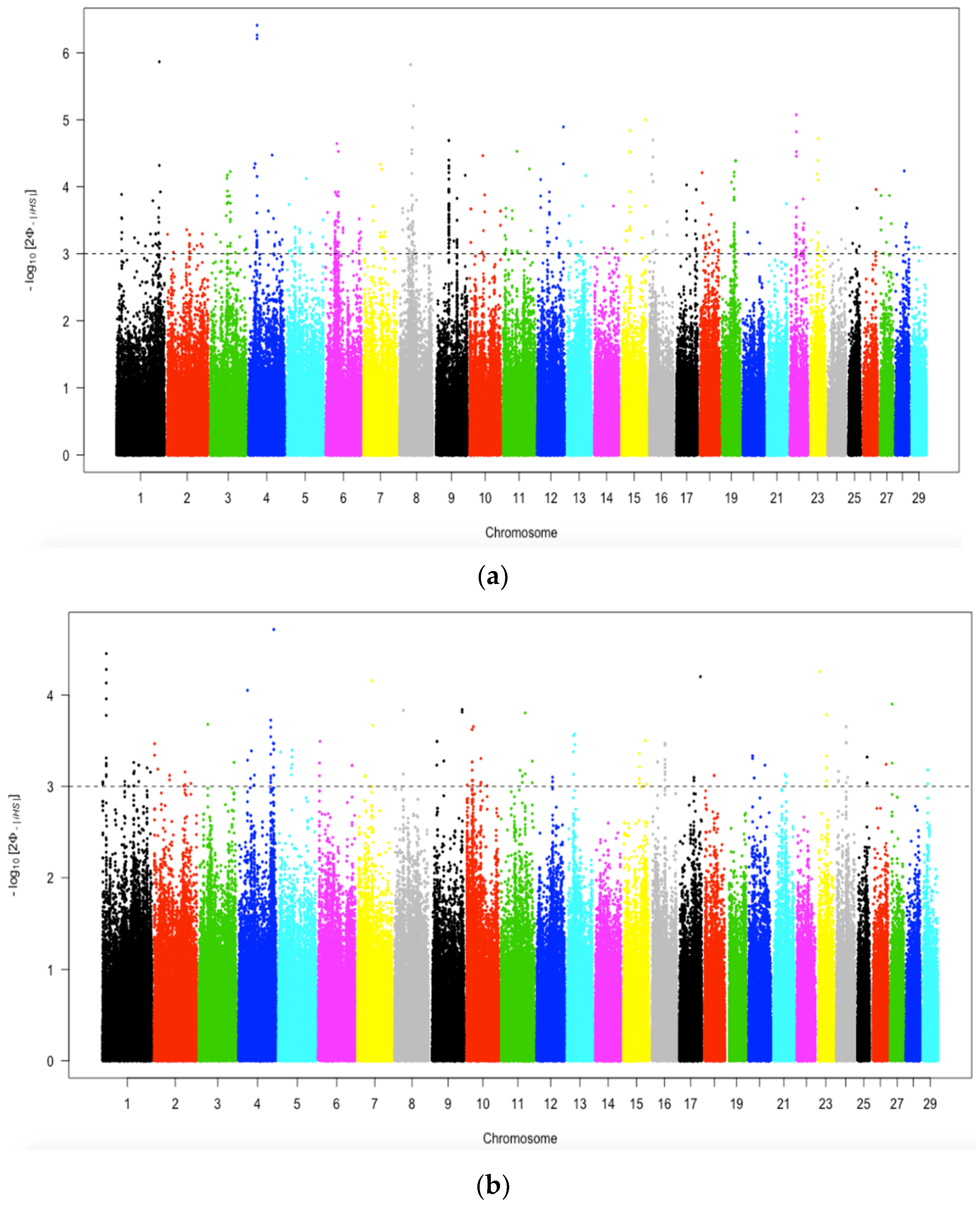
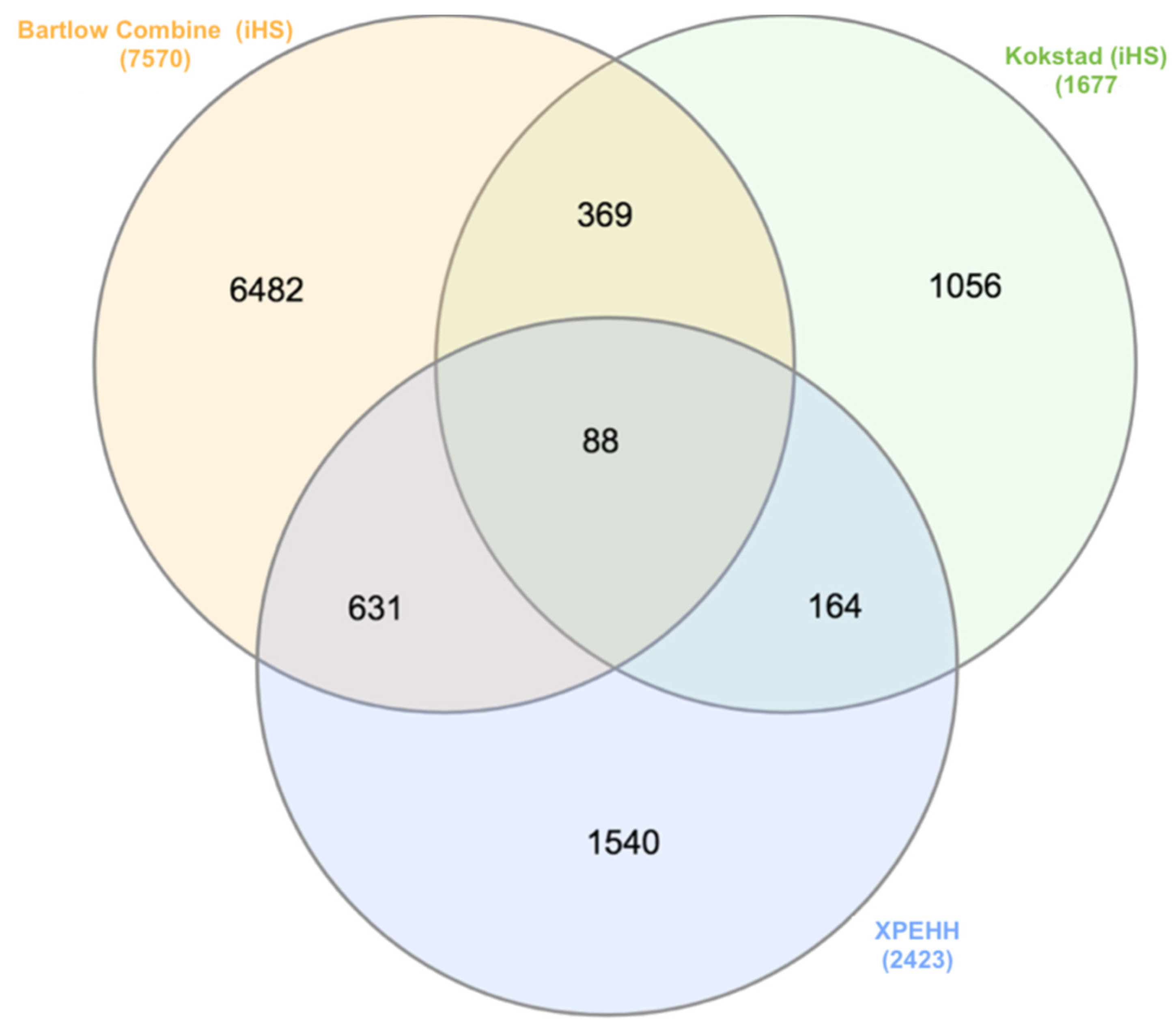
3.6.1. Recent Positive Selection Identified by iHS
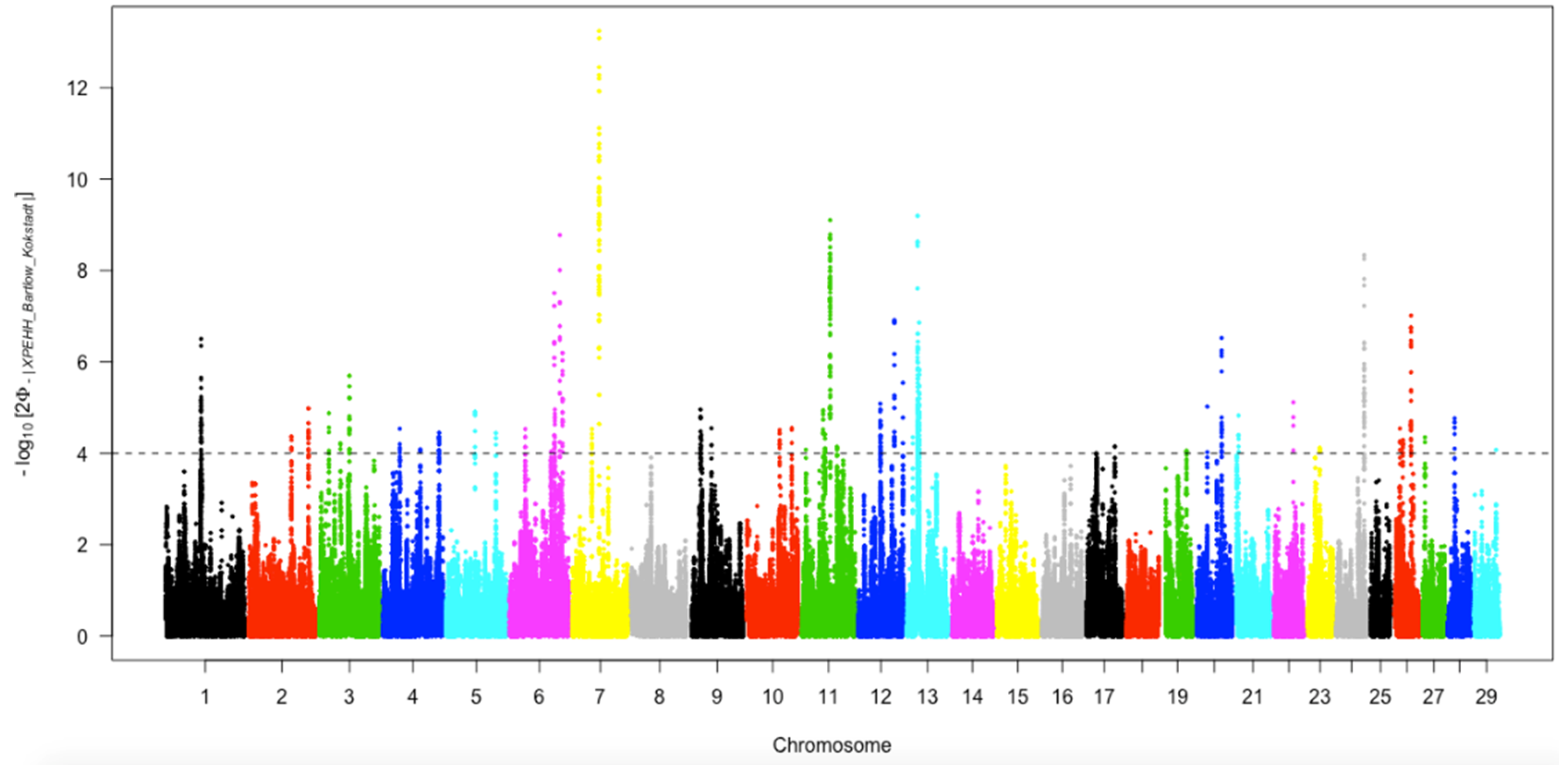
3.6.2. Positive Selection Identified by Cross-Population Extended Haplotype Homozygosity
3.6.3. Genomic Annotation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bester, J.; Matjuda, L.E.; Rust, J.M.; Fourie, H.J. The Nguni: A Case Study; Animal Improvement Institute: Irene, South Africa, 2003; pp. 1–36. [Google Scholar]
- Mwai, O.; Hanotte, O.; Kwon, Y.-J.; Cho, S. African Indigenous Cattle: Unique Genetic Resources in a Rapidly Changing World. Asian-Australas. J. Anim. Sci. 2015, 28, 911–921. [Google Scholar] [CrossRef]
- Makina, S.O.; Muchadeyi, F.C.; van Marle-Köster, E.; Taylor, J.F.; Makgahlela, M.L.; Maiwashe, A. Genome-Wide Scan for Selection Signatures in Six Cattle Breeds in South Africa. Genet. Sel. Evol. 2015, 47, 92. [Google Scholar] [CrossRef] [PubMed]
- Rechav, Y.; Kostrzewski, M.W. Relative Resistance of Six Cattle Breeds to the Tick Boophilus Decoloratus in South Africa. Onderstepoort J. Vet. Res. 1991, 58, 181–186. [Google Scholar] [PubMed]
