
Genomic Imprinting at the Porcine DIRAS3 Locus


Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection and Data Used
2.2. Whole-Genome Bisulfite Sequencing (WGBS) and RNA Sequencing (RNA-seq)
2.3. Bioinformatics and Statistical Analysis
3. Results
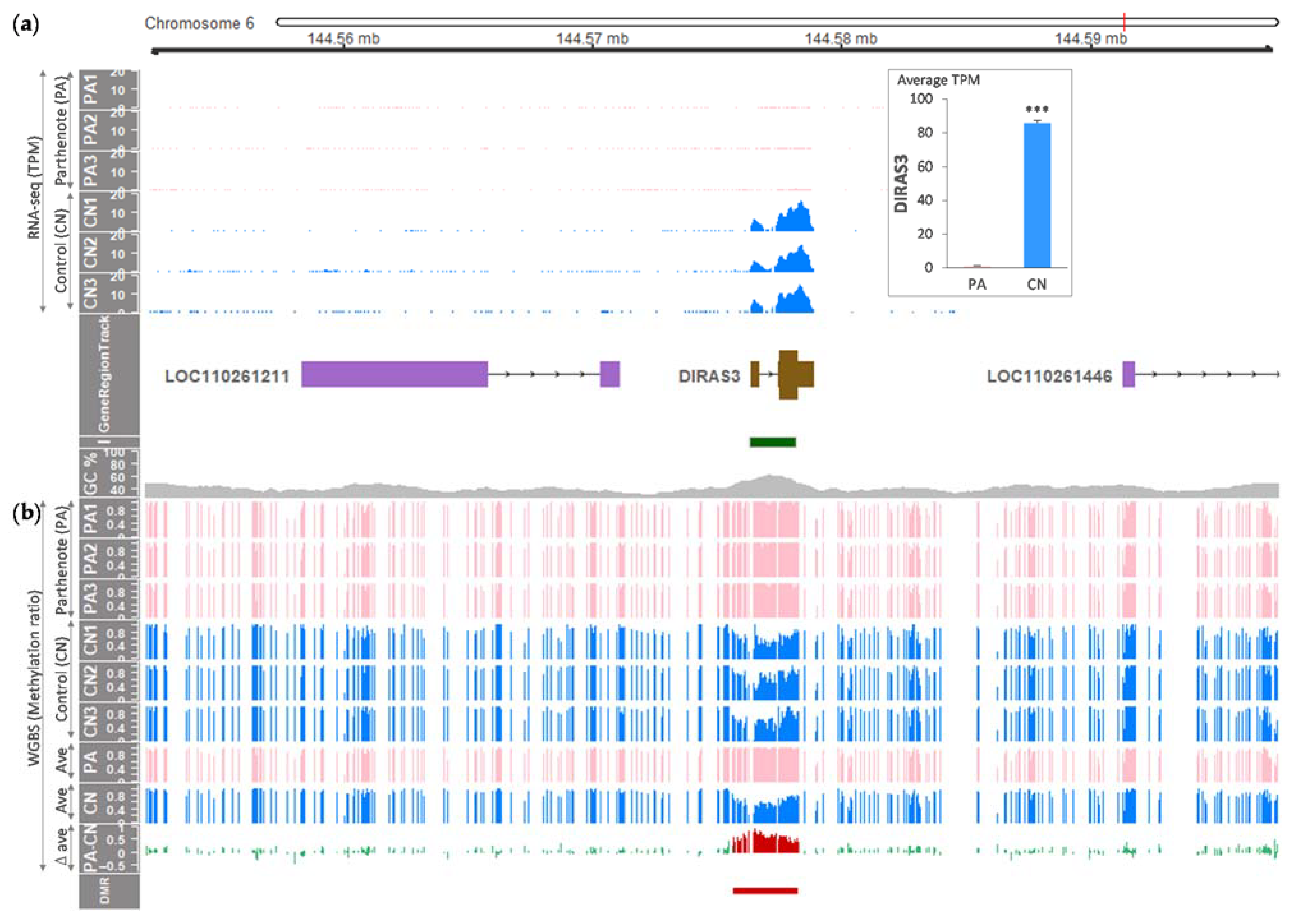
3.1. Differential Expression and Methylation at the DIRAS3 Locus of Porcine Embryos
3.2. Sequence Elements and Differentially Methylated CpGs within the Porcine DIRAS3 Locus
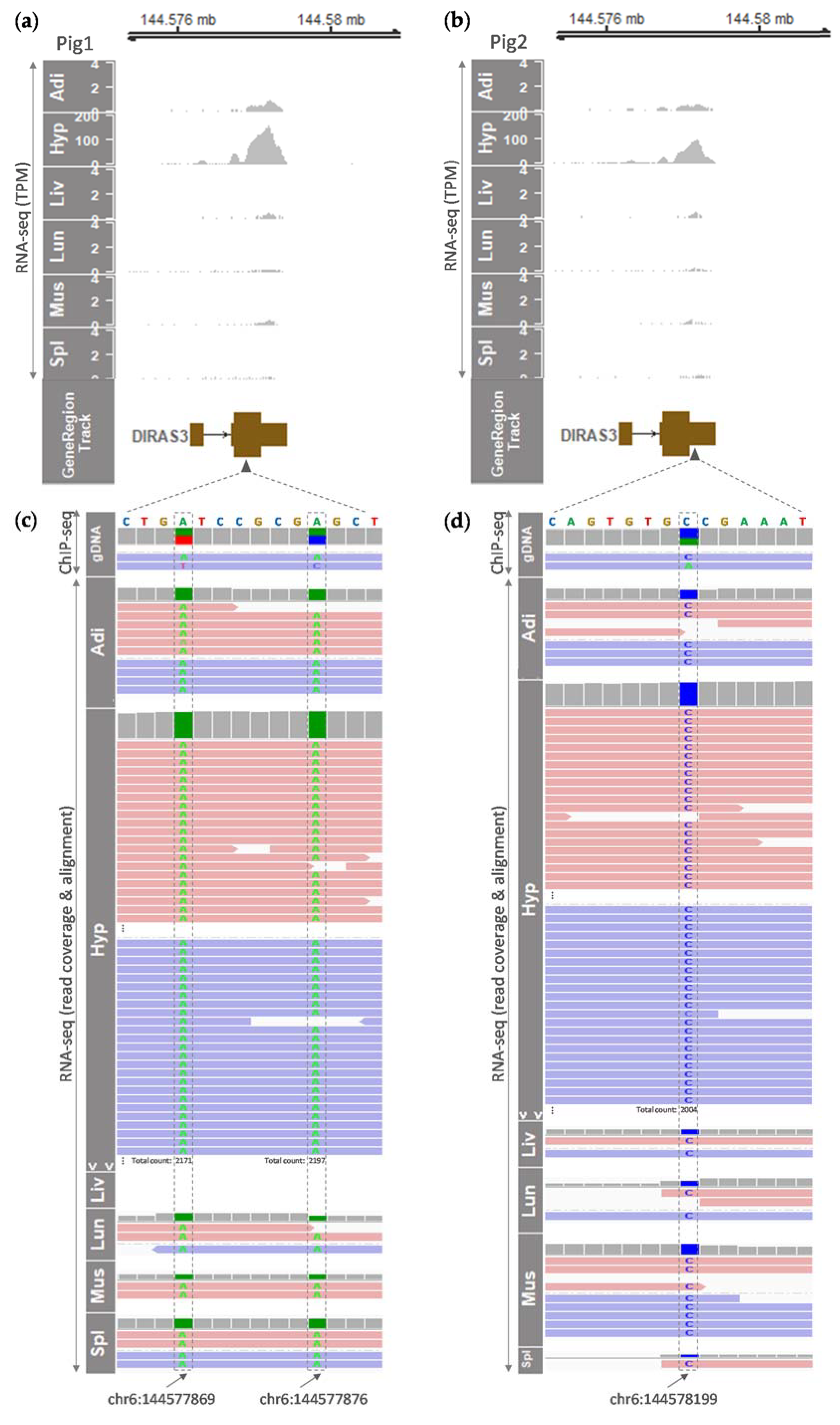
3.3. Tissue-Specific and Monoallelic Expression of the DIRAS3 Gene in Adult Pigs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Franklin, G.C.; Adam, G.I.; Ohlsson, R. Genomic imprinting and mammalian development. Placenta 1996, 17, 3–14. [Google Scholar] [CrossRef]
- Greenberg, M.V.C.; Bourc’his, D. The diverse roles of DNA methylation in mammalian development and disease. Nat. Rev. Mol. Cell Biol 2019, 20, 590–607. [Google Scholar] [CrossRef]
- Zeng, Y.; Chen, T. DNA Methylation Reprogramming during Mammalian Development. Genes 2019, 10, 257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- da Rocha, S.T.; Gendrel, A.V. The influence of DNA methylation on monoallelic expression. Essays Biochem. 2019, 63, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Ono, R.; Kobayashi, S.; Wagatsuma, H.; Aisaka, K.; Kohda, T.; Kaneko-Ishino, T.; Ishino, F. A retrotransposon-derived gene, PEG10, is a novel imprinted gene located on human chromosome 7q21. Genomics 2001, 73, 232–237. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, M.; Zimprich, A.; Lorenz-Depiereux, B.; Kalscheuer, V.; Asmus, F.; Gasser, T.; Meitinger, T.; Strom, T.M. The epsilon-sarcoglycan gene (SGCE), mutated in myoclonus-dystonia syndrome, is maternally imprinted. Eur. J. Hum. Genet. 