
Optimizing Antimicrobial Drug Dosing in Critically Ill Patients


Abstract
:1. Introduction
2. Pharmacokinetic and Pharmacodynamic Characteristics of Antimicrobials
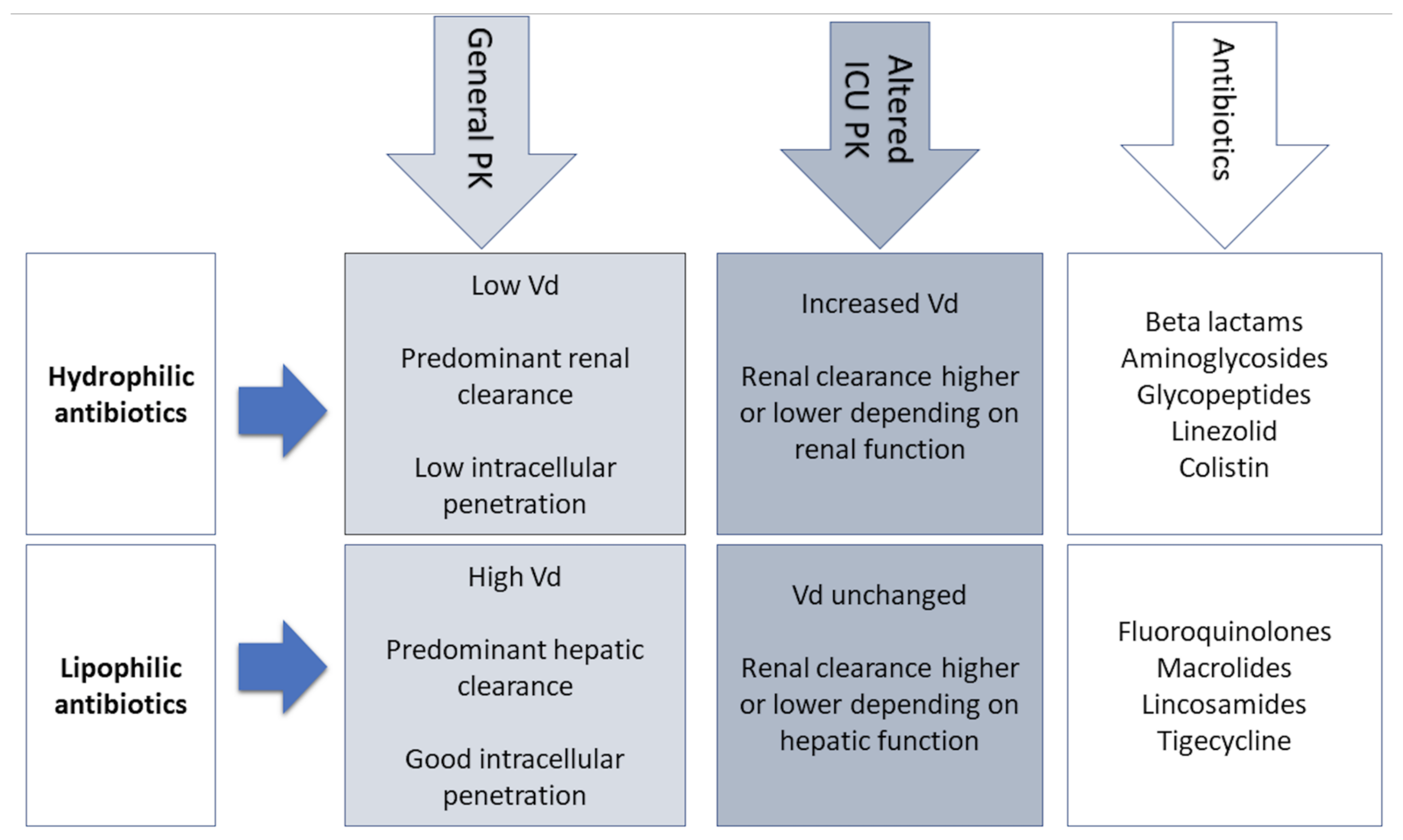
3. PK Changes in Critically Ill Patients
4. Antibiotics in the ICU
4.1. Β-Lactams
4.2. Aminoglycosides
4.3. Glycopeptides
Vancomycin
4.4. Colistin
4.5. Fluoroquinolones
5. Strategies to Optimize Dosing
6. Other Approaches to Optimize Dosing–Nebulization
7. Impact of Dosing Strategies on Outcomes
8. Future Directions and Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rhodes, A.; Evans, L.E.; Alhazzani, W.; Levy, M.M.; Antonelli, M.; Ferrer, R.; Kumar, A.; Sevransky, J.E.; Sprung, C.L.; Nunnally, M.E.; et al. Surviving Sepsis Campaign: International Guidelines for Management of Sepsis and Septic Shock: 2016. Intensive Care Med. 2017, 43, 304–377. [Google Scholar] [CrossRef] [PubMed]
- Goncalves-Pereira, J.; Paiva, J.A. Dose modulation: A new concept of antibiotic therapy in the critically ill patient? J. Crit. Care 2013, 28, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Roberts, J.A.; Abdul-Aziz, M.H.; Lipman, J.; Mouton, J.W.; Vinks, A.A.; Felton, T.W.; Hope, W.W.; Farkas, A.; Neely, M.N.; Schentag, J.J.; et al. Individualised antibiotic dosing for patients who are critically ill: Challenges and potential solutions. Lancet Infect. Dis. 2014, 14, 498–509. [Google Scholar] [CrossRef] [Green Version]
- Ulldemolins, M.; Roberts, J.A.; Lipman, J.; Rello, J. Antibiotic dosing in multiple organ dysfunction syndrome. Chest 2011, 139, 1210–1220. [Google Scholar] [CrossRef] [Green Version]
- Jamal, W.; Al Roomi, E.; AbdulAziz, L.R.; Rotimi, V.O. Evaluation of Curetis Unyvero, a multiplex PCR-based testing system, for rapid detection of bacteria and antibiotic resistance and impact of the assay on management of severe nosocomial pneumonia. J. Clin. Microbiol. 2014, 52, 2487–2492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blot, S.I.; Pea, F.; Lipman, J. The effect of pathophysiology on pharmacokinetics in the critically ill patient--concepts appraised by the example of antimicrobial agents. Adv. Drug Deliv. Rev. 2014, 77, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.S.; Yogaratnam, D.; Levasseur-Franklin, K.E.; Forni, A.; Fong, J. Introduction to drug pharmacokinetics in the critically ill patient. Chest 2012, 141, 1327–1336. [Google Scholar] [CrossRef] [PubMed]
- Roberts, D.J.; Hall, R.I. Drug absorption, distribution, metabolism and excretion considerations in critically ill adults. Expert Opin. Drug Metab. Toxicol. 2013, 9, 1067–1084. [Google Scholar] [CrossRef]
- Levison, M.E.; Levison, J.H. Pharmacokinetics and pharmacodynamics of antibacterial agents. Infect. Dis. Clin. N. Am. 2009, 23, 791–815. [Google Scholar] [CrossRef] [Green Version]
- Craig, W.A. Pharmacokinetic/pharmacodynamic parameters: Rationale for antibacterial dosing of mice and men. Clin. Infect. Dis. 1998, 26, 1–10. [Google Scholar] [CrossRef]
- Moniz, P.; Coelho, L.; Povoa, P. Antimicrobial Stewardship in the Intensive Care Unit: The Role of Biomarkers, Pharmacokinetics, and Pharmacodynamics. Adv. Ther. 2021, 38, 164–179. [Google Scholar] [CrossRef]
- Roberts, J.A.; Lipman, J. Pharmacokinetic issues for antibiotics in the critically ill patient. Crit. Care Med. 2009, 37, 840–851. [Google Scholar] [CrossRef] [Green Version]
- Mouton, J.W.; Punt, N.; Vinks, A.A. Concentration-effect relationship of ceftazidime explains why the time above the MIC is 40 percent for a static effect in vivo. Antimicrob. Agents Chemother. 2007, 51, 3449–3451. [Google Scholar] [CrossRef] [Green Version]
- Williams, P.; Cotta, M.O.; Roberts, J.A. Pharmacokinetics/Pharmacodynamics of beta-Lactams and Therapeutic Drug Monitoring: From Theory to Practical Issues in the Intensive Care Unit. Semin Respir Crit. Care Med. 2019, 40, 476–487. [Google Scholar]
- Abdul-Aziz, M.H.; Alffenaar, J.C.; Bassetti, M.; Bracht, H.; Dimopoulos, G.; Marriott, D.; Neely, M.N.; Paiva, J.A.; Pea, F.; Sjovall, F.; et al. Antimicrobial therapeutic drug monitoring in critically ill adult patients: A Position Paper. Intensive Care Med. 2020, 46, 1127–1153. [Google Scholar] [CrossRef]
- Pea, F.; Viale, P.; Furlanut, M. Antimicrobial therapy in critically ill patients: A review of pathophysiological conditions responsible for altered disposition and pharmacokinetic variability. Clin. Pharmacokinet. 2005, 44, 1009–1034. [Google Scholar] [CrossRef]
- Estes, L. Review of pharmacokinetics and pharmacodynamics of antimicrobial agents. Mayo Clin. Proc. 1998, 73, 1114–1122. [Google Scholar] [CrossRef]
- Varghese, J.M.; Roberts, J.A.; Lipman, J. Antimicrobial pharmacokinetic and pharmacodynamic issues in the critically ill with severe sepsis and septic shock. Crit. Care Clin. 2011, 27, 19–34. [Google Scholar] [CrossRef]
- Gous, A.; Lipman, J.; Scribante, J.; Tshukutsoane, S.; Hon, H.; Pinder, M.; Mathivha, R.; Verhoef, L.; Stass, H. Fluid shifts have no influence on ciprofloxacin pharmacokinetics in intensive care patients with intra-abdominal sepsis. Int. J. Antimicrob. Agents 2005, 26, 50–55. [Google Scholar] [CrossRef]
- Ulldemolins, M.; Roberts, J.A.; Rello, J.; Paterson, D.L.; Lipman, J. The effects of hypoalbuminaemia on optimizing antibacterial dosing in critically ill patients. Clin. Pharmacokinet. 2011, 50, 99–110. [Google Scholar] [CrossRef]
- SAFE Study Investigators; Finfer, S.; Bellomo, R.; McEvoy, S.; Lo, S.K.; Myburgh, J.; Neal, B.; Norton, R. Effect of baseline serum albumin concentration on outcome of resuscitation with albumin or saline in patients in intensive care units: Analysis of data from the saline versus albumin fluid evaluation (SAFE) study. BMJ 2006, 333, 1044. [Google Scholar]
- Brochard, L.; Abroug, F.; Brenner, M.; Broccard, A.F.; Danner, R.L.; Ferrer, M.; Laghi, F.; Magder, S.; Papazian, L.; Pelosi, P.; et al. An Official ATS/ERS/ESICM/SCCM/SRLF Statement: Prevention and Management of Acute Renal Failure in the ICU Patient: An international consensus conference in intensive care medicine. Am. J. Respir. Crit. Care Med. 2010, 181, 1128–1155. [Google Scholar] [CrossRef] [PubMed]
