Analysis of Transcriptome and miRNAome in the Muscle of Bamei Pigs at Different Developmental Stages

{kind=link}
{kind=link}
{kind=link}
{kind=link}
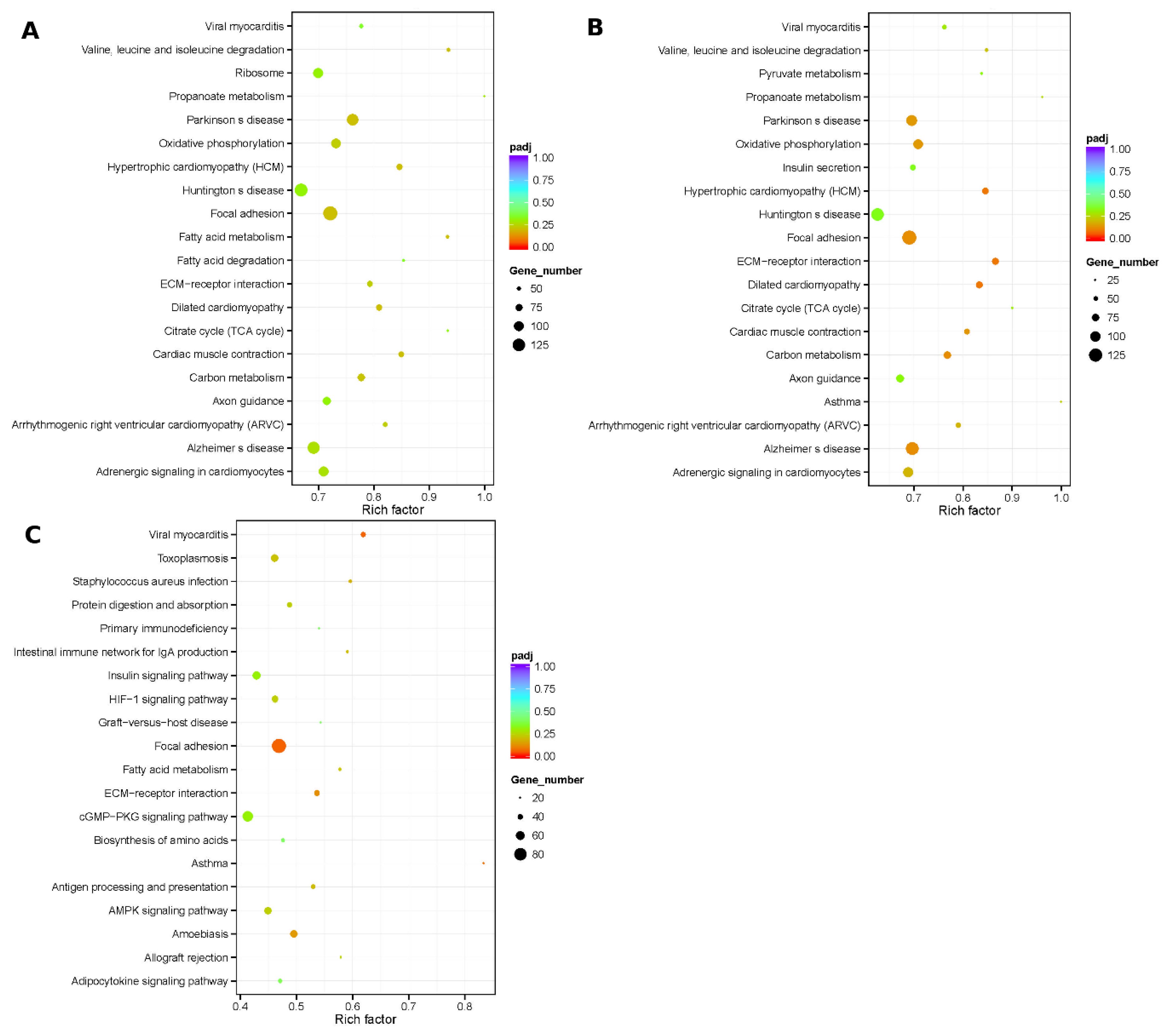{kind=link}
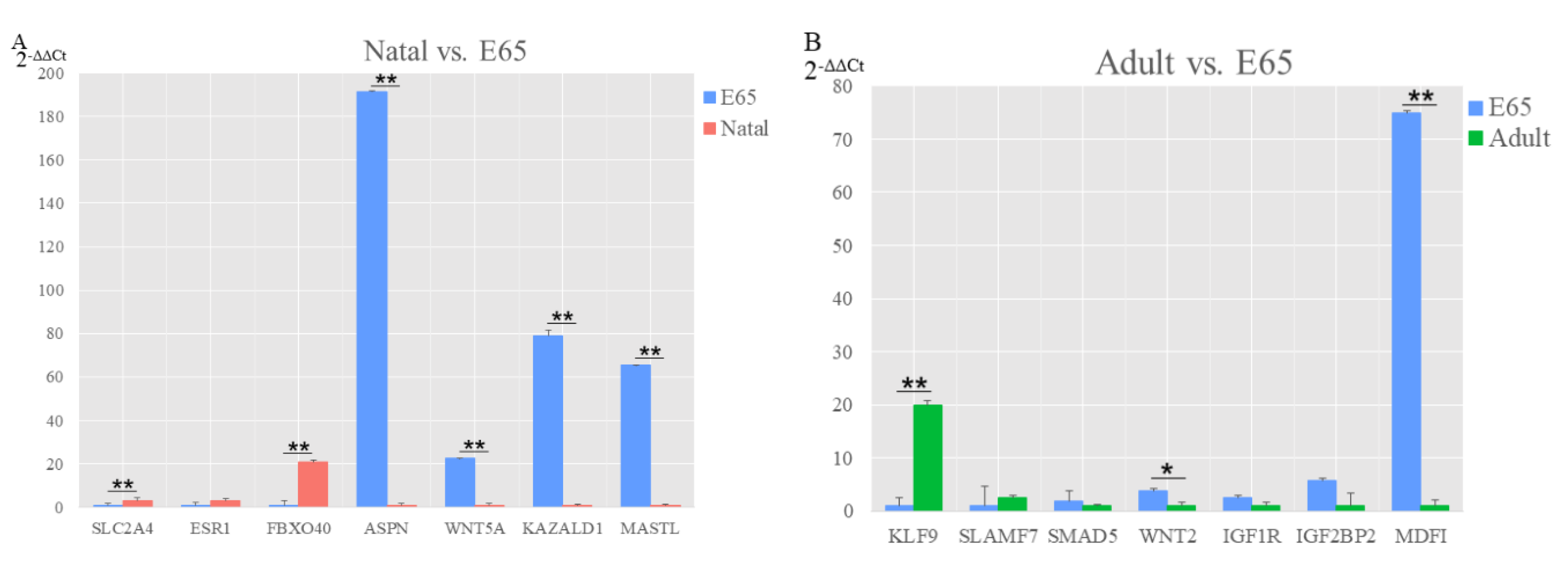{kind=link}
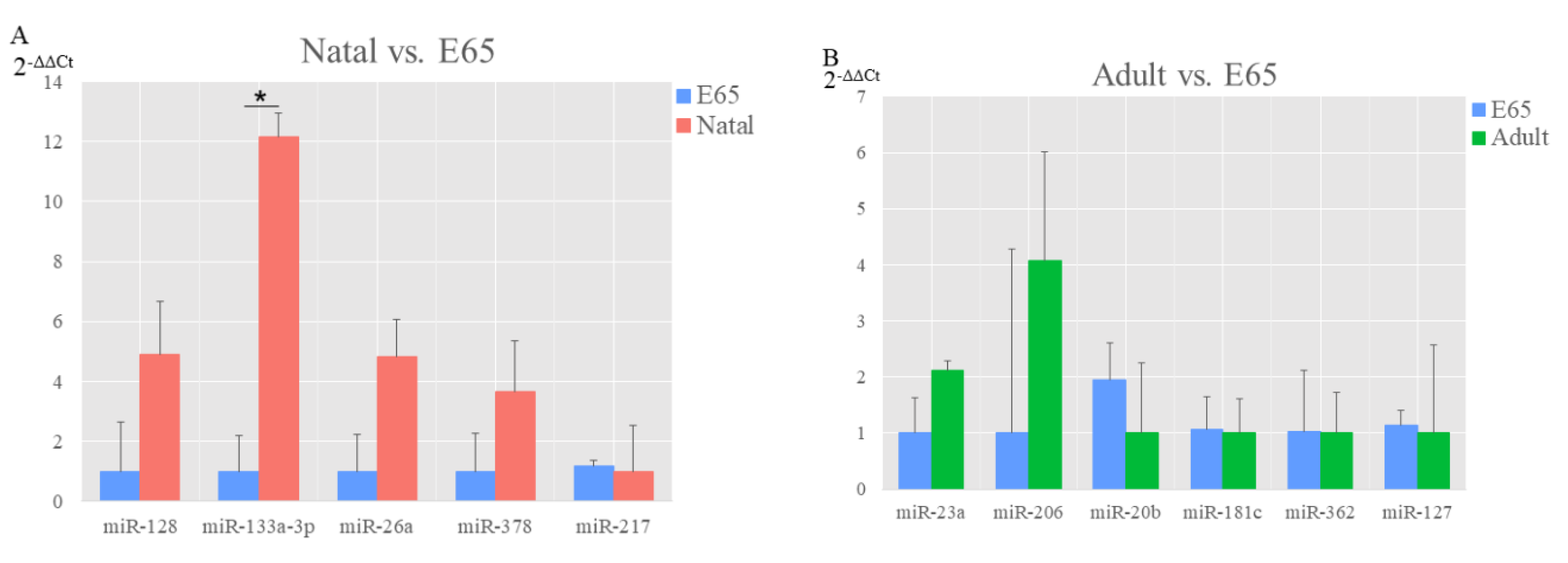{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animal and Longissimus Dorsi Muscle Tissue Collection
2.2. RNA Extraction and Quality Assessment
2.3. Construction of cDNA Library and Illumina Sequencing
2.4. Construction of Small RNA Library and Illumina Sequencing
2.5. Analyses of RNA-Seq Data
2.6. Analyses of sRNA-Seq Data
2.7. Integrated Analysis of DE Genes and miRNA
2.8. Validation of Sequencing Results by Quantitative Real-Time PCR (qRT-PCR)
3. Results
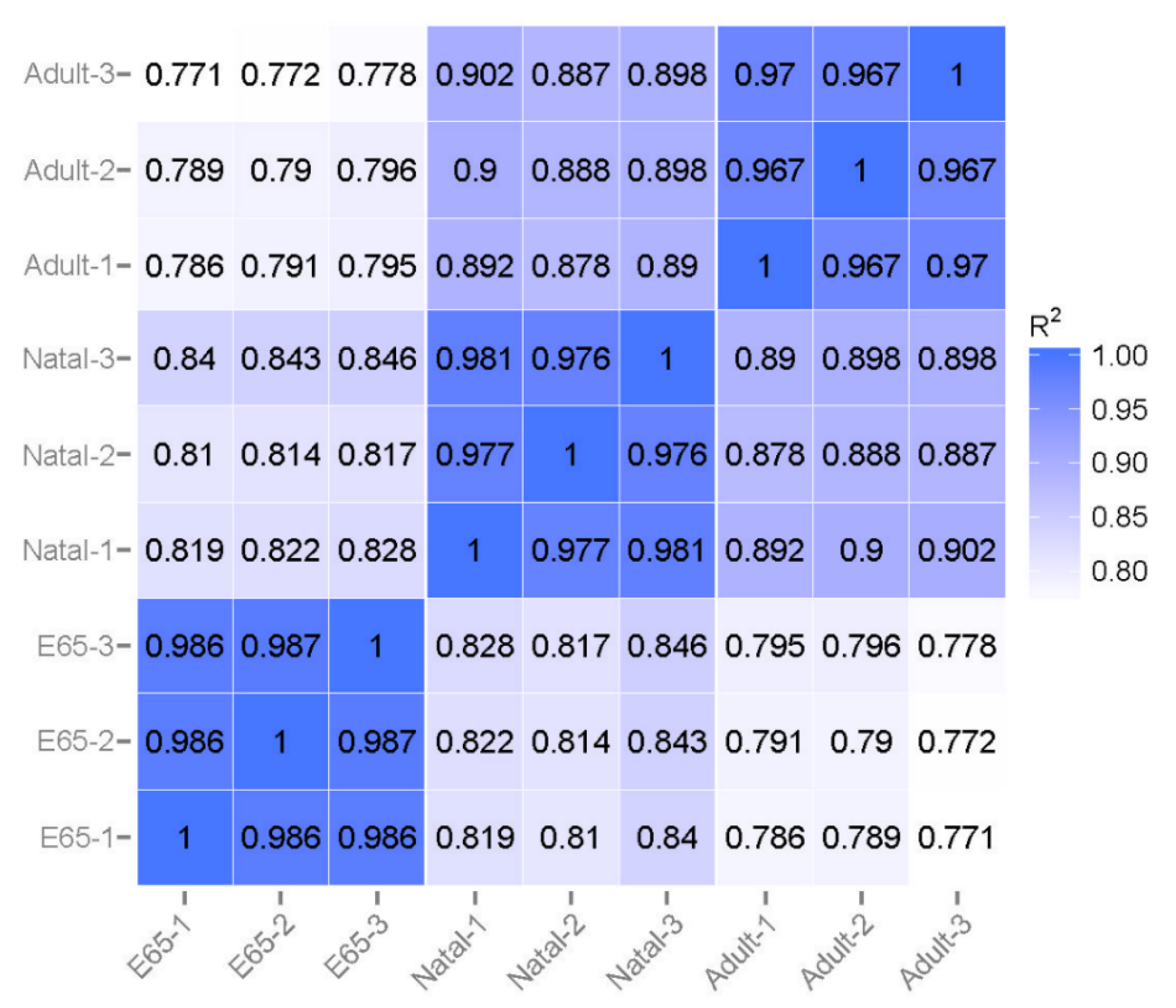
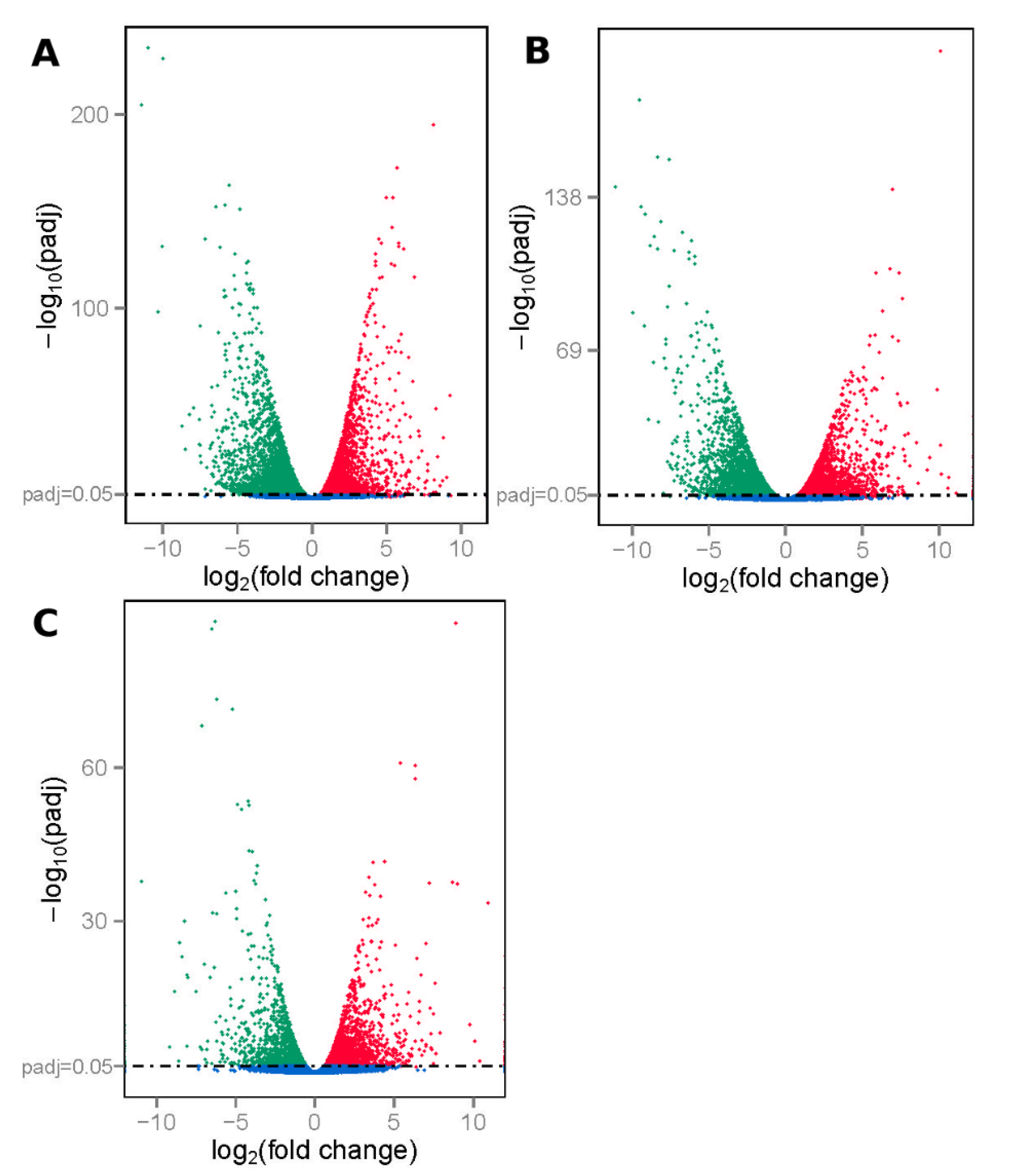
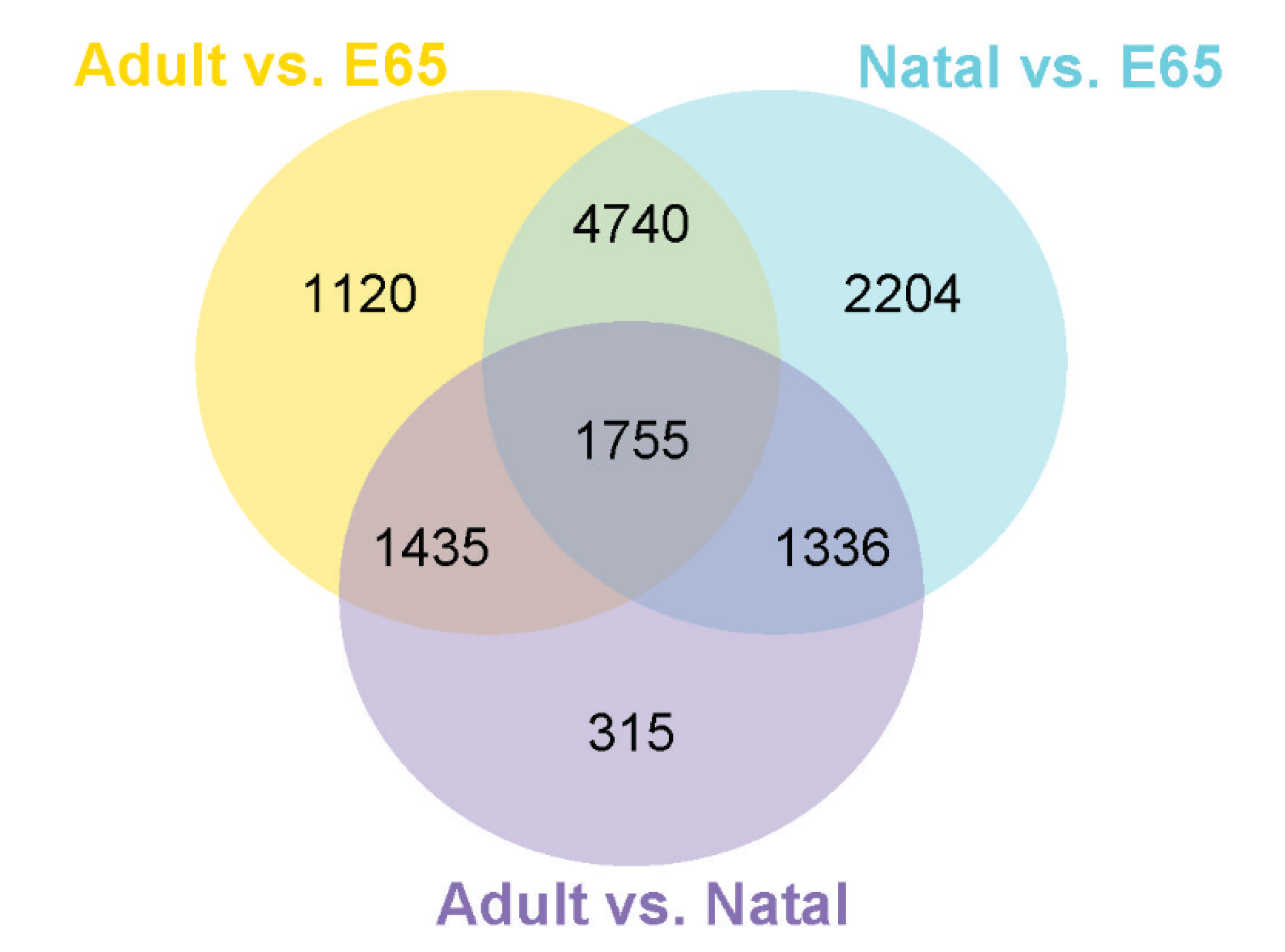
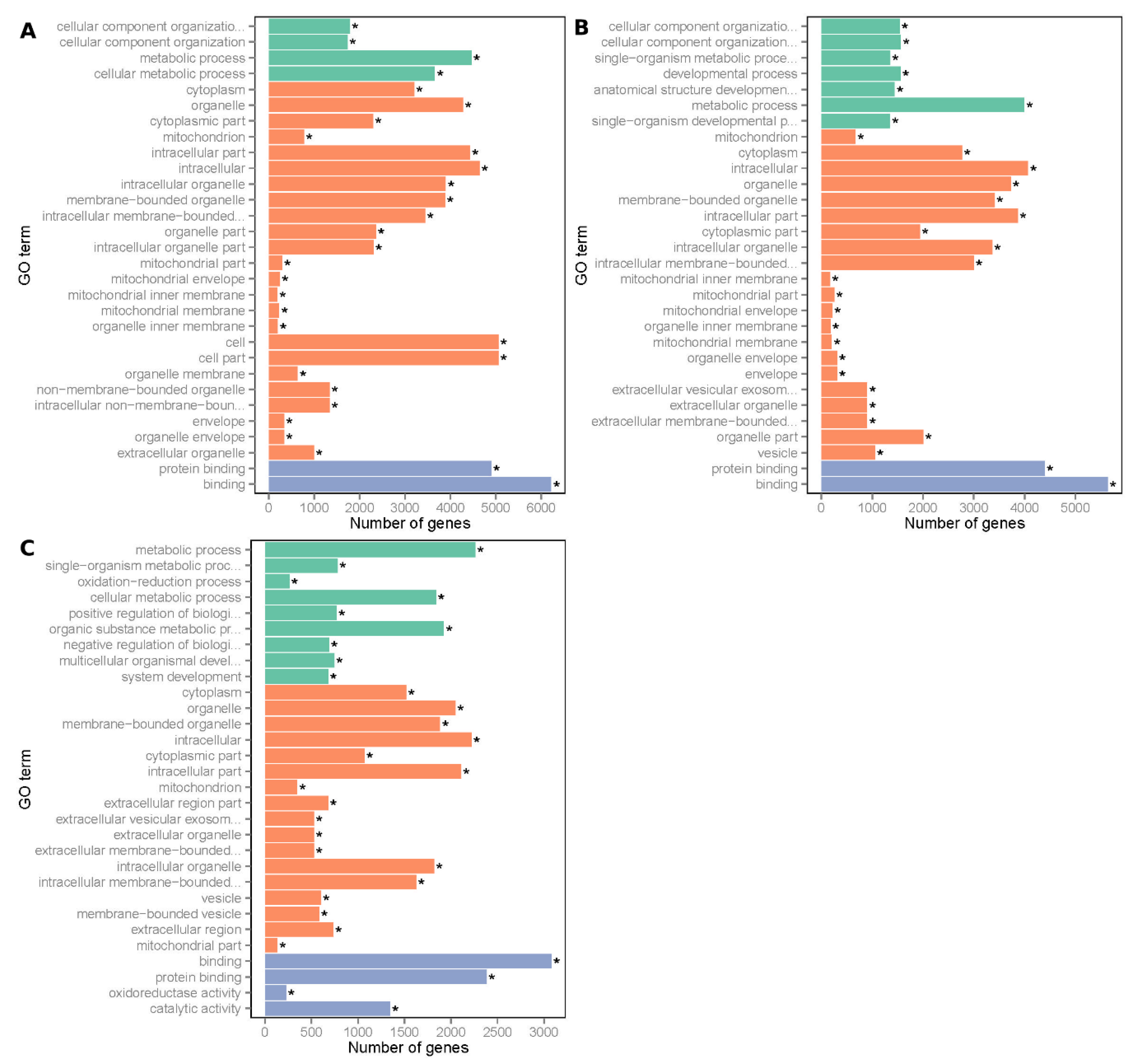
3.1. Summary of RNA Sequencing Data Mapping and Annotation
3.2. Summary of sRNA Sequencing Data Mapping and Annotation
3.3. Identification of Potential Target Genes of DE miRNA
3.4. Integrated Analysis of DE Genes and miRNA
3.5. Validation of Sequencing Data by qRT-PCR
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| adult | 10-month postnatal pigs |
| ASPN | Asporin |
| DEGs | DE genes |
| E65 | 65-day embryonic pigs |
| FDR | False discovery rate |
| FPKM | Fragments per kilobase of the exon model per million mapped reads |
| GO | Gene ontology |
| IGF1R | Insulin-like growth factor 1 receptor |
| IGF2BP2 | Insulin-like growth factor 2 mRNA binding protein 2 |
| KAZALD1 | Kazal-type serine peptidase inhibitor domain 1 |
| KEGG | Kyoto encyclopedia of genes and genomes |
| LDM | Longissimus dorsi muscle |
| MDFI | MyoD family inhibitor |
| MYF6 | Myogenic factor 6 |
| natal | 0-day postnatal pigs |
| nt | Nucleotides |
| padj | Adjusted p-value |
| RIN | RNA integrity numbers |
| RNA-Seq | RNA sequencing |
| rRNA | Ribosomal RNA |
| SMAD5 | SMAD family member 5 |
| snRNA | Small nuclear RNA |
| snoRNA | Small nucleolar RNA |
| sRNA | Small RNA |
| TPM | Transcript per million |
| tRNA | Transfer RNA |
| vs. | Versus |
| WNT2 | Wnt family member 2 |
| WNT5A | Wnt family member 5A |