2003, 11, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Piras, G.; El Kharroubi, A.; Kozlov, S.; Escalante-Alcalde, D.; Hernandez, L.; Copeland, N.G.; Gilbert, D.J.; Jenkins, N.A.; Stewart, C.L. Zac1 (Lot1), a potential tumor suppressor gene, and the gene for epsilon-sarcoglycan are maternally imprinted genes: Identification by a subtractive screen of novel uniparental fibroblast lines. Mol. Cell. Biol. 2000, 20, 3308–3315. [Google Scholar] [CrossRef]
- Ono, R.; Shiura, H.; Aburatani, H.; Kohda, T.; Kaneko-Ishino, T.; Ishino, F. Identification of a large novel imprinted gene cluster on mouse proximal chromosome 6. Genome Res. 2003, 13, 1696–1705. [Google Scholar] [CrossRef] [Green Version]
- Ahn, J.; Hwang, I.S.; Park, M.R.; Cho, I.C.; Hwang, S.; Lee, K. The Landscape of Genomic Imprinting at the Porcine SGCE/PEG10 Locus from Methylome and Transcriptome of Parthenogenetic Embryos. G3 (Bethesda) 2020. [Google Scholar] [CrossRef]
- Duan, J.E.; Zhang, M.; Flock, K.; Seesi, S.A.; Mandoiu, I.; Jones, A.; Johnson, E.; Pillai, S.; Hoffman, M.; McFadden, K.; et al. Effects of maternal nutrition on the expression of genomic imprinted genes in ovine fetuses. Epigenetics 2018, 13, 793–807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Liu, S.; Fang, X.; He, C.; Hu, X. The mechanisms of DIRAS family members in role of tumor suppressor. J. Cell. Physiol. 2019, 234, 5564–5577. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Bast, R.C., Jr. The tumor suppressor gene ARHI (DIRAS3) inhibits ovarian cancer cell migration through multiple mechanisms. Cell Adh. Migr. 2013, 7, 232–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Y.; Xu, F.; Peng, H.; Fang, X.; Zhao, S.; Li, Y.; Cuevas, B.; Kuo, W.L.; Gray, J.W.; Siciliano, M.; et al. NOEY2 (ARHI), an imprinted putative tumor suppressor gene in ovarian and breast carcinomas. Proc. Natl. Acad. Sci. USA 1999, 96, 214–219. [Google Scholar] [CrossRef] [Green Version]