- Seller-Perez, G.; Herrera-Gutierrez, M.E.; Banderas-Bravo, E.; Olalla-Sanchez, R.; Lozano-Saez, R.; Quesada-Garcia, G. Concordance in critical patients between the equations designed for the calculation of glomerular filtration rate and 24-hour creatinine clearance. Med. Intensiva 2010, 34, 294–302. [Google Scholar] [PubMed] [Green Version]
- Bidell, M.R.; Lodise, T.P. Suboptimal Clinical Response Rates with Newer Antibiotics Among Patients with Moderate Renal Impairment: Review of the Literature and Potential Pharmacokinetic and Pharmacodynamic Considerations for Observed Findings. Pharmacotherapy 2018, 38, 1205–1215. [Google Scholar] [CrossRef] [PubMed]
- Chen, S. Retooling the creatinine clearance equation to estimate kinetic GFR when the plasma creatinine is changing acutely. J. Am. Soc. Nephrol. 2013, 24, 877–888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seller-Perez, G.; Herrera-Gutierrez, M.E.; Maynar-Moliner, J.; Sanchez-Izquierdo-Riera, J.A.; Marinho, A.; do Pico, J.L. Estimating kidney function in the critically ill patients. Crit. Care Res. Pract. 2013, 2013, 721810. [Google Scholar] [CrossRef]
- Heintz, B.H.; Matzke, G.R.; Dager, W.E. Antimicrobial dosing concepts and recommendations for critically ill adult patients receiving continuous renal replacement therapy or intermittent hemodialysis. Pharmacotherapy 2009, 29, 562–577. [Google Scholar] [CrossRef] [Green Version]
- Pea, F.; Viale, P.; Pavan, F.; Furlanut, M. Pharmacokinetic considerations for antimicrobial therapy in patients receiving renal replacement therapy. Clin. Pharmacokinet. 2007, 46, 997–1038. [Google Scholar] [CrossRef]
- Udy, A.A.; Roberts, J.A.; Boots, R.J.; Paterson, D.L.; Lipman, J. Augmented renal clearance: Implications for antibacterial dosing in the critically ill. Clin. Pharmacokinet. 2010, 49, 1–16. [Google Scholar] [CrossRef]
- Roberts, J.A.; Webb, S.; Paterson, D.; Ho, K.M.; Lipman, J. A systematic review on clinical benefits of continuous administration of beta-lactam antibiotics. Crit. Care Med. 2009, 37, 2071–2078. [Google Scholar] [CrossRef] [Green Version]
- Goncalves-Pereira, J.; Povoa, P. Antibiotics in critically ill patients: A systematic review of the pharmacokinetics of beta-lactams. Crit. Care 2011, 15, R206. [Google Scholar] [CrossRef] [Green Version]
- Andes, D.; Craig, W.A. In vivo activities of amoxicillin and amoxicillin-clavulanate against Streptococcus pneumoniae: Application to breakpoint determinations. Antimicrob. Agents Chemother. 1998, 42, 2375–2379. [Google Scholar] [CrossRef] [Green Version]
- Mouton, J.W.; Punt, N. Use of the t > MIC to choose between different dosing regimens of beta-lactam antibiotics. J. Antimicrob. Chemother. 2001, 47, 500–501. [Google Scholar] [CrossRef] [Green Version]
- Craig, W.A. Basic pharmacodynamics of antibacterials with clinical applications to the use of beta-lactams, glycopeptides, and linezolid. Infect. Dis. Clin. N. Am. 2003, 17, 479–501. [Google Scholar] [CrossRef]
- Drusano, G.L. Antimicrobial pharmacodynamics: Critical interactions of ‘bug and drug’. Nat. Rev. Microbiol. 2004, 2, 289–300. [Google Scholar] [CrossRef]
- Taccone, F.S.; Laterre, P.F.; Dugernier, T.; Spapen, H.; Delattre, I.; Wittebole, X.; De Backer, D.; Layeux, B.; Wallemacq, P.; Vincent, J.L.; et al. Insufficient beta-lactam concentrations in the early phase of severe sepsis and septic shock. Crit. Care 2010, 14, R126. [Google Scholar] [CrossRef] [Green Version]
- Roberts, J.A.; Pea, F.; Lipman, J. The clinical relevance of plasma protein binding changes. Clin. Pharmacokinet. 2013, 52, 1–8. [Google Scholar] [CrossRef]
- Georges, B.; Conil, J.M.; Ruiz, S.; Seguin, T.; Cougot, P.; Fourcade, O.; Houin, G.; Saivin, S. Ceftazidime dosage regimen in intensive care unit patients: From a population pharmacokinetic approach to clinical practice via Monte Carlo simulations. Br. J. Clin. Pharmacol. 2012, 73, 588–596. [Google Scholar] [CrossRef] [Green Version]
- Seyler, L.; Cotton, F.; Taccone, F.S.; De Backer, D.; Macours, P.; Vincent, J.L.; Jacobs, F. Recommended beta-lactam regimens are inadequate in septic patients treated with continuous renal replacement therapy. Crit. Care 2011, 15, R137. [Google Scholar] [CrossRef] [Green Version]
- Vardakas, K.Z.; Voulgaris, G.L.; Maliaros, A.; Samonis, G.; Falagas, M.E. Prolonged versus short-term intravenous infusion of antipseudomonal beta-lactams for patients with sepsis: A systematic review and meta-analysis of randomised trials. Lancet Infect. Dis. 2018, 18, 108–120. [Google Scholar] [CrossRef]
- Crandon, J.L.; Ariano, R.E.; Zelenitsky, S.A.; Nicasio, A.M.; Kuti, J.L.; Nicolau, D.P. Optimization of meropenem dosage in the critically ill population based on renal function. Intensive Care Med. 2011, 37, 632–638. [Google Scholar] [CrossRef] [PubMed]
- Lodise, T.P., Jr.; Lomaestro, B.; Rodvold, K.A.; Danziger, L.H.; Drusano, G.L. Pharmacodynamic profiling of piperacillin in the presence of tazobactam in patients through the use of population pharmacokinetic models and Monte Carlo simulation. Antimicrob. Agents Chemother. 2004, 48, 4718–4724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lomaestro, B.M.; Drusano, G.L. Pharmacodynamic evaluation of extending the administration time of meropenem using a Monte Carlo simulation. Antimicrob. Agents Chemother. 2005, 49, 461–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicasio, A.M.; Ariano, R.E.; Zelenitsky, S.A.; Kim, A.; Crandon, J.L.; Kuti, J.L.; Nicolau, D.P. Population pharmacokinetics of high-dose, prolonged-infusion cefepime in adult critically ill patients with ventilator-associated pneumonia. Antimicrob. Agents Chemother. 2009, 53, 1476–1481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicolau, D.P.; McNabb, J.; Lacy, M.K.; Quintiliani, R.; Nightingale, C.H. Continuous versus intermittent administration of ceftazidime in intensive care unit patients with nosocomial pneumonia. Int. J. Antimicrob. Agents 2001, 17, 497–504. [Google Scholar] [CrossRef]