References
- Cardoso, T.F.; Quintanilla, R.; Tibau, J.; Gil, M.; Marmol-Sanchez, E.; Gonzalez-Rodriguez, O.; Gonzalez-Prendes, R.; Amills, M. Nutrient supply affects the mRNA expression profile of the porcine skeletal muscle. BMC Genom. 2017, 18, 603. [Google Scholar] [CrossRef] [Green Version]
- Tang, Z.; Li, Y.; Wan, P.; Li, X.; Zhao, S.; Liu, B.; Fan, B.; Zhu, M.; Yu, M.; Li, K. LongSAGE analysis of skeletal muscle at three prenatal stages in Tongcheng and Landrace pigs. Genome Biol. 2007, 8, R115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, G.H.; Lu, J.X.; Chen, Y.; Zhao, Y.Q.; Guo, P.H.; Yang, J.T.; Zang, R.X. Comparison of the adipogenesis in intramuscular and subcutaneous adipocytes from Bamei and Landrace pigs. Biochem. Cell Biol. 2014, 92, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Li, Z.; Cai, Y.; Xu, H.; Yang, R.; Lan, X. Genetic variants in fat- and short-tailed sheep from high-throughput RNA-sequencing data. Anim. Genet. 2018, 49, 483–487. [Google Scholar] [CrossRef]
- Ma, L.; Zhang, M.; Jin, Y.; Erdenee, S.; Hu, L.; Chen, H.; Cai, Y.; Lan, X. Comparative transcriptome profiling of mRNA and lncRNA related to tail adipose tissues of sheep. Front. Genet. 2018, 9, 365. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Sun, X.; Cai, H.; Sun, Y.; Plath, M.; Li, C.; Lan, X.; Lei, C.; Lin, F.; Bai, Y.; et al. Long non-coding RNA ADNCR suppresses adipogenic differentiation by targeting miR-204. Biochim. Biophys. Acta 2016, 1859, 871–882. [Google Scholar] [CrossRef]
- Cai, H.; Li, M.; Sun, X.; Plath, M.; Li, C.; Lan, X.; Lei, C.; Huang, Y.; Bai, Y.; Qi, X.; et al. Global transcriptome analysis during adipogenic differentiation and involvement of transthyretin gene in adipogenesis in cattle. Front. Genet. 2018, 9, 463. [Google Scholar] [CrossRef]
- Xu, X.; Mishra, B.; Qin, N.; Sun, X.; Zhang, S.; Yang, J.; Xu, R. Differential transcriptome analysis of early postnatal developing longissimus dorsi muscle from two pig breeds characterized in divergent myofiber traits and fatness. Anim. Biotechnol. 2019, 30, 63–74. [Google Scholar] [CrossRef]
- Xu, J.; Wang, C.; Jin, E.; Gu, Y.; Li, S.; Li, Q. Identification of differentially expressed genes in longissimus dorsi muscle between Wei and Yorkshire pigs using RNA sequencing. Genes Genom. 2018, 40, 413–421. [Google Scholar] [CrossRef]