- Marufu, M.C. Prevalence of Ticks and Tick-Borne Diseases in Cattle on Communal Rangelands in the Highland Areas of the Eastern Cape Province, South Africa. Ph.D. Thesis, University of Fort Hare, Alice, South Africa, 2008. [Google Scholar]
- Ndlovu, T.; Chimonyo, M.; Okoh, A.I. A Comparison of Stress Hormone Concentrations at Slaughter in Nguni, Bonsmara and Angus Steers. Afr. J. Agric. Res. 2008, 3, 96–100. [Google Scholar]
- Mapiye, C.; Chimonyo, M.; Muchenje, V. Potential for Value-Addition of Nguni Cattle Products in the Communal Areas of South Africa: A Review. Afr. J. Agric. Res. 2007, 2, 488–495. [Google Scholar]
- Katiyatiya, C.L.F.; Muchenje, V. Hair Coat Characteristics and Thermophysiological Stress Response of Nguni and Boran Cows Raised under Hot Environmental Conditions. Int. J. Biometeorol. 2017, 61, 2183–2194. [Google Scholar] [CrossRef]
- Sanarana, Y.; Visser, C.; Bosman, L.; Nephawe, K.; Maiwashe, A.; van Marle-Köster, E. Genetic Diversity in South African Nguni Cattle Ecotypes Based on Microsatellite Markers. Trop. Anim. Health Prod. 2016, 48, 379–385. [Google Scholar] [CrossRef] [PubMed]
- Schoeman, S.J. Recent Research into the Production Potential of Indigenous Cattle with Special Reference to the Sanga. S. Afr. J. Anim. Sci. 1989, 19, 55–61. [Google Scholar]