- Roberts, J.A.; Kirkpatrick, C.M.; Roberts, M.S.; Dalley, A.J.; Lipman, J. First-dose and steady-state population pharmacokinetics and pharmacodynamics of piperacillin by continuous or intermittent dosing in critically ill patients with sepsis. Int. J. Antimicrob. Agents 2010, 35, 156–163. [Google Scholar] [CrossRef]
- Dulhunty, J.M.; Roberts, J.A.; Davis, J.S.; Webb, S.A.; Bellomo, R.; Gomersall, C.; Shirwadkar, C.; Eastwood, G.M.; Myburgh, J.; Paterson, D.L.; et al. A Multicenter Randomized Trial of Continuous versus Intermittent beta-Lactam Infusion in Severe Sepsis. Am. J. Respir. Crit. Care Med. 2015, 192, 1298–1305. [Google Scholar] [CrossRef]
- Korbila, I.P.; Tansarli, G.S.; Karageorgopoulos, D.E.; Vardakas, K.Z.; Falagas, M.E. Extended or continuous versus short-term intravenous infusion of cephalosporins: A meta-analysis. Expert Rev. Anti-Infect. Ther. 2013, 11, 585–595. [Google Scholar] [CrossRef]
- Ogutlu, A.; Guclu, E.; Karabay, O.; Utku, A.C.; Tuna, N.; Yahyaoglu, M. Effects of Carbapenem consumption on the prevalence of Acinetobacter infection in intensive care unit patients. Ann. Clin. Microbiol. Antimicrob 2014, 13, 7. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Kuti, J.L.; Nightingale, C.H.; Nicolau, D.P. Population pharmacokinetic analysis and dosing regimen optimization of meropenem in adult patients. J. Clin. Pharmacol. 2006, 46, 1171–1178. [Google Scholar] [CrossRef]
- Wong, G.; Briscoe, S.; McWhinney, B.; Ally, M.; Ungerer, J.; Lipman, J.; Roberts, J.A. Therapeutic drug monitoring of beta-lactam antibiotics in the critically ill: Direct measurement of unbound drug concentrations to achieve appropriate drug exposures. J. Antimicrob. Chemother. 2018, 73, 3087–3094. [Google Scholar] [CrossRef] [Green Version]
- Tabah, A.; De Waele, J.; Lipman, J.; Zahar, J.R.; Cotta, M.O.; Barton, G.; Timsit, J.F.; Roberts, J.A. The ADMIN-ICU survey: A survey on antimicrobial dosing and monitoring in ICUs. J. Antimicrob. Chemother. 2015, 70, 2671–2677. [Google Scholar] [CrossRef] [Green Version]
- Heil, E.L.; Nicolau, D.P.; Farkas, A.; Roberts, J.A.; Thom, K.A. Pharmacodynamic Target Attainment for Cefepime, Meropenem, and Piperacillin-Tazobactam Using a Pharmacokinetic/Pharmacodynamic-Based Dosing Calculator in Critically Ill Patients. Antimicrob. Agents Chemother. 2018. [Google Scholar] [CrossRef] [Green Version]
- Vial, T.; Bailly, H.; Perault-Pochat, M.C.; Default, A.; Boulay, C.; Chouchana, L.; Kassai, B.; French Network of Pharmacovigilance C. Beta-lactam-induced severe neutropaenia: A descriptive study. Fundam. Clin. Pharmacol. 2019, 33, 225–231. [Google Scholar] [CrossRef]
- Finkelsztejn, A.; Cabral, L.; Bragatti, J.A.; Silva, A.V.; Schuh, A.F. Imipenem-associated encephalopathy: Alert to physicians. Arq. Neuropsiquiatr. 2010, 68, 137–139. [Google Scholar] [CrossRef] [Green Version]
- Fugate, J.E.; Kalimullah, E.A.; Hocker, S.E.; Clark, S.L.; Wijdicks, E.F.; Rabinstein, A.A. Cefepime neurotoxicity in the intensive care unit: A cause of severe, underappreciated encephalopathy. Crit. Care 2013, 17, R264. [Google Scholar] [CrossRef] [Green Version]
- Shaheen, T.; Volles, D.; Calland, F.; Sifri, C.D.; Mytinger, J.; Hagspiel, K.; Sawyer, R.; Bonatti, H. Cefepime-associated status epilepticus in an ICU patient with renal failure. J. Chemother. 2009, 21, 452–454. [Google Scholar] [CrossRef]
- Chandorkar, G.; Xiao, A.; Mouksassi, M.S.; Hershberger, E.; Krishna, G. Population pharmacokinetics of ceftolozane/tazobactam in healthy volunteers, subjects with varying degrees of renal function and patients with bacterial infections. J. Clin. Pharmacol. 2015, 55, 230–239. [Google Scholar] [CrossRef]
- Van Duin, D.; Bonomo, R.A. Ceftazidime/Avibactam and Ceftolozane/Tazobactam: Second-generation beta-Lactam/beta-Lactamase Inhibitor Combinations. Clin. Infect. Dis. 2016, 63, 234–241. [Google Scholar] [CrossRef] [Green Version]
- Xiao, A.J.; Caro, L.; Popejoy, M.W.; Huntington, J.A.; Kullar, R. PK/PD Target Attainment With Ceftolozane/Tazobactam Using Monte Carlo Simulation in Patients With Various Degrees of Renal Function, Including Augmented Renal Clearance and End-Stage Renal Disease. Infect. Dis. Ther. 2017, 6, 137–148. [Google Scholar] [CrossRef] [Green Version]
- Roger, C.; Louart, B.; Elotmani, L.; Barton, G.; Escobar, L.; Koulenti, D.; Lipman, J.; Leone, M.; Muller, L.; Boutin, C.; et al. An international survey on aminoglycoside practices in critically ill patients: The AMINO III study. Ann. Intensive Care 2021, 11, 49. [Google Scholar] [CrossRef]
- Giamarelloum, H. Aminoglycosides plus beta-lactams against gram-negative organisms. Evaluation of in vitro synergy and chemical interactions. Am. J. Med. 1986, 80, 126–137. [Google Scholar] [CrossRef]
- Giamarellou, H.; Zissis, N.P.; Tagari, G.; Bouzos, J. In vitro synergistic activities of aminoglycosides and new beta-lactams against multiresistant Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 1984, 25, 534–536. [Google Scholar] [CrossRef] [Green Version]
- Klastersky, J.; Meunier-Carpentier, F.; Prevost, J.M.; Staquet, M. Synergism between amikacin and cefazolin against Klebsiella: In vitro studies and effect on the bactericidal activity of serum. J. Infect. Dis. 1976, 134, 271–276. [Google Scholar] [CrossRef]
- Paul, M.; Lador, A.; Grozinsky-Glasberg, S.; Leibovici, L. Beta lactam antibiotic monotherapy versus beta lactam-aminoglycoside antibiotic combination therapy for sepsis. Cochrane Database Syst. Rev. 2014. [Google Scholar] [CrossRef]
- Craig, W.A.; Ebert, S.C. Killing and regrowth of bacteria in vitro: A review. Scand. J. Infect. Dis. Suppl. 1990, 74, 63–70. [Google Scholar]
- Moore, R.D.; Lietman, P.S.; Smith, C.R. Clinical response to aminoglycoside therapy: Importance of the ratio of peak concentration to minimal inhibitory concentration. J. Infect. Dis. 1987, 155, 93–99. [Google Scholar] [CrossRef]
- Gonzalez, L.S., 3rd; Spencer, J.P. Aminoglycosides: A practical review. Am. Fam. Physician 1998, 58, 1811–1820. [Google Scholar]
- MacKenzie, F.M.; Gould, I.M. The post-antibiotic effect. J. Antimicrob. Chemother. 1993, 32, 519–537. [Google Scholar] [CrossRef]