- Xu, F.; Xia, W.; Luo, R.Z.; Peng, H.; Zhao, S.; Dai, J.; Long, Y.; Zou, L.; Le, W.; Liu, J.; et al. The human ARHI tumor suppressor gene inhibits lactation and growth in transgenic mice. Cancer Res. 2000, 60, 4913–4920. [Google Scholar]
- Bischoff, S.R.; Tsai, S.; Hardison, N.; Motsinger-Reif, A.A.; Freking, B.A.; Nonneman, D.; Rohrer, G.; Piedrahita, J.A. Characterization of conserved and nonconserved imprinted genes in swine. Biol. Reprod. 2009, 81, 906–920. [Google Scholar] [CrossRef] [Green Version]
- Cheng, H.C.; Zhang, F.W.; Deng, C.Y.; Jiang, C.D.; Xiong, Y.Z.; Li, F.E.; Lei, M.G. NNAT and DIRAS3 genes are paternally expressed in pigs. Genet. Sel. Evol. 2007, 39, 599–607. [Google Scholar] [CrossRef]
- Ahn, J.; Wu, H.; Lee, J.; Hwang, I.S.; Yu, D.; Ahn, J.S.; Lee, J.W.; Hwang, S.; Lee, K. Identification of a Novel Imprinted Transcript in the Porcine GNAS Complex Locus Using Methylome and Transcriptome of Parthenogenetic Fetuses. Genes 2020, 11, 96. [Google Scholar] [CrossRef] [Green Version]
- Xi, Y.; Li, W. BSMAP: Whole genome bisulfite sequence MAPping program. BMC Bioinform. 2009, 10, 232. [Google Scholar] [CrossRef] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, F.; Dundar, F.; Diehl, S.; Gruning, B.A.; Manke, T. deepTools: A flexible platform for exploring deep-sequencing data. Nucleic Acids Res. 2014, 42, W187–W191. [Google Scholar] [CrossRef] [Green Version]
- Hahne, F.; Ivanek, R. Visualizing Genomic Data Using Gviz and Bioconductor. Methods Mol. Biol. 2016, 1418, 335–351. [Google Scholar] [CrossRef] [PubMed]
- Thorvaldsdottir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef] [Green Version]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juhling, F.; Kretzmer, H.; Bernhart, S.H.; Otto, C.; Stadler, P.F.; Hoffmann, S. metilene: Fast and sensitive calling of differentially methylated regions from bisulfite sequencing data. Genome Res. 2016, 26, 256–262. [Google Scholar] [CrossRef] [Green Version]