- Rege, J.E.O.; Tawah, C.L. The State of African Cattle Genetic Resources II. Geographical Distribution, Characteristics and Uses of Present-Day Breeds and Strains. Anim. Genet. Resour. 1999, 26, 1–25. [Google Scholar] [CrossRef]
- Rischkowsky, B.; Food and Agriculture Organization of the United Nations. The State of the World’s Animal Genetic Resources for Food and Agriculture—In Brief; Food and Agriculture Organization: Rome, Italy, 2007; ISBN 9789251057636. [Google Scholar]
- Nyamushamba, G.B.; Mapiye, C.; Tada, O. Conservation of Indigenous Cattle Genetic Resources in Southern Africa’s Smallholder Areas: Turning Threats into opportunities—A Review. Asian-Australas. J. Anim. Sci. 2017, 30, 603. [Google Scholar] [CrossRef]
- Marufu, M.C.; Qokweni, L.; Chimonyo, M.; Dzama, K. Relationships between Tick Counts and Coat Characteristics in Nguni and Bonsmara Cattle Reared on Semiarid Rangelands in South Africa. Ticks Tick Borne Dis. 2011, 2, 172–177. [Google Scholar] [CrossRef] [PubMed]
- Research Stations. Available online: https://www.kzndard.gov.za/research-stations (accessed on 21 February 2022).