- De Montmollin, E.; Bouadma, L.; Gault, N.; Mourvillier, B.; Mariotte, E.; Chemam, S.; Massias, L.; Papy, E.; Tubach, F.; Wolff, M.; et al. Predictors of insufficient amikacin peak concentration in critically ill patients receiving a 25 mg/kg total body weight regimen. Intensive Care Med. 2014, 40, 998–1005. [Google Scholar] [CrossRef]
- Roger, C.; Nucci, B.; Louart, B.; Friggeri, A.; Knani, H.; Evrard, A.; Lavigne, J.P.; Allaouchiche, B.; Lefrant, J.Y.; Roberts, J.A.; et al. Impact of 30 mg/kg amikacin and 8 mg/kg gentamicin on serum concentrations in critically ill patients with severe sepsis. J. Antimicrob. Chemother. 2016, 71, 208–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goncalves-Pereira, J.; Martins, A.; Povoa, P. Pharmacokinetics of gentamicin in critically ill patients: Pilot study evaluating the first dose. Clin. Infect. Dis. 2010, 16, 1258–1263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rea, R.S.; Capitano, B.; Bies, R.; Bigos, K.L.; Smith, R.; Lee, H. Suboptimal aminoglycoside dosing in critically ill patients. Ther. Drug Monit. 2008, 30, 674–681. [Google Scholar] [CrossRef] [PubMed]
- Bellomo, R.; Kellum, J.A.; Ronco, C.; Wald, R.; Martensson, J.; Maiden, M.; Bagshaw, S.M.; Glassford, N.J.; Lankadeva, Y.; Vaara, S.T.; et al. Acute kidney injury in sepsis. Intensive Care Med. 2017, 43, 816–828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rybak, M.J.; Le, J.; Lodise, T.P.; Levine, D.P.; Bradley, J.S.; Liu, C.; Mueller, B.A.; Pai, M.P.; Wong-Beringer, A.; Rotschafer, J.C.; et al. Therapeutic monitoring of vancomycin for serious methicillin-resistant Staphylococcus aureus infections: A revised consensus guideline and review by the American Society of Health-System Pharmacists, the Infectious Diseases Society of America, the Pediatric Infectious Diseases Society, and the Society of Infectious Diseases Pharmacists. Am. J. Health Syst. Pharm. 2020, 77, 835–864. [Google Scholar] [PubMed] [Green Version]
- Singh, N.B.; Yim, J.; Jahanbakhsh, S.; Sakoulas, G.; Rybak, M.J. Impact of cefazolin co-administration with vancomycin to reduce development of vancomycin-intermediate Staphylococcus aureus. Diagn. Microbiol. Infect. Dis. 2018, 91, 363–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vidaillac, C.; Gardete, S.; Tewhey, R.; Sakoulas, G.; Kaatz, G.W.; Rose, W.E.; Tomasz, A.; Rybak, M.J. Alternative mutational pathways to intermediate resistance to vancomycin in methicillin-resistant Staphylococcus aureus. J. Infect. Dis. 2013, 208, 67–74. [Google Scholar] [CrossRef]
- Neely, M.N.; Youn, G.; Jones, B.; Jelliffe, R.W.; Drusano, G.L.; Rodvold, K.A.; Lodise, T.P. Are vancomycin trough concentrations adequate for optimal dosing? Antimicrob. Agents Chemother. 2014, 58, 309–316. [Google Scholar] [CrossRef] [Green Version]
- Kullar, R.; Davis, S.L.; Levine, D.P.; Rybak, M.J. Impact of vancomycin exposure on outcomes in patients with methicillin-resistant Staphylococcus aureus bacteremia: Support for consensus guidelines suggested targets. Clin. Infect. Dis. 2011, 52, 975–981. [Google Scholar] [CrossRef] [Green Version]
- Moise-Broder, P.A.; Forrest, A.; Birmingham, M.C.; Schentag, J.J. Pharmacodynamics of vancomycin and other antimicrobials in patients with Staphylococcus aureus lower respiratory tract infections. Clin. Pharmacokinet. 2004, 43, 925–942. [Google Scholar] [CrossRef]
- Ghosh, N.; Chavada, R.; Maley, M.; van Hal, S.J. Impact of source of infection and vancomycin AUC0-24/MICBMD targets on treatment failure in patients with methicillin-resistant Staphylococcus aureus bacteraemia. Clin. Microbiol. Infect. 2014, 20, O1098–O1105. [Google Scholar] [CrossRef] [Green Version]
- Martirosov, D.M.; Bidell, M.R.; Pai, M.P.; Scheetz, M.H.; Rosenkranz, S.L.; Lodise, T.P. Relationship between vancomycin exposure and outcomes among patients with MRSA bloodstream infections with vancomycin Etest(R) MIC values of 1.5mg/L: A pilot study. Diagn. Microbiol. Infect. Dis. 2017, 88, 259–263. [Google Scholar] [CrossRef]
- Zelenitsky, S.; Rubinstein, E.; Ariano, R.; Iacovides, H.; Dodek, P.; Mirzanejad, Y.; Kumar, A. Vancomycin pharmacodynamics and survival in patients with methicillin-resistant Staphylococcus aureus-associated septic shock. Int. J. Antimicrob. Agents 2013, 41, 255–260. [Google Scholar] [CrossRef]
- Wysocki, M.; Delatour, F.; Faurisson, F.; Rauss, A.; Pean, Y.; Misset, B.; Thomas, F.; Timsit, J.F.; Similowski, T.; Mentec, H.; et al. Continuous versus intermittent infusion of vancomycin in severe Staphylococcal infections: Prospective multicenter randomized study. Antimicrob. Agents Chemother. 2001, 45, 2460–2467. [Google Scholar] [CrossRef] [Green Version]
- Hao, J.J.; Chen, H.; Zhou, J.X. Continuous versus intermittent infusion of vancomycin in adult patients: A systematic review and meta-analysis. Int. J. Antimicrob. Agents 2016, 47, 28–35. [Google Scholar] [CrossRef]
- Rybak, M.; Lomaestro, B.; Rotschafer, J.C.; Moellering, R., Jr.; Craig, W.; Billeter, M.; Dalovisio, J.R.; Levine, D.P. Therapeutic monitoring of vancomycin in adult patients: A consensus review of the American Society of Health-System Pharmacists, the Infectious Diseases Society of America, and the Society of Infectious Diseases Pharmacists. Am. J. Health Syst. Pharm. 2009, 66, 82–98. [Google Scholar] [CrossRef]
- Finch, N.A.; Zasowski, E.J.; Murray, K.P.; Mynatt, R.P.; Zhao, J.J.; Yost, R.; Pogue, J.M.; Rybak, M.J. A Quasi-Experiment To Study the Impact of Vancomycin Area under the Concentration-Time Curve-Guided Dosing on Vancomycin-Associated Nephrotoxicity. Antimicrob. Agents Chemother. 2017. [Google Scholar] [CrossRef] [Green Version]
- Hale, C.M.; Seabury, R.W.; Steele, J.M.; Darko, W.; Miller, C.D. Are Vancomycin Trough Concentrations of 15 to 20 mg/L Associated with Increased Attainment of an AUC/MIC >/= 400 in Patients With Presumed MRSA Infection? J. Pharm. Pract. 2017, 30, 329–335. [Google Scholar] [CrossRef]
- Turner, R.B.; Kojiro, K.; Won, R.; Chang, E.; Chan, D.; Elbarbry, F. Prospective evaluation of vancomycin pharmacokinetics in a heterogeneous critically ill population. Diagn Microbiol Infect Dis 2018, 92, 346–351. [Google Scholar] [CrossRef]