- Ayuso, M.; Fernandez, A.; Nunez, Y.; Benitez, R.; Isabel, B.; Barragan, C.; Fernandez, A.I.; Rey, A.I.; Medrano, J.F.; Canovas, A.; et al. Comparative analysis of muscle transcriptome between pig genotypes identifies genes and regulatory mechanisms associated to growth, fatness and metabolism. PLoS ONE 2015, 10, e0145162. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Chen, X.; Qin, J.; Liu, S.; Zhao, R.; Yu, T.; Chu, G.; Yang, G.; Pang, W. Comparative analysis of long noncoding RNAs expressed during intramuscular adipocytes adipogenesis in fat-type and lean-type pigs. J. Agric. Food Chem. 2018, 66, 12122–12130. [Google Scholar] [CrossRef] [PubMed]
- Piorkowska, K.; Zukowski, K.; Ropka-Molik, K.; Tyra, M.; Gurgul, A. A comprehensive transcriptome analysis of skeletal muscles in two Polish pig breeds differing in fat and meat quality traits. Genet. Mol. Biol. 2018, 41, 125–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Xi, Y.; Liu, G.; Zhao, Y.; Li, J.; Lei, M. Comparative transcriptomic analysis of skeletal muscle tissue during prenatal stages in Tongcheng and Yorkshire pig using RNA-seq. Funct. Integr. Genom. 2018, 18, 195–209. [Google Scholar] [CrossRef]
- Xi, Y.; Liu, H.; Zhao, Y.; Li, J.; Li, W.; Liu, G.; Lin, J.; Liu, W.; Zhang, J.; Lei, M.; et al. Comparative analyses of longissimus muscle miRNAomes reveal microRNAs associated with differential regulation of muscle fiber development between Tongcheng and Yorkshire pigs. PLoS ONE 2018, 13, e0200445. [Google Scholar] [CrossRef] [Green Version]
- Liang, Y.; Wang, Y.; Ma, L.; Zhong, Z.; Yang, X.; Tao, X.; Chen, X.; He, Z.; Yang, Y.; Zeng, K.; et al. Comparison of microRNAs in adipose and muscle tissue from seven indigenous Chinese breeds and Yorkshire pigs. Anim. Genet. 2019, 50, 439–448. [Google Scholar] [CrossRef]
- Shang, P.; Wang, Z.; Chamba, Y.; Zhang, B.; Zhang, H.; Wu, C. A comparison of prenatal muscle transcriptome and proteome profiles between pigs with divergent growth phenotypes. J. Cell. Biochem. 2019, 120, 5277–5286. [Google Scholar] [CrossRef]