- Xu, L.; Zhu, B.; Wang, Z.; Xu, L.; Liu, Y.; Chen, Y.; Zhang, L.; Gao, X.; Gao, H.; Zhang, S.; et al. Evaluation of Linkage Disequilibrium, Effective Population Size and Haplotype Block Structure in Chinese Cattle. Animals 2019, 9, 83. [Google Scholar] [CrossRef]
- Jasielczuk, I.; Gurgul, A.; Szmatoła, T.; Semik-Gurgul, E.; Pawlina-Tyszko, K.; Stefaniuk-Szmukier, M.; Polak, G.; Tomczyk-Wrona, I.; Bugno-Poniewierska, M. Linkage Disequilibrium, Haplotype Blocks and Historical Effective Population Size in Arabian Horses and Selected Polish Native Horse Breeds. Livest. Sci. 2020, 239, 104095. [Google Scholar] [CrossRef]
- Badke, Y.M.; Bates, R.O.; Ernst, C.W.; Schwab, C.; Steibel, J.P. Estimation of Linkage Disequilibrium in Four US Pig Breeds. BMC Genom. 2012, 13, 24. [Google Scholar] [CrossRef]
- Jasielczuk, I.; Gurgul, A.; Szmatoła, T.; Ząbek, T.; Pawlina, K.; Semik, E.; Bugno-Poniewierska, M. Linkage Disequilibrium and Haplotype Block Structure in Limousin, Simmental and Native Polish Red Cattle. Livest. Sci. 2016, 191, 57–63. [Google Scholar] [CrossRef]
- Qanbari, S. On the Extent of Linkage Disequilibrium in the Genome of Farm Animals. Front. Genet. 2019, 10, 1304. [Google Scholar] [CrossRef] [PubMed]
- Palamara, P.F.; Lencz, T.; Darvasi, A.; Pe’er, I. Length Distributions of Identity by Descent Reveal Fine-Scale Demographic History. Am. J. Hum. Genet. 2012, 91, 809–822. [Google Scholar] [CrossRef]
- Browning, S.R.; Browning, B.L.; Daviglus, M.L.; Durazo-Arvizu, R.A.; Schneiderman, N.; Kaplan, R.C.; Laurie, C.C. Ancestry-Specific Recent Effective Population Size in the Americas. PLoS Genet. 2018, 14, e1007385. [Google Scholar] [CrossRef]
- Zhao, F.; McParland, S.; Kearney, F.; Du, L.; Berry, D.P. Detection of Selection Signatures in Dairy and Beef Cattle Using High-Density Genomic Information. Genet. Sel. Evol. 2015, 47, 49. [Google Scholar] [CrossRef]
- Chen, M.; Pan, D.; Ren, H.; Fu, J.; Li, J.; Su, G.; Wang, A.; Jiang, L.; Zhang, Q.; Liu, J.-F. Identification of Selective Sweeps Reveals Divergent Selection between Chinese Holstein and Simmental Cattle Populations. Genet. Sel. Evol. 2016, 48, 76. [Google Scholar] [CrossRef]
- Cesarani, A.; Sorbolini, S.; Criscione, A.; Bordonaro, S.; Pulina, G.; Battacone, G.; Marletta, D.; Gaspa, G.; Macciotta, N.P.P. Genome-Wide Variability and Selection Signatures in Italian Island Cattle Breeds. Anim. Genet. 2018, 49, 371–383. [Google Scholar] [CrossRef] [PubMed]
- South African Weather Service. Available online: https://www.weathersa.co.za/ (accessed on 25 March 2022).
- Gertenbach, W.D.; Kars, A.A. Towards the Conservation of the Indigenous Cattle of KwaZulu-Natal. S. Afr. J. Anim. Sci. 2009, 29. [Google Scholar] [CrossRef]
- Peard, R.H. The Agricultural Research Stations of the Natal Region: A General Guide; Department of Agriculture and Fisheries: Pietermaritzburg, South Africa, 1980.
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2021; Available online: https://www.R-project.org/ (accessed on 5 February 2021).
- Hill, W.G. Estimation of Linkage Disequilibrium in Randomly Mating Populations. Heredity 1974, 33, 229–239. [Google Scholar] [CrossRef]
- Ardlie, K.G.; Kruglyak, L.; Seielstad, M. Patterns of Linkage Disequilibrium in the Human Genome. Nat. Rev. Genet. 2002, 3, 299–309. [Google Scholar] [CrossRef]
- Zhao, H.; Nettleton, D.; Soller, M.; Dekkers, J.C.M. Evaluation of Linkage Disequilibrium Measures between Multi-Allelic Markers as Predictors of Linkage Disequilibrium between Markers and QTL. Genet. Res. 2005, 86, 77–87. [Google Scholar] [CrossRef]
- Barrett, J.C.; Fry, B.; Maller, J.; Daly, M.J. Haploview: Analysis and Visualization of LD and Haplotype Maps. Bioinformatics 2005, 21, 263–265. [Google Scholar] [CrossRef]
- Gabriel, S.B.; Schaffner, S.F.; Nguyen, H.; Moore, J.M.; Roy, J.; Blumenstiel, B.; Higgins, J.; DeFelice, M.; Lochner, A.; Faggart, M.; et al. The structure of haplotype blocks in the human genome. Science 2002, 296, 2225–2229. [Google Scholar] [CrossRef]
- Heberle, H.; Meirelles, G.V.; da Silva, F.R.; Telles, G.P.; Minghim, R. InteractiVenn: A Web-Based Tool for the Analysis of Sets through Venn Diagrams. BMC Bioinform. 2015, 16, 169. [Google Scholar] [CrossRef]
- Barbato, M.; Orozcoter Wengel, P.; Tapio, M.; Bruford, M.W. SNeP: A Tool to Estimate Trends in Recent Effective Population Size Trajectories Using Genome-Wide SNP Data. Front. Genet. 2015, 6, 109. [Google Scholar] [CrossRef]
- Ohta, T.; Kimura, M. Linkage disequilibrium between two segregating nucleotide sites under the steady flux of mutations in a finite population. Genetics 1971, 68, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Sabeti, P.C.; Varilly, P.; Fry, B.; Lohmueller, J.; Hostetter, E.; Cotsapas, C.; Xie, X.; Byrne, E.H.; McCarroll, S.A.; Gaudet, R.; et al. Genome-Wide Detection and Characterization of Positive Selection in Human Populations. Nature 2007, 449, 913–918. [Google Scholar] [CrossRef] [PubMed]
- Voight, B.F.; Kudaravalli, S.; Wen, X.; Pritchard, J.K. A Map of Recent Positive Selection in the Human Genome. PLoS Biol. 2006, 4, e72. [Google Scholar]
- Kasarda, R.; Moravčíková, N.; Trakovická, A.; Mészáros, G.; Kadlečík, O. Genome-Wide Selection Signatures in Pinzgau Cattle. Potravin. Slovak J. Food Sci. 2015, 9, 268–274. [Google Scholar] [CrossRef]
- Scheet, P.; Stephens, M. A Fast and Flexible Statistical Model for Large-Scale Population Genotype Data: Applications to Inferring Missing Genotypes and Haplotypic Phase. Am. J. Hum. Genet. 2006, 78, 629–644. [Google Scholar] [CrossRef]
- Gautier, M.; Klassmann, A.; Vitalis, R. rehh2.0: A Reimplementation of the R Packagerehhto Detect Positive Selection from Haplotype Structure. Mol. Ecol. Resour. 2017, 17, 78–90. [Google Scholar] [CrossRef]
- National Library of Medicine. Available online: https://www.ncbi.nlm.nih.gov/assembly/GCF_002263795.1/ (accessed on 10 March 2019).