- Patel, N.; Pai, M.P.; Rodvold, K.A.; Lomaestro, B.; Drusano, G.L.; Lodise, T.P. Vancomycin: We can’t get there from here. Clin. Infect. Dis. 2011, 52, 969–974. [Google Scholar] [CrossRef] [Green Version]
- Van Hal, S.J.; Paterson, D.L.; Lodise, T.P. Systematic review and meta-analysis of vancomycin-induced nephrotoxicity associated with dosing schedules that maintain troughs between 15 and 20 milligrams per liter. Antimicrob. Agents Chemother. 2013, 57, 734–744. [Google Scholar] [CrossRef] [Green Version]
- Neely, M.N.; Kato, L.; Youn, G.; Kraler, L.; Bayard, D.; van Guilder, M.; Schumitzky, A.; Yamada, W.; Jones, B.; Minejima, E. Prospective Trial on the Use of Trough Concentration versus Area under the Curve To Determine Therapeutic Vancomycin Dosing. Antimicrob. Agents Chemother. 2018, 62. [Google Scholar] [CrossRef] [Green Version]
- Kalil, A.C.; Metersky, M.L.; Klompas, M.; Muscedere, J.; Sweeney, D.A.; Palmer, L.B.; Napolitano, L.M.; O’Grady, N.P.; Bartlett, J.G.; Carratala, J.; et al. Management of Adults with Hospital-acquired and Ventilator-associated Pneumonia: 2016 Clinical Practice Guidelines by the Infectious Diseases Society of America and the American Thoracic Society. Clin. Infect. Dis. 2016, 63, e61–e111. [Google Scholar] [CrossRef]
- Torres, A.; Niederman, M.S.; Chastre, J.; Ewig, S.; Fernandez-Vandellos, P.; Hanberger, H.; Kollef, M.; Li Bassi, G.; Luna, C.M.; Martin-Loeches, I.; et al. International ERS/ESICM/ESCMID/ALAT guidelines for the management of hospital-acquired pneumonia and ventilator-associated pneumonia: Guidelines for the management of hospital-acquired pneumonia (HAP)/ventilator-associated pneumonia (VAP) of the European Respiratory Society (ERS), European Society of Intensive Care Medicine (ESICM), European Society of Clinical Microbiology and Infectious Diseases (ESCMID) and Asociacion Latinoamericana del Torax (ALAT). Eur. Respir. J. 2017. [Google Scholar] [CrossRef]
- Gu, W.J.; Wang, F.; Tang, L.; Bakker, J.; Liu, J.C. Colistin for the treatment of ventilator-associated pneumonia caused by multidrug-resistant Gram-negative bacteria: A systematic review and meta-analysis. Int. J. Antimicrob. Agents 2014, 44, 477–485. [Google Scholar] [CrossRef]
- Tulli, G.; Messori, A.; Trippoli, S.; Marinai, C. Non-inferiority of colistin compared with standard care for the treatment of ventilator-associated pneumonia. Int. J. Antimicrob. Agents 2017, 49, 638–641. [Google Scholar] [CrossRef]
- Florescu, D.F.; Qiu, F.; McCartan, M.A.; Mindru, C.; Fey, P.D.; Kalil, A.C. What is the efficacy and safety of colistin for the treatment of ventilator-associated pneumonia? A systematic review and meta-regression. Clin. Infect. Dis. 2012, 54, 670–680. [Google Scholar] [CrossRef]
- Cisneros, J.M.; Rosso-Fernandez, C.M.; Roca-Oporto, C.; De Pascale, G.; Jimenez-Jorge, S.; Fernandez-Hinojosa, E.; Matthaiou, D.K.; Ramirez, P.; Diaz-Miguel, R.O.; Estella, A.; et al. Colistin versus meropenem in the empirical treatment of ventilator-associated pneumonia (Magic Bullet study): An investigator-driven, open-label, randomized, noninferiority controlled trial. Crit. Care 2019, 23, 383. [Google Scholar] [CrossRef] [Green Version]
- Cheah, S.E.; Wang, J.; Nguyen, V.T.; Turnidge, J.D.; Li, J.; Nation, R.L. New pharmacokinetic/pharmacodynamic studies of systemically administered colistin against Pseudomonas aeruginosa and Acinetobacter baumannii in mouse thigh and lung infection models: Smaller response in lung infection. J. Antimicrob. Chemother. 2015, 70, 3291–3297. [Google Scholar]
- Mohamed, A.F.; Karaiskos, I.; Plachouras, D.; Karvanen, M.; Pontikis, K.; Jansson, B.; Papadomichelakis, E.; Antoniadou, A.; Giamarellou, H.; Armaganidis, A.; et al. Application of a loading dose of colistin methanesulfonate in critically ill patients: Population pharmacokinetics, protein binding, and prediction of bacterial kill. Antimicrob. Agents Chemother. 2012, 56, 4241–4249. [Google Scholar] [CrossRef] [Green Version]
- Nation, R.L.; Garonzik, S.M.; Li, J.; Thamlikitkul, V.; Giamarellos-Bourboulis, E.J.; Paterson, D.L.; Turnidge, J.D.; Forrest, A.; Silveira, F.P. Updated US and European Dose Recommendations for Intravenous Colistin: How Do They Perform? Clin. Infect. Dis. 2016, 62, 552–558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nation, R.L.; Garonzik, S.M.; Thamlikitkul, V.; Giamarellos-Bourboulis, E.J.; Forrest, A.; Paterson, D.L.; Li, J.; Silveira, F.P. Dosing guidance for intravenous colistin in critically-ill patients. Clin. Infect. Dis. 2017, 64, 565–571. [Google Scholar] [CrossRef] [PubMed]
- Forrest, A.; Garonzik, S.M.; Thamlikitkul, V.; Giamarellos-Bourboulis, E.J.; Paterson, D.L.; Li, J.; Silveira, F.P.; Nation, R.L. Pharmacokinetic/Toxicodynamic Analysis of Colistin-Associated Acute Kidney Injury in Critically Ill Patients. Antimicrob. Agents Chemother. 2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phe, K.; Johnson, M.L.; Palmer, H.R.; Tam, V.H. Validation of a model to predict the risk of nephrotoxicity in patients receiving colistin. Antimicrob. Agents Chemother. 2014, 58, 6946–6948. [Google Scholar] [CrossRef] [Green Version]
- Pogue, J.M.; Lee, J.; Marchaim, D.; Yee, V.; Zhao, J.J.; Chopra, T.; Lephart, P.; Kaye, K.S. Incidence of and risk factors for colistin-associated nephrotoxicity in a large academic health system. Clin. Infect. Dis. 2011, 53, 879–884. [Google Scholar] [CrossRef] [Green Version]
- Rattanaumpawan, P.; Ungprasert, P.; Thamlikitkul, V. Risk factors for colistin-associated nephrotoxicity. J. Infect. 2011, 62, 187–190. [Google Scholar] [CrossRef]
- Rotschafer, J.C.; Ullman, M.A.; Sullivan, C.J. Optimal use of fluoroquinolones in the intensive care unit setting. Crit. Care Clin. 2011, 27, 95–106. [Google Scholar] [CrossRef]
- Mihu, C.N.; Rhomberg, P.R.; Jones, R.N.; Coyle, E.; Prince, R.A.; Rolston, K.V. Escherichia coli resistance to quinolones at a comprehensive cancer center. Diagn. Microbiol. Infect. Dis. 2010, 67, 266–269. [Google Scholar] [CrossRef]
- Graffunder, E.M.; Venezia, R.A. Risk factors associated with nosocomial methicillin-resistant Staphylococcus aureus (MRSA) infection including previous use of antimicrobials. J. Antimicrob. Chemother. 2002, 49, 999–1005. [Google Scholar] [CrossRef]