- RCoreTeam. R: A language and environment for statistical computing. R Foundation for Statistical Computing. Vienna, Austria, 2021. Available online: https://www.R-project.org/ (accessed on 29 April 2021).
- Klett, H.; Balavarca, Y.; Toth, R.; Gigic, B.; Habermann, N.; Scherer, D.; Schrotz-King, P.; Ulrich, A.; Schirmacher, P.; Herpel, E.; et al. Robust prediction of gene regulation in colorectal cancer tissues from DNA methylation profiles. Epigenetics 2018, 13, 386–397. [Google Scholar] [CrossRef]
- Niemczyk, M.; Ito, Y.; Huddleston, J.; Git, A.; Abu-Amero, S.; Caldas, C.; Moore, G.E.; Stojic, L.; Murrell, A. Imprinted chromatin around DIRAS3 regulates alternative splicing of GNG12-AS1, a long noncoding RNA. Am. J. Hum. Genet. 2013, 93, 224–235. [Google Scholar] [CrossRef] [Green Version]
- Barlow, D.P.; Bartolomei, M.S. Genomic imprinting in mammals. Cold Spring Harb. Perspect. Biol. 2014, 6. [Google Scholar] [CrossRef] [Green Version]
- Tian, X.C. Genomic imprinting in farm animals. Annu. Rev. Anim. Biosci. 2014, 2, 23–40. [Google Scholar] [CrossRef]
- Tucci, V.; Isles, A.R.; Kelsey, G.; Ferguson-Smith, A.C.; Erice Imprinting, G. Genomic Imprinting and Physiological Processes in Mammals. Cell 2019, 176, 952–965. [Google Scholar] [CrossRef] [Green Version]
- Feng, W.; Marquez, R.T.; Lu, Z.; Liu, J.; Lu, K.H.; Issa, J.P.; Fishman, D.M.; Yu, Y.; Bast, R.C., Jr. Imprinted tumor suppressor genes ARHI and PEG3 are the most frequently down-regulated in human ovarian cancers by loss of heterozygosity and promoter methylation. Cancer 2008, 112, 1489–1502. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Luo, R.Z.; Fujii, S.; Wang, L.; Hu, W.; Andreeff, M.; Pan, Y.; Kadota, M.; Oshimura, M.; Sahin, A.A.; et al. Aberrant methylation and silencing of ARHI, an imprinted tumor suppressor gene in which the function is lost in breast cancers. Cancer Res. 2003, 63, 4174–4180. [Google Scholar] [PubMed]