- Quinlan, A.R.; Hall, I.M. BEDTools: A Flexible Suite of Utilities for Comparing Genomic Features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef]
- Liu, L.L.; Fang, C.; Liu, W.J. Identification on Novel Locus of Dairy Traits of Kazakh Horse in Xinjiang. Gene 2018, 677, 105–110. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Sato, Y.; Furumichi, M.; Tanabe, M. KEGG for Integration and Interpretation of Large-Scale Molecular Data Sets. Nucleic Acids Res. 2012, 40, D109–D114. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene Ontology: Tool for the Unification of Biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and Integrative Analysis of Large Gene Lists Using DAVID Bioinformatics Resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.-L.; Park, C.A.; Reecy, J.M. Bringing the Animal QTLdb and CorrDB into the Future: Meeting New Challenges and Providing Updated Services. Nucleic Acids Res. 2022, 50, D956–D961. [Google Scholar] [CrossRef] [PubMed]
- Zwane, A.A.; Maiwashe, A.; Makgahlela, M.L.; Choudhury, A.; Taylor, J.F.; Van Marle-Köster, E. Genome-Wide Identification of Breed-Informative Single-Nucleotide Polymorphisms in Three South African Indigenous Cattle Breeds. S. Afr. J. Anim. Sci. 2016, 46, 302–312. [Google Scholar] [CrossRef]
- Slarkin, M. Gene Flow in Natural Populations. Annu. Rev. Ecol. Syst. 1985, 16, 393–430. [Google Scholar] [CrossRef]
- McKay, S.D.; Schnabel, R.D.; Murdoch, B.M.; Matukumalli, L.K.; Aerts, J.; Coppieters, W.; Crews, D.; Neto, E.D.; Gill, C.A.; Gao, C.; et al. An assessment of population structure in eight breeds of cattle using a whole genome SNP panel. BMC Genet. 2008, 9, 37. [Google Scholar] [CrossRef]
- Matukumalli, L.K.; Lawley, C.T.; Schnabel, R.D.; Taylor, J.F.; Allan, M.F.; Heaton, M.P.; O’Connell, J.; Moore, S.S.; Smith, T.P.; Sonstegard, T.S.; et al. Development and characterization of a high density SNP genotyping assay for cattle. PLoS ONE 2009, 4, e5350. [Google Scholar] [CrossRef]
- O’Brien, A.M.P.; Pérez O’Brien, A.M.; Mészáros, G.; Utsunomiya, Y.T.; Sonstegard, T.S.; Fernando Garcia, J.; Van Tassell, C.P.; Carvalheiro, R.; da Silva, M.V.B.; Sölkner, J. Linkage Disequilibrium Levels in Bos Indicus and Bos Taurus Cattle Using Medium and High Density SNP Chip Data and Different Minor Allele Frequency Distributions. Livest. Sci. 2014, 166, 121–132. [Google Scholar] [CrossRef]
- Karimi, K.; Esmailizadeh Koshkoiyeh, A.; Gondro, C. Comparison of Linkage Disequilibrium Levels in Iranian Indigenous Cattle Using Whole Genome SNPs Data. Hanguk Tongmul Chawon Kwahakhoe Chi 2015, 57, 47. [Google Scholar] [CrossRef]
- Toro Ospina, A.M.; Maiorano, A.M.; Curi, R.A.; Pereira, G.L.; Zerlotti-Mercadante, M.E.; Dos Santos Gonçalves Cyrillo, J.N.; Aspilcueta-Borquis, R.R.; de V. Silva, J.A., II. Linkage Disequilibrium and Effective Population Size in Gir Cattle Selected for Yearling Weight. Reprod. Domest. Anim. 2019, 54, 1524–1531. [Google Scholar] [CrossRef]
- Bonsma, F.N.; Bisschop, J.H.R.; Barnnrd, W.G.; Van Rensburg, J.A.; Duvenhage, J.J.; Van Wyk, H.P.D.; Watermeyer, F. Nguni cattle. Bull. Dept. Agric. 1950, 311. [Google Scholar]
- Rogers, A.R. How Population Growth Affects Linkage Disequilibrium. Genetics 2014, 197, 1329–1341. [Google Scholar] [CrossRef] [PubMed]