- Wang, J.; Ren, Q.; Hua, L.; Chen, J.; Zhang, J.; Bai, H.; Li, H.; Xu, B.; Shi, Z.; Cao, H.; et al. Comprehensive analysis of differentially expressed mRNA, lncRNA and circRNA and their ceRNA networks in the longissimus dorsi muscle of two different pig breeds. Int. J. Mol. Sci. 2019, 20, 1107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trapnell, C.; Pachter, L.; Salzberg, S.L. TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics 2009, 25, 1105–1111. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef] [Green Version]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq-a Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; Van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [Green Version]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [Green Version]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [Green Version]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, X.; Cai, T.; Olyarchuk, J.G.; Wei, L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [Green Version]
- Friedlander, M.R.; Mackowiak, S.D.; Li, N.; Chen, W.; Rajewsky, N. miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res. 2012, 40, 37–52. [Google Scholar] [CrossRef]
- Kozomara, A.; Griffiths-Jones, S. miRBase: Annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014, 42, D68–D73. [Google Scholar] [CrossRef] [Green Version]
- Kalvari, I.; Argasinska, J.; Quinones-Olvera, N.; Nawrocki, E.P.; Rivas, E.; Eddy, S.R.; Bateman, A.; Finn, R.D.; Petrov, A.I. Rfam 13.0: Shifting to a genome-centric resource for non-coding RNA families. Nucleic Acids Res. 2018, 46, D335–D342. [Google Scholar] [CrossRef]
- Kalvari, I.; Nawrocki, E.P.; Argasinska, J.; Quinones-Olvera, N.; Finn, R.D.; Bateman, A.; Petrov, A.I. Non-coding RNA analysis using the Rfam database. Curr. Protoc. Bioinform. 2018, 62, e51. [Google Scholar] [CrossRef] [PubMed]
- Wen, M.; Shen, Y.; Shi, S.; Tang, T. miREvo: An integrative microRNA evolutionary analysis platform for next-generation sequencing experiments. BMC Bioinform. 2012, 13, 140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, G.P.; Kin, K.; Lynch, V.J. Measurement of mRNA abundance using RNA-seq data: RPKM measure is inconsistent among samples. Theory Biosci. 2012, 131, 281–285. [Google Scholar] [CrossRef]