- Pepin, J.; Saheb, N.; Coulombe, M.A.; Alary, M.E.; Corriveau, M.P.; Authier, S.; Leblanc, M.; Rivard, G.; Bettez, M.; Primeau, V.; et al. Emergence of fluoroquinolones as the predominant risk factor for Clostridium difficile-associated diarrhea: A cohort study during an epidemic in Quebec. Clin. Infect. Dis. 2005, 41, 1254–1260. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.C.; Tsuji, B.T.; Forrest, A. Optimizing use of quinolones in the critically ill. Semin Respir Crit. Care Med. 2007, 28, 586–595. [Google Scholar] [CrossRef]
- Wright, D.H.; Brown, G.H.; Peterson, M.L.; Rotschafer, J.C. Application of fluoroquinolone pharmacodynamics. J. Antimicrob. Chemother. 2000, 46, 669–683. [Google Scholar] [CrossRef]
- Forrest, A.; Nix, D.E.; Ballow, C.H.; Goss, T.F.; Birmingham, M.C.; Schentag, J.J. Pharmacodynamics of intravenous ciprofloxacin in seriously ill patients. Antimicrob. Agents Chemother. 1993, 37, 1073–1081. [Google Scholar] [CrossRef] [Green Version]
- Zelenitsky, S.A.; Ariano, R.E. Support for higher ciprofloxacin AUC 24/MIC targets in treating Enterobacteriaceae bloodstream infection. J. Antimicrob. Chemother. 2010, 65, 1725–1732. [Google Scholar] [CrossRef] [Green Version]
- Bellon, A.; Perez-Garcia, G.; Coverdale, J.H.; Chacko, R.C. Seizures associated with levofloxacin: Case presentation and literature review. Eur J. Clin. Pharmacol. 2009, 65, 959–962. [Google Scholar] [CrossRef] [Green Version]
- Cone, C.; Horowitz, B. Convulsions associated with moxifloxacin. Am. J. Health Syst. Pharm. 2015, 72, 910–912. [Google Scholar] [CrossRef]
- Mazzei, D.; Accardo, J.; Ferrari, A.; Primavera, A. Levofloxacin neurotoxicity and non-convulsive status epilepticus (NCSE): A case report. Clin. Neurol. Neurosurg. 2012, 114, 1371–1373. [Google Scholar] [CrossRef]
- Kollef, M.H. Antibiotics for the critically ill: More than just selecting appropriate initial therapy. Crit. Care 2013, 17, 146. [Google Scholar] [CrossRef] [Green Version]
- Konig, C.; Simmen, H.P.; Blaser, J. Bacterial concentrations in pus and infected peritoneal fluid--implications for bactericidal activity of antibiotics. J. Antimicrob. Chemother. 1998, 42, 227–232. [Google Scholar] [CrossRef]
- Udekwu, K.I.; Parrish, N.; Ankomah, P.; Baquero, F.; Levin, B.R. Functional relationship between bacterial cell density and the efficacy of antibiotics. J. Antimicrob. Chemother. 2009, 63, 745–757. [Google Scholar] [CrossRef]
- Kumar, A. Optimizing antimicrobial therapy in sepsis and septic shock. Crit. Care Nurs. Clin. N. Am. 2011, 23, 79–97. [Google Scholar] [CrossRef] [PubMed]
- Udy, A.A.; Roberts, J.A.; Lipman, J. Clinical implications of antibiotic pharmacokinetic principles in the critically ill. Intensive Care Med. 2013, 39, 2070–2082. [Google Scholar] [CrossRef] [PubMed]
- Pea, F.; Bertolissi, M.; Di Silvestre, A.; Poz, D.; Giordano, F.; Furlanut, M. TDM coupled with Bayesian forecasting should be considered an invaluable tool for optimizing vancomycin daily exposure in unstable critically ill patients. Int. J. Antimicrob. Agents 2002, 20, 326–332. [Google Scholar] [CrossRef]
- Galar, A.; Munoz, P.; Valerio, M.; Cercenado, E.; Garcia-Gonzalez, X.; Burillo, A.; Sanchez-Somolinos, M.; Juarez, M.; Verde, E.; Bouza, E. Current use of daptomycin and systematic therapeutic drug monitoring: Clinical experience in a tertiary care institution. Int. J. Antimicrob. Agents 2019, 53, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Galar, A.; Valerio, M.; Munoz, P.; Alcala, L.; Garcia-Gonzalez, X.; Burillo, A.; Sanjurjo, M.; Grau, S.; Bouza, E. Systematic Therapeutic Drug Monitoring for Linezolid: Variability and Clinical Impact. Antimicrob. Agents Chemother. 2017, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, G.; Sime, F.B.; Lipman, J.; Roberts, J.A. How do we use therapeutic drug monitoring to improve outcomes from severe infections in critically ill patients? BMC Infect Dis 2014, 14, 288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhaese, S.A.M.; Thooft, A.D.J.; Farkas, A.; Lipman, J.; Verstraete, A.G.; Stove, V.; Roberts, J.A.; De Waele, J.J. Early target attainment of continuous infusion piperacillin/tazobactam and meropenem in critically ill patients: A prospective observational study. J. Crit. Care 2019, 52, 75–79. [Google Scholar] [CrossRef]
- Ambrose, P.G.; Bhavnani, S.M.; Rubino, C.M.; Louie, A.; Gumbo, T.; Forrest, A.; Drusano, G.L. Pharmacokinetics-pharmacodynamics of antimicrobial therapy: It’s not just for mice anymore. Clin. Infect. Dis. 2007, 44, 79–86. [Google Scholar] [CrossRef]
- Drusano, G.L.; Louie, A. Optimization of aminoglycoside therapy. Antimicrob. Agents Chemother. 2011, 55, 2528–2531. [Google Scholar] [CrossRef] [Green Version]
- Shekar, K.; Fraser, J.F.; Taccone, F.S.; Welch, S.; Wallis, S.C.; Mullany, D.V.; Lipman, J.; Roberts, J.A.; Investigators AES. The combined effects of extracorporeal membrane oxygenation and renal replacement therapy on meropenem pharmacokinetics: A matched cohort study. Crit. Care 2014, 18, 565. [Google Scholar] [CrossRef] [Green Version]
- Donadello, K.; Roberts, J.A.; Cristallini, S.; Beumier, M.; Shekar, K.; Jacobs, F.; Belhaj, A.; Vincent, J.L.; de Backer, D.; Taccone, F.S. Vancomycin population pharmacokinetics during extracorporeal membrane oxygenation therapy: A matched cohort study. Crit. Care 2014, 18, 632. [Google Scholar] [CrossRef] [Green Version]
- Phe, K.; Heil, E.L.; Tam, V.H. Optimizing Pharmacokinetics-Pharmacodynamics of Antimicrobial Management in Patients with Sepsis: A Review. J. Infect. Dis. 2020, 222, S132–S141. [Google Scholar] [CrossRef]
- Delattre, I.K.; Musuamba, F.T.; Nyberg, J.; Taccone, F.S.; Laterre, P.F.; Verbeeck, R.K.; Jacobs, F.; Wallemacq, P.E. Population pharmacokinetic modeling and optimal sampling strategy for Bayesian estimation of amikacin exposure in critically ill septic patients. Ther. Drug. Monit. 2010, 32, 749–756. [Google Scholar] [CrossRef] [Green Version]
- Lipman, J.; Wallis, S.C.; Boots, R.J. Cefepime versus cefpirome: The importance of creatinine clearance. Anesth. Analg. 2003, 97, 1149–1154. [Google Scholar] [CrossRef] [Green Version]