- Meuwissen, T. Genomic Selection: Marker Assisted Selection on a Genome Wide Scale. J. Anim. Breed. Genet. 2007, 124, 321–322. [Google Scholar] [CrossRef]
- Gasparin, G.; Miyata, M.; Coutinho, L.L.; Martinez, M.L.; Teodoro, R.L.; Furlong, J.; Machado, M.A.; Silva, M.V.G.B.; Sonstegard, T.S.; Regitano, L.C.A. Mapping of Quantitative Trait Loci Controlling Tick [Riphicephalus (Boophilus) microplus] Resistance on Bovine Chromosomes 5, 7 and 14. Anim. Genet. 2007, 38, 453–459. [Google Scholar] [CrossRef] [PubMed]
- Jia, C.; Wang, H.; Li, C.; Wu, X.; Zan, L.; Ding, X.; Guo, X.; Bao, P.; Pei, J.; Chu, M.; et al. Genome-Wide Detection of Copy Number Variations in Polled Yak Using the Illumina BovineHD BeadChip. BMC Genom. 2019, 20, 376. [Google Scholar] [CrossRef] [PubMed]
- Twomey, A.J.; Berry, D.P.; Evans, R.D.; Doherty, M.L.; Graham, D.A.; Purfield, D.C. Genome-Wide Association Study of Endo-Parasite Phenotypes Using Imputed Whole-Genome Sequence Data in Dairy and Beef Cattle. Genet. Sel. Evol. 2019, 51, 15. [Google Scholar] [CrossRef] [PubMed]
- Muchenje, V.; Dzama, K.; Chimonyo, M.; Raats, J.G.; Strydom, P.E. Tick Susceptibility and Its Effects on Growth Performance and Carcass Characteristics of Nguni, Bonsmara and Angus Steers Raised on Natural Pasture. Animal 2008, 2, 298–304. [Google Scholar] [CrossRef]
- Mokry, F.B.; Buzanskas, M.E.; de Alvarenga Mudadu, M.; do Amaral Grossi, D.; Higa, R.H.; Ventura, R.V.; de Lima, A.O.; Sargolzaei, M.; Meirelles, S.L.C.; Schenkel, F.S.; et al. Linkage Disequilibrium and Haplotype Block Structure in a Composite Beef Cattle Breed. BMC Genom. 2014, 15, S6. [Google Scholar] [CrossRef]
- Wang, M.D.; Dzama, K.; Hefer, C.A.; Muchadeyi, F.C. Genomic Population Structure and Prevalence of Copy Number Variations in South African Nguni Cattle. BMC Genom. 2015, 16, 894. [Google Scholar] [CrossRef]
- Shifman, S.; Kuypers, J.; Kokoris, M.; Yakir, B.; Darvasi, A. Linkage Disequilibrium Patterns of the Human Genome across Populations. Hum. Mol. Genet. 2003, 12, 771–776. [Google Scholar] [CrossRef]
- Khanyile, K.S.; Dzomba, E.F.; Muchadeyi, F.C. Haplo-Block Structure of Southern African Village Chicken Populations Inferred Using Genome-Wide SNP Data. Genet. Mol. Res. 2015, 14, 12276–12287. [Google Scholar] [CrossRef]
- Clark, A.G.; Weiss, K.M.; Nickerson, D.A.; Taylor, S.L.; Buchanan, A.; Stengård, J.; Salomaa, V.; Vartiainen, E.; Perola, M.; Boerwinkle, E.; et al. Haplotype Structure and Population Genetic Inferences from Nucleotide-Sequence Variation in Human Lipoprotein Lipase. Am. J. Hum. Genet. 1998, 63, 595–612. [Google Scholar] [CrossRef]
- Templeton, A.R.; Clark, A.G.; Weiss, K.M.; Nickerson, D.A.; Boerwinkle, E.; Sing, C.F. Recombinational and Mutational Hotspots within the Human Lipoprotein Lipase Gene. Am. J. Hum. Genet. 2000, 66, 69–83. [Google Scholar] [CrossRef] [PubMed]
- De Roos, A.P.W.; Hayes, B.J.; Spelman, R.J.; Goddard, M.E. Linkage Disequilibrium and Persistence of Phase in Holstein-Friesian, Jersey and Angus Cattle. Genetics 2008, 179, 1503–1512. [Google Scholar] [CrossRef] [PubMed]
- Food and Agriculture Organization of the United Nations. In Vivo Conservation of Animal Genetic Resources; Food and Agriculture Organization of the United Nations: Rome, Italy, 2013; ISBN 9789251077252. [Google Scholar]