- Wang, L.; Feng, Z.; Wang, X.; Wang, X.; Zhang, X. DEGseq: An R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 2010, 26, 136–138. [Google Scholar] [CrossRef] [PubMed]
- Storey, J.D. The positive false discovery rate: A Bayesian interpretation and the q-value. Ann. Stat. 2003, 31, 2013–2035. [Google Scholar] [CrossRef]
- Enright, A.J.; John, B.; Gaul, U.; Tuschl, T.; Sander, C.; Marks, D.S. MicroRNA targets in Drosophila. Genome Biol. 2003, 5, R1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Hou, L.; Xu, J.; Jiao, Y.; Li, H.; Pan, Z.; Duan, J.; Gu, T.; Hu, C.; Wang, C. MiR-27b promotes muscle development by inhibiting MDFI expression. Cell. Physiol. Biochem. 2018, 46, 2271–2283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taipale, M.; Solovieva, S.; Leino-Arjas, P.; Mannikko, M. Functional polymorphisms in asporin and CILP together with joint loading predispose to hand osteoarthritis. BMC Genet. 2017, 18, 108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Qi, R.; Wang, J.; Huang, W.; Wu, Y.; Huang, X.; Yang, F.; Huang, J. Differential expression profile of miRNAs in porcine muscle and adipose tissue during development. Gene 2017, 618, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Otto, A.; Schmidt, C.; Luke, G.; Allen, S.; Valasek, P.; Muntoni, F.; Lawrence-Watt, D.; Patel, K. Canonical Wnt signalling induces satellite-cell proliferation during adult skeletal muscle regeneration. J. Cell Sci. 2008, 121, 2939–2950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, L.; Chen, Y.; Liu, X.; Ye, S.; Yu, K.; Huang, Z.; Yu, J.; Zhou, X.; Chen, H.; Mo, D. Integrative analysis of porcine microRNAome during skeletal muscle development. PLoS ONE 2013, 8, e72418. [Google Scholar] [CrossRef] [PubMed]
- He, D.; Zou, T.; Gai, X.; Ma, J.; Li, M.; Huang, Z.; Chen, D. MicroRNA expression profiles differ between primary myofiber of lean and obese pig breeds. PLoS ONE 2017, 12, e0181897. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Tang, Z.; Liu, H.; Wang, N.; Ju, H.; Li, K. Discovery of microRNAs associated with myogenesis by deep sequencing of serial developmental skeletal muscles in pigs. PLoS ONE 2012, 7, e52123. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Chen, X.; Zheng, Y.; Li, F.; Lu, Z.; Chen, C.; Liu, J.; Wang, Y.; Peng, Y.; Shen, Z.; et al. MiR-23a inhibits myogenic differentiation through down regulation of fast myosin heavy chain isoforms. Exp. Cell Res. 2012, 318, 2324–2334. [Google Scholar] [CrossRef]