- Goncalves-Pereira, J.; Silva, N.E.; Mateus, A.; Pinho, C.; Povoa, P. Assessment of pharmacokinetic changes of meropenem during therapy in septic critically ill patients. BMC Pharmacol. Toxicol. 2014, 15, 21. [Google Scholar] [CrossRef] [Green Version]
- De Waele, J.J.; Carrette, S.; Carlier, M.; Stove, V.; Boelens, J.; Claeys, G.; Leroux-Roels, I.; Hoste, E.; Depuydt, P.; Decruyenaere, J.; et al. Therapeutic drug monitoring-based dose optimisation of piperacillin and meropenem: A randomised controlled trial. Intensive Care Med. 2014, 40, 380–387. [Google Scholar] [CrossRef]
- Richter, D.C.; Frey, O.; Rohr, A.; Roberts, J.A.; Koberer, A.; Fuchs, T.; Papadimas, N.; Heinzel-Gutenbrunner, M.; Brenner, T.; Lichtenstern, C.; et al. Therapeutic drug monitoring-guided continuous infusion of piperacillin/tazobactam significantly improves pharmacokinetic target attainment in critically ill patients: A retrospective analysis of four years of clinical experience. Infection 2019, 47, 1001–1011. [Google Scholar] [CrossRef] [Green Version]
- Aubert, G.; Carricajo, A.; Coudrot, M.; Guyomarc’h, S.; Auboyer, C.; Zeni, F. Prospective determination of serum ceftazidime concentrations in intensive care units. Ther. Drug. Monit. 2010, 32, 517–519. [Google Scholar] [CrossRef]
- Chapuis, T.M.; Giannoni, E.; Majcherczyk, P.A.; Chiolero, R.; Schaller, M.D.; Berger, M.M.; Bolay, S.; Decosterd, L.A.; Bugnon, D.; Moreillon, P. Prospective monitoring of cefepime in intensive care unit adult patients. Crit. Care 2010, 14, R51. [Google Scholar] [CrossRef] [Green Version]
- De Waele, J.J.; Lipman, J.; Akova, M.; Bassetti, M.; Dimopoulos, G.; Kaukonen, M.; Koulenti, D.; Martin, C.; Montravers, P.; Rello, J.; et al. Risk factors for target non-attainment during empirical treatment with beta-lactam antibiotics in critically ill patients. Intensive Care Med. 2014, 40, 1340–1351. [Google Scholar] [CrossRef]
- Povoa, P.; Spriet, I.; Zahar, J.R. Antibiotic dosing in the critically ill: Asking the same questions but expecting different answers. Intensive Care Med. 2014, 40, 1780–1782. [Google Scholar] [CrossRef] [Green Version]
- Nicolau, D.P.; Freeman, C.D.; Belliveau, P.P.; Nightingale, C.H.; Ross, J.W.; Quintiliani, R. Experience with a once-daily aminoglycoside program administered to 2,184 adult patients. Antimicrob. Agents Chemother. 1995, 39, 650–655. [Google Scholar] [CrossRef] [Green Version]
- McKinnon, P.S.; Paladino, J.A.; Schentag, J.J. Evaluation of area under the inhibitory curve (AUIC) and time above the minimum inhibitory concentration (T>MIC) as predictors of outcome for cefepime and ceftazidime in serious bacterial infections. Int. J. Antimicrob. Agents 2008, 31, 345–351. [Google Scholar] [CrossRef]
- Mouton, J.W.; Meletiadis, J.; Voss, A.; Turnidge, J. Variation of MIC measurements: The contribution of strain and laboratory variability to measurement precision. J. Antimicrob. Chemother. 2018, 73, 2374–2379. [Google Scholar] [CrossRef] [Green Version]
- May, F.; El-Helali, N.; Timsit, J.F.; Misset, B. Absence of obvious link between supra-therapeutic serum levels of beta lactams and clinical toxicity in ICU patients with acute renal failure treated with intermittent hemodialysis. Crit. Care 2016, 20, 220. [Google Scholar] [CrossRef] [Green Version]
- Joukhadar, C.; Frossard, M.; Mayer, B.X.; Brunner, M.; Klein, N.; Siostrzonek, P.; Eichler, H.G.; Muller, M. Impaired target site penetration of beta-lactams may account for therapeutic failure in patients with septic shock. Crit. Care Med. 2001, 29, 385–391. [Google Scholar] [CrossRef]
- Roberts, J.A.; Roberts, M.S.; Robertson, T.A.; Dalley, A.J.; Lipman, J. Piperacillin penetration into tissue of critically ill patients with sepsis--bolus versus continuous administration? Crit. Care Med. 2009, 37, 926–933. [Google Scholar] [CrossRef] [Green Version]
- Rawson, T.M.; O’Hare, D.; Herrero, P.; Sharma, S.; Moore, L.S.P.; de Barra, E.; Roberts, J.A.; Gordon, A.C.; Hope, W.; Georgiou, P.; et al. Delivering precision antimicrobial therapy through closed-loop control systems. J. Antimicrob. Chemother. 2018, 73, 835–843. [Google Scholar] [CrossRef] [Green Version]
- Scaglione, F.; Esposito, S.; Leone, S.; Lucini, V.; Pannacci, M.; Ma, L.; Drusano, G.L. Feedback dose alteration significantly affects probability of pathogen eradication in nosocomial pneumonia. Eur. Respir. J. 2009, 34, 394–400. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Roberts, D.; Wood, K.E.; Light, B.; Parrillo, J.E.; Sharma, S.; Suppes, R.; Feinstein, D.; Zanotti, S.; Taiberg, L.; et al. Duration of hypotension before initiation of effective antimicrobial therapy is the critical determinant of survival in human septic shock. Crit. Care Med. 2006, 34, 1589–1596. [Google Scholar] [CrossRef]
- Leisman, D.; Huang, V.; Zhou, Q.; Gribben, J.; Bianculli, A.; Bernshteyn, M.; Ward, M.F.; Schneider, S.M. Delayed Second Dose Antibiotics for Patients Admitted from the Emergency Department with Sepsis: Prevalence, Risk Factors, and Outcomes. Crit. Care Med. 2017, 45, 956–965. [Google Scholar] [CrossRef] [PubMed]
- Lipman, J.; Roberts, J. Does Appropriate Antibiotic Therapy Mean Only Adequate Spectrum and Timing? Crit. Care Med. 2015, 43, 1773–1774. [Google Scholar] [CrossRef] [PubMed]
- Arulkumaran, N.; Routledge, M.; Schlebusch, S.; Lipman, J.; Conway Morris, A. Antimicrobial-associated harm in critical care: A narrative review. Intensive Care Med. 2020, 46, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Beumier, M.; Casu, G.S.; Hites, M.; Wolff, F.; Cotton, F.; Vincent, J.L.; Jacobs, F.; Taccone, F.S. Elevated beta-lactam concentrations associated with neurological deterioration in ICU septic patients. Minerva Anestesiol. 2015, 81, 497–506. [Google Scholar]
- Burgess, D.S. Pharmacodynamic principles of antimicrobial therapy in the prevention of resistance. Chest 1999, 115, 19S–23S. [Google Scholar] [CrossRef] [Green Version]
- Karam, G.; Chastre, J.; Wilcox, M.H.; Vincent, J.L. Antibiotic strategies in the era of multidrug resistance. Crit. Care 2016, 20, 136. [Google Scholar] [CrossRef] [Green Version]