- Bang, J.; Zippin, J.H. Cyclic Adenosine Monophosphate (cAMP) Signaling in Melanocyte Pigmentation and Melanomagenesis. Pigment Cell Melanoma Res. 2021, 34, 28–43. [Google Scholar] [CrossRef] [PubMed]
- Cuello, F.; Herberg, F.W.; Stathopoulou, K.; Henning, P.; Diering, S. Regulation of Cardiac PKA Signaling by cAMP and Oxidants. Antioxidants 2021, 10, 663. [Google Scholar] [CrossRef]
- Mkize, L.S.; Zishiri, O.T. Genetic Variation and Population Differentiation in the Bovine Lymphocyte Antigen DRB3.2 Locus of South African Nguni Crossbred Cattle. Animals 2021, 11, 1651. [Google Scholar] [CrossRef]
- Aeschlimann, P.B.; Häberli, M.A.; Reusch, T.B.H.; Boehm, T.; Milinski, M. Female Sticklebacks Gasterosteus Aculeatus Use Self-Reference to Optimize MHC Allele Number during Mate Selection. Behav. Ecol. Sociobiol. 2003, 54, 119–126. [Google Scholar] [CrossRef]
- Hamilton, C.A.; Young, R.; Jayaraman, S.; Sehgal, A.; Paxton, E.; Thomson, S.; Katzer, F.; Hope, J.; Innes, E.; Morrison, L.J.; et al. Development of in Vitro Enteroids Derived from Bovine Small Intestinal Crypts. Vet. Res. 2018, 49, 54. [Google Scholar] [CrossRef]
- Beauchemin, K.A.; Eriksen, L.; Nørgaard, P.; Rode, L.M. Salivary Secretion during Meals in Lactating Dairy Cattle. J. Dairy Sci. 2008, 91, 2077–2081. [Google Scholar] [CrossRef]
- Rojas-Downing, M.M.; Nejadhashemi, A.P.; Harrigan, T.; Woznicki, S.A. Climate change and livestock: Impacts, adaptation, and mitigation. Clim. Risk Manag. 2017, 16, 145–163. [Google Scholar] [CrossRef]











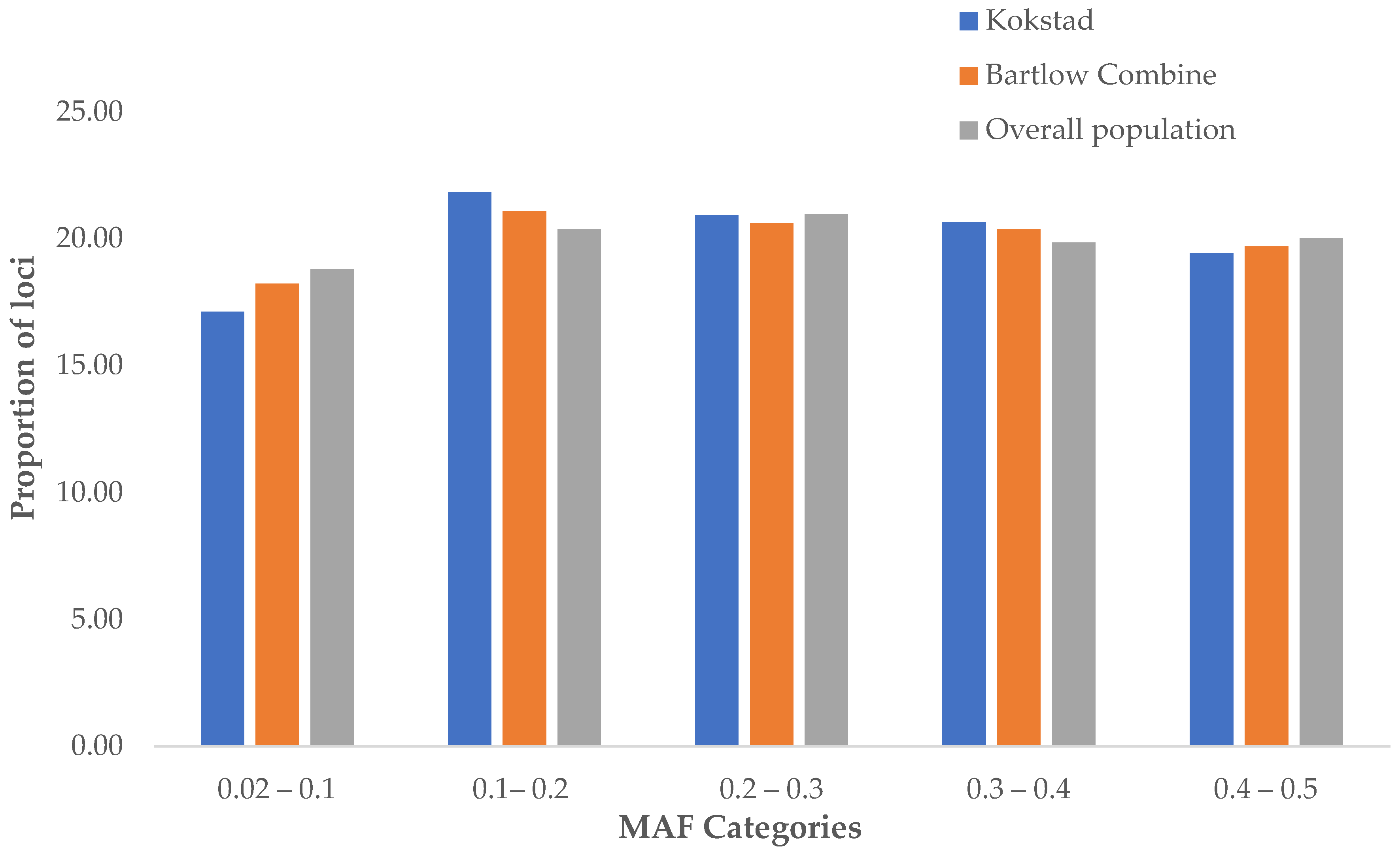{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Population | # Animals Pre-QC | # SNPs Pre-QC | # SNPs Post-QC | #Animals Post-QC | Genotyping Rate | Mean IBD | FIS | Mean MAF |
|---|---|---|---|---|---|---|---|---|
| Bartlow Combine | 99 | 777,962 | 643,275 | 85 | 0.998 | 0.041 | −0.014 | 0.25 ± 0.14 |
| Kokstad | 44 | 777,962 | 650,317 | 42 | 0.999 | 0.069 | −0.028 | 0.26 ± 0.14 |
| Overall | 143 | 777,962 | 650,430 | 127 | 0.998 | 0.032 | −0.011 | 0.25 ± 0.14 |
| Bartlow Combine | Kokstad | Overall Population | ||||
|---|---|---|---|---|---|---|
| Chr | Mean LD ± SD (r2) | Median (r2) | Mean LD ± SD (r2) | Median (r2) | Mean LD ± SD (r2) | Median (r2) |
| 1 | 0.419 ± 0.221 | 0.337 | 0.413 ± 0.214 | 0.335 | 0.422 ± 0.223 | 0.338 |
| 2 | 0.423 ± 0.225 | 0.338 | 0.412 ± 0.215 | 0.335 | 0.422 ± 0.225 | 0.338 |
| 3 | 0.418 ± 0.223 | 0.332 | 0.401 ± 0.209 | 0.324 | 0.420 ± 0.224 | 0.334 |
| 4 | 0.410 ± 0.213 | 0.332 | 0.408 ± 0.206 | 0.337 | 0.412 ± 0.213 | 0.335 |
| 5 | 0.419 ± 0.222 | 0.336 | 0.411 ± 0.213 | 0.334 | 0.421 ± 0.222 | 0.339 |
| 6 | 0.418 ± 0.220 | 0.338 | 0.413 ± 0.214 | 0.335 | 0.421 ± 0.223 | 0.337 |
| 7 | 0.436 ± 0.232 | 0.347 | 0.417 ± 0.216 | 0.339 | 0.433 ± 0.228 | 0.348 |
| 8 | 0.429 ± 0.221 | 0.350 | 0.417 ± 0.215 | 0.340 | 0.428 ± 0.221 | 0.349 |
| 9 | 0.417 ± 0.224 | 0.331 | 0.409 ± 0.214 | 0.330 | 0.421 ± 0.225 | 0.334 |
| 10 | 0.410 ± 0.217 | 0.328 | 0.398 ± 0.206 | 0.323 | 0.412 ± 0.219 | 0.330 |
| 11 | 0.419 ± 0.225 | 0.332 | 0.408 ± 0.213 | 0.331 | 0.422 ± 0.227 | 0.335 |
| 12 | 0.420 ± 0.220 | 0.338 | 0.402 ± 0.210 | 0.324 | 0.424 ± 0.223 | 0.341 |
| 13 | 0.414 ± 0.216 | 0.335 | 0.406 ± 0.207 | 0.332 | 0.418 ± 0.216 | 0.338 |