- Hidestrand, M.; Richards-Malcolm, S.; Gurley, C.M.; Nolen, G.; Grimes, B.; Waterstrat, A.; Zant, G.V.; Peterson, C.A. Sca-1-expressing nonmyogenic cells contribute to fibrosis in aged skeletal muscle. J. Gerontol. A Biol. Sci. Med. Sci. 2008, 63, 566–579. [Google Scholar] [CrossRef] [Green Version]
- Wu, T.; Zhang, X.; Tian, M.; Tao, Q.; Zhang, L.; Ding, Y.; Zhang, X.; Yin, Z. Transcriptome analysis reveals candidate genes involved in splay leg syndrome in piglets. J. Appl. Genet. 2018, 59, 475–483. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, N.; Akiyama, S.; Katagiri, T.; Namiki, M.; Kurokawa, T.; Suda, T. Smad1 and smad5 act downstream of intracellular signalings of BMP-2 that inhibits myogenic differentiation and induces osteoblast differentiation in C2C12 myoblasts. Biochem. Biophys. Res. Commun. 1997, 238, 574–580. [Google Scholar] [CrossRef]
- Li, Z.; Hassan, M.Q.; Volinia, S.; van Wijnen, A.J.; Stein, J.L.; Croce, C.M.; Lian, J.B.; Stein, G.S. A microRNA signature for a BMP2-induced osteoblast lineage commitment program. Proc. Natl. Acad. Sci. USA 2008, 105, 13906–13911. [Google Scholar] [CrossRef] [Green Version]
- Punch, V.G.; Jones, A.E.; Rudnicki, M.A. Transcriptional networks that regulate muscle stem cell function. Wiley Interdiscip. Rev. Syst. Biol. Med. 2009, 1, 128–140. [Google Scholar] [CrossRef]
- Zuo, J.; Wu, F.; Liu, Y.; Xiao, J.; Xu, M.; Yu, Q.; Xia, M.; He, X.; Zou, S.; Tan, H.; et al. MicroRNA transcriptome profile analysis in porcine muscle and the effect of miR-143 on the MYH7 gene and protein. PLoS ONE 2015, 10, e0124873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, L.; Du, J.; Xia, Y.; Tan, Z.; Fu, Y.; Yang, Q.; Li, X.; Tang, G.; Jiang, Y.; Wang, J.; et al. Genome-wide landscape of DNA methylomes and their relationship with mRNA and miRNA transcriptomes in oxidative and glycolytic skeletal muscles. Sci. Rep. 2016, 6, 32186. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.H.; Zhu, M.J.; Li, X.Y.; Zhao, S.H. Discovery of porcine microRNAs and profiling from skeletal muscle tissues during development. PLoS ONE 2008, 3, e3225. [Google Scholar] [CrossRef]