- Drusano, G.L. Prevention of resistance: A goal for dose selection for antimicrobial agents. Clin. Infect. Dis. 2003, 36, S42–S50. [Google Scholar] [CrossRef]
- Tam, V.H.; Louie, A.; Fritsche, T.R.; Deziel, M.; Liu, W.; Brown, D.L.; Deshpande, L.; Leary, R.; Jones, R.N.; Drusano, G.L. Impact of drug-exposure intensity and duration of therapy on the emergence of Staphylococcus aureus resistance to a quinolone antimicrobial. J. Infect. Dis. 2007, 195, 1818–1827. [Google Scholar] [CrossRef] [Green Version]
- Tam, V.H.; Schilling, A.N.; Neshat, S.; Poole, K.; Melnick, D.A.; Coyle, E.A. Optimization of meropenem minimum concentration/MIC ratio to suppress in vitro resistance of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2005, 49, 4920–4927. [Google Scholar] [CrossRef] [Green Version]
- Heffernan, A.J.; Sime, F.B.; Lipman, J.; Roberts, J.A. Individualising Therapy to Minimize Bacterial Multidrug Resistance. Drugs 2018, 78, 621–641. [Google Scholar] [CrossRef]
- Thomas, J.K.; Forrest, A.; Bhavnani, S.M.; Hyatt, J.M.; Cheng, A.; Ballow, C.H.; Schentag, J.J. Pharmacodynamic evaluation of factors associated with the development of bacterial resistance in acutely ill patients during therapy. Antimicrob. Agents Chemother. 1998, 42, 521–527. [Google Scholar] [CrossRef] [Green Version]
- Havey, T.C.; Fowler, R.A.; Daneman, N. Duration of antibiotic therapy for bacteremia: A systematic review and meta-analysis. Crit. Care 2011, 15, R267. [Google Scholar] [CrossRef] [Green Version]
- Chastre, J.; Wolff, M.; Fagon, J.Y.; Chevret, S.; Thomas, F.; Wermert, D.; Clementi, E.; Gonzalez, J.; Jusserand, D.; Asfar, P.; et al. Comparison of 8 vs 15 days of antibiotic therapy for ventilator-associated pneumonia in adults: A randomized trial. JAMA 2003, 290, 2588–2598. [Google Scholar] [CrossRef]
- Drusano, G.L. What are the properties that make an antibiotic acceptable for therapy of community-acquired pneumonia? J. Antimicrob. Chemother. 2011, 66 (Suppl. S3), iii61–iii67. [Google Scholar] [CrossRef]
- Sweeney, D.A.; Kalil, A.C. Why don’t we have more inhaled antibiotics to treat ventilator-associated pneumonia? Clin. Microbiol. Infect. 2019, 25, 1195–1199. [Google Scholar] [CrossRef]
- Sweeney, D.A.; Kalil, A.C. The last breath for inhaled antibiotics and VAP? Not so fast. Lancet Infect. Dis. 2020, 20, 265–266. [Google Scholar] [CrossRef]
- Ferrari, F.; Goldstein, I.; Nieszkowszka, A.; Elman, M.; Marquette, C.H.; Rouby, J.J.; Experimental ICUSG. Lack of lung tissue and systemic accumulation after consecutive daily aerosols of amikacin in ventilated piglets with healthy lungs. Anesthesiology 2003, 98, 1016–1019. [Google Scholar] [CrossRef]
- Ferrari, F.; Liu, Z.H.; Lu, Q.; Becquemin, M.H.; Louchahi, K.; Aymard, G.; Marquette, C.H.; Rouby, J.J. Comparison of lung tissue concentrations of nebulized ceftazidime in ventilated piglets: Ultrasonic versus vibrating plate nebulizers. Intensive Care Med. 2008, 34, 1718–1723. [Google Scholar] [CrossRef]
- Lu, Q.; Girardi, C.; Zhang, M.; Bouhemad, B.; Louchahi, K.; Petitjean, O.; Wallet, F.; Becquemin, M.H.; Le Naour, G.; Marquette, C.H.; et al. Nebulized and intravenous colistin in experimental pneumonia caused by Pseudomonas aeruginosa. Intensive Care Med. 2010, 36, 1147–1155. [Google Scholar] [CrossRef]
- Goldstein, I.; Wallet, F.; Nicolas-Robin, A.; Ferrari, F.; Marquette, C.H.; Rouby, J.J. Lung deposition and efficiency of nebulized amikacin during Escherichia coli pneumonia in ventilated piglets. Am. J. Respir. Crit. Care Med. 2002, 166, 1375–1381. [Google Scholar] [CrossRef]
- Rouby, J.J.; Bouhemad, B.; Monsel, A.; Brisson, H.; Arbelot, C.; Lu, Q. Aerosolized antibiotics for ventilator-associated pneumonia: Lessons from experimental studies. Anesthesiology 2012, 117, 1364–1380. [Google Scholar] [CrossRef] [Green Version]
- Niederman, M.S. Adjunctive Nebulized Antibiotics: What Is Their Place in ICU Infections? Front. Med. 2019, 6, 99. [Google Scholar] [CrossRef]
- Niederman, M.S.; Chastre, J.; Corkery, K.; Fink, J.B.; Luyt, C.E.; Garcia, M.S. BAY41-6551 achieves bactericidal tracheal aspirate amikacin concentrations in mechanically ventilated patients with Gram-negative pneumonia. Intensive Care Med. 2012, 38, 263–271. [Google Scholar] [CrossRef]
- Athanassa, Z.E.; Markantonis, S.L.; Fousteri, M.Z.; Myrianthefs, P.M.; Boutzouka, E.G.; Tsakris, A.; Baltopoulos, G.J. Pharmacokinetics of inhaled colistimethate sodium (CMS) in mechanically ventilated critically ill patients. Intensive Care Med. 2012, 38, 1779–1786. [Google Scholar] [CrossRef]
- Ehrmann, S.; Chastre, J.; Diot, P.; Lu, Q. Nebulized antibiotics in mechanically ventilated patients: A challenge for translational research from technology to clinical care. Ann. Intensive Care 2017, 7, 78. [Google Scholar] [CrossRef]
- Wood, G.C.; Swanson, J.M. An Update on Aerosolized Antibiotics for Treating Hospital-Acquired and Ventilator-Associated Pneumonia in Adults. Ann. Pharmacother. 2017, 51, 1112–1121. [Google Scholar] [CrossRef]
- Luyt, C.E.; Hekimian, G.; Brechot, N.; Chastre, J. Aerosol Therapy for Pneumonia in the Intensive Care Unit. Clin. Chest Med. 2018, 39, 823–836. [Google Scholar] [CrossRef]
- Montgomery, A.B.; Rhomberg, P.R.; Abuan, T.; Walters, K.A.; Flamm, R.K. Amikacin-fosfomycin at a five-to-two ratio: Characterization of mutation rates in microbial strains causing ventilator-associated pneumonia and interactions with commonly used antibiotics. Antimicrob. Agents Chemother. 2014, 58, 3708–3713. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.X.; Blaskovich, M.A.; Pelingon, R.; Ramu, S.; Kavanagh, A.; Elliott, A.G.; Butler, M.S.; Montgomery, A.B.; Cooper, M.A. Mucin Binding Reduces Colistin Antimicrobial Activity. Antimicrob. Agents Chemother. 2015, 59, 5925–5931. [Google Scholar] [CrossRef] [Green Version]
- Lu, Q.; Yang, J.; Liu, Z.; Gutierrez, C.; Aymard, G.; Rouby, J.J. Nebulized ceftazidime and amikacin in ventilator-associated pneumonia caused by Pseudomonas aeruginosa. Am. J. Respir. Crit. Care Med. 2011, 184, 106–115. [Google Scholar] [CrossRef] [Green Version]
- Coates, A.L.; Green, M.; Leung, K.; Chan, J.; Ribeiro, N.; Ratjen, F.; Charron, M. A