| 14 | 0.407 ± 0.211 | 0.330 | 0.405 ± 0.208 | 0.330 | 0.409 ± 0.212 | 0.330 |
| 15 | 0.415 ± 0.217 | 0.334 | 0.407 ± 0.214 | 0.329 | 0.417 ± 0.220 | 0.335 |
| 16 | 0.416 ± 0.224 | 0.330 | 0.406 ± 0.216 | 0.325 | 0.423 ± 0.227 | 0.339 |
| 17 | 0.410 ± 0.221 | 0.325 | 0.400 ± 0.210 | 0.322 | 0.417 ± 0.225 | 0.330 |
| 18 | 0.407 ± 0.221 | 0.319 | 0.387 ± 0.198 | 0.314 | 0.413 ± 0.223 | 0.326 |
| 19 | 0.408 ± 0.215 | 0.327 | 0.397 ± 0.204 | 0.322 | 0.415 ± 0.218 | 0.334 |
| 20 | 0.409 ± 0.217 | 0.327 | 0.409 ± 0.213 | 0.330 | 0.417 ± 0.222 | 0.332 |
| 21 | 0.424 ± 0.224 | 0.341 | 0.410 ± 0.216 | 0.328 | 0.428 ± 0.227 | 0.344 |
| 22 | 0.422 ± 0.225 | 0.337 | 0.400 ± 0.208 | 0.323 | 0.421 ± 0.225 | 0.334 |
| 23 | 0.399 ± 0.217 | 0.314 | 0.384 ± 0.203 | 0.307 | 0.409 ± 0.222 | 0.320 |
| 24 | 0.405 ± 0.214 | 0.325 | 0.396 ± 0.205 | 0.321 | 0.410 ± 0.218 | 0.327 |
| 25 | 0.410 ± 0.223 | 0.322 | 0.387 ± 0.207 | 0.378 | 0.421 ± 0.230 | 0.330 |
| 26 | 0.390 ± 0.208 | 0.310 | 0.379 ± 0.198 | 0.306 | 0.399 ± 0.215 | 0.314 |
| 27 | 0.406 ± 0.220 | 0.321 | 0.399 ± 0.207 | 0.324 | 0.416 ± 0.225 | 0.329 |
| 28 | 0.398 ± 0.211 | 0.318 | 0.385 ± 0.203 | 0.310 | 0.406 ± 0.216 | 0.323 |
| 29 | 0.401 ± 0.212 | 0.320 | 0.391 ± 0.201 | 0.316 | 0.406 ± 0.215 | 0.324 |
| Mean | 0.413 ± 0.219 | 0.330 | 0.402 ± 0.209 | 0.328 | 0.417 ± 0.222 | 0.333 |
| Populations | Bartlow Combine | Kokstad | Overall Population |
|---|---|---|---|
| Blocks (n) | 77,305 | 66,237 | 84,182 |
| Total block lengtha (Mb) | 1570.09 | 1367.42 | 1619.33 |
| % Coverage | 61.99 | 53.96 | 65.05 |
| Mean block length (kb) | 20.31 | 20.64 | 19.24 |
| Haplotype frequency | 0.23 | 0.23 | 0.23 |
| Median block length (kb) | 10.08 | 10.10 | 9.82 |
| Max block length (kb) | 999.05 | 999.37 | 999.05 |
| No. of SNPs in blocks | 485,025 | 429,462 | 508,313 |
| SNP % in blocks | 74.88 | 65.44 | 78.27 |
| Mean num of SNPs in blocks | 6.27 | 6.48 | 6.04 |
| Max num of SNPs in blocks | 185.00 | 193.00 | 186.00 |
| Populations | SNPs under Selection | Genes | GO-Terms | KEGG Pathways |
|---|---|---|---|---|
| iHS test | ||||
| Bartlow Combine | 553 | 7570 | 183 | 61 |
| Kokstad | 166 | 1677 | 68 | 11 |
| Overall population | 505 | 94 | 23 | 0 |
| XPEHH | ||||
| Bartlow Combine vs Kokstad | 57 | 2423 | 137 | 37 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dlamini, N.M.; Dzomba, E.F.; Magawana, M.; Ngcamu, S.; Muchadeyi, F.C. Linkage Disequilibrium, Haplotype Block Structures, Effective Population Size and Genome-Wide Signatures of Selection of Two Conservation Herds of the South African Nguni Cattle. Animals 2022, 12, 2133. https://doi.org/10.3390/ani12162133
Dlamini NM, Dzomba EF, Magawana M, Ngcamu S, Muchadeyi FC. Linkage Disequilibrium, Haplotype Block Structures, Effective Population Size and Genome-Wide Signatures of Selection of Two Conservation Herds of the South African Nguni Cattle. Animals. 2022; 12(16):2133. https://doi.org/10.3390/ani12162133
Chicago/Turabian StyleDlamini, Njabulo M., Edgar F. Dzomba, Mpumelelo Magawana, Sphamandla Ngcamu, and Farai C. Muchadeyi. 2022. "Linkage Disequilibrium, Haplotype Block Structures, Effective Population Size and Genome-Wide Signatures of Selection of Two Conservation Herds of the South African Nguni Cattle" Animals 12, no. 16: 2133. https://doi.org/10.3390/ani12162133