- Zhou, Q.Y.; Fang, M.D.; Huang, T.H.; Li, C.C.; Yu, M.; Zhao, S.H. Detection of differentially expressed genes between Erhualian and Large White placentas on day 75 and 90 of gestation. BMC Genom. 2009, 10, 337. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Gene/Locus | Coordinate (chr6) Start & End | Embryo | Methylation (Ratio, ave) | Expression (TPM, ave) | p-Value | Adjusted p-Value |
|---|---|---|---|---|---|---|
| LOC110261211 | 144,558,280 144,571,021 (12,741 bp) | PA | 0.88 ± 0.01 | 0.16 ± 0.00 | 0.69 | 0.89 |
| CN | 0.86 ± 0.01 | 0.18 ± 0.05 | ||||
| DMR | 144,575,647 144,578,475 (2828 bp) | PA | 0.95 ± 0.00 | NA | NA | NA |
| CN | 0.50 ± 0.01 | NA | ||||
| DIRAS3 | 144,576,342 144,578,820 (2478 bp) | PA | 0.96 ± 0.00 | 1.00 ± 0.17 | <0.05 | <0.05 |
| CN | 0.49 ± 0.01 | 85.78 ± 1.55 | ||||
| LOC110261446 | 144,591,277 144,721,840 (130,563 bp) | PA | 0.81 ± 0.00 | 0.00 ± 0.00 | NA | NA |
| CN | 0.77 ± 0.00 | 0.00 ± 0.00 |
| Gene | Expression (TPM, ave) | |||||
|---|---|---|---|---|---|---|
| Adipose Tissue | Hypothalamus | Liver | Lung | Muscle | Spleen | |
| LOC110261211 | 0.07 ± 0.03 | 0.17 ± 0.09 | 0.05 ± 0.01 | 0.12 ± 0.01 | 0.02 ± 0.00 | 0.11 ± 0.02 |
| DIRAS3 | 1.60 ± 0.30 | 468.30 ± 160.39 | 0.26 ± 0.04 | 0.57 ± 0.06 | 0.34 ± 0.09 | 0.31 ± 0.06 |
| LOC110261446 | 0.00 ± 0.00 | 0.00 ± 0.00 | 0.00 ± 0.00 | 0.00 ± 0.00 | 0.00 ± 0.00 | 0.00 ± 0.00 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahn, J.; Hwang, I.-S.; Park, M.-R.; Hwang, S.; Lee, K. Genomic Imprinting at the Porcine DIRAS3 Locus. Animals 2021, 11, 1315. https://doi.org/10.3390/ani11051315
Ahn J, Hwang I-S, Park M-R, Hwang S, Lee K. Genomic Imprinting at the Porcine DIRAS3 Locus. Animals. 2021; 11(5):1315. https://doi.org/10.3390/ani11051315
Chicago/Turabian StyleAhn, Jinsoo, In-Sul Hwang, Mi-Ryung Park, Seongsoo Hwang, and Kichoon Lee. 2021. "Genomic Imprinting at the Porcine DIRAS3 Locus" Animals 11, no. 5: 1315. https://doi.org/10.3390/ani11051315