- Haugk, K.L.; Wilson, H.M.; Swisshelm, K.; Quinn, L.S. Insulin-like growth factor (IGF)-binding protein-related protein-1: An autocrine/paracrine factor that inhibits skeletal myoblast differentiation but permits proliferation in response to IGF. Endocrinology 2000, 141, 100–110. [Google Scholar] [CrossRef] [PubMed]
- Gagnon, A.; Dods, P.; Roustan-Delatour, N.; Chen, C.S.; Sorisky, A. Phosphatidylinositol-3,4,5-trisphosphate is required for insulin-like growth factor 1-mediated survival of 3T3-L1 preadipocytes. Endocrinology 2001, 142, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Mai, M.; Jin, L.; Tian, S.; Liu, R.; Huang, W.; Tang, Q.; Ma, J.; Jiang, A.; Wang, X.; Hu, Y.; et al. Deciphering the microRNA transcriptome of skeletal muscle during porcine development. PeerJ 2016, 4, e1504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, X.; Yang, Y.; Zhu, S.; Hua, C.; Zhou, R.; Mu, Y.; Tang, Z.; Li, K. Comparison of skeletal muscle miRNA and mRNA profiles among three pig breeds. Mol. Genet. Genom. 2016, 291, 559–573. [Google Scholar] [CrossRef]
- Cai, Z.; Zhang, L.; Jiang, X.; Sheng, Y.; Xu, N. Differential miRNA expression profiles in the longissimus dorsi muscle between intact and castrated male pigs. Res. Vet. Sci. 2015, 99, 99–104. [Google Scholar] [CrossRef]
- Kim, H.K.; Lee, Y.S.; Sivaprasad, U.; Malhotra, A.; Dutta, A. Muscle-specific microRNA miR-206 promotes muscle differentiation. J. Cell Biol. 2006, 174, 677–687. [Google Scholar] [CrossRef]
- Jiao, Y.; Huang, B.; Chen, Y.; Hong, G.; Xu, J.; Hu, C.; Wang, C. Integrated analyses reveal overexpressed Notch1 promoting porcine satellite cells’ proliferation through regulating the cell cycle. Int. J. Mol. Sci. 2018, 19, 271. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Qiangba, Y.; Shang, P.; Wang, Z.; Ma, J.; Wang, L.; Zhang, H. A comprehensive microRNA expression profile related to hypoxia adaptation in the Tibetan pig. PLoS ONE 2015, 10, e0143260. [Google Scholar] [CrossRef] [PubMed]
- Hamrick, M.W.; Herberg, S.; Arounleut, P.; He, H.Z.; Shiver, A.; Qi, R.Q.; Zhou, L.; Isales, C.M.; Mi, Q.S. The adipokine leptin increases skeletal muscle mass and significantly alters skeletal muscle miRNA expression profile in aged mice. Biochem. Biophys. Res. Commun. 2010, 400, 379–383. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Gilbert, J.A.; Zhang, Y.; Zhang, M.; Qiu, Q.; Ramanujan, K.; Shavlakadze, T.; Eash, J.K.; Scaramozza, A.; Goddeeris, M.M.; et al. An HMGA2-IGF2BP2 axis regulates myoblast proliferation and myogenesis. Dev. Cell 2012, 23, 1176–1188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goto, Y.; Shinjo, K.; Kondo, Y.; Shen, L.; Toyota, M.; Suzuki, H.; Gao, W.; An, B.; Fujii, M.; Murakami, H.; et al. Epigenetic profiles distinguish malignant pleural mesothelioma from lung adenocarcinoma. Cancer Res. 2009, 69, 9073–9082. [Google Scholar] [CrossRef] [Green Version]
- Chou, C.H.; Shrestha, S.; Yang, C.D.; Chang, N.W.; Lin, Y.L.; Liao, K.W.; Huang, W.C.; Sun, T.H.; Tu, S.J.; Lee, W.H.; et al. miRTarBase update 2018: A resource for experimentally validated microRNA-target interactions. Nucleic Acids Res. 2018, 46, D296–D302. [Google Scholar] [CrossRef] [PubMed]
- Seale, P.; Bjork, B.; Yang, W.; Kajimura, S.; Chin, S.; Kuang, S.; Scime, A.; Devarakonda, S.; Conroe, H.M.; Erdjument-Bromage, H.; et al. PRDM16 controls a brown fat/skeletal muscle switch. Nature 2008, 454, 961–967. [Google Scholar] [CrossRef] [Green Version]
- Takada, I.; Kouzmenko, A.P.; Kato, S. Wnt and PPARgamma signaling in osteoblastogenesis and adipogenesis. Nat. Rev. Rheumatol. 2009, 5, 442–447. [Google Scholar] [CrossRef]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, G.; Ma, L.; Wang, L.; Zhou, J.; Ma, Y.; Yang, C. Analysis of Transcriptome and miRNAome in the Muscle of Bamei Pigs at Different Developmental Stages. Animals 2020, 10, 1198. https://doi.org/10.3390/ani10071198
Wu G, Ma L, Wang L, Zhou J, Ma Y, Yang C. Analysis of Transcriptome and miRNAome in the Muscle of Bamei Pigs at Different Developmental Stages. Animals. 2020; 10(7):1198. https://doi.org/10.3390/ani10071198
Chicago/Turabian StyleWu, Guofang, Lin Ma, Lei Wang, Jiping Zhou, Yuhong Ma, and Chen Yang. 2020. "Analysis of Transcriptome and miRNAome in the Muscle of Bamei Pigs at Different Developmental Stages" Animals 10, no. 7: 1198. https://doi.org/10.3390/ani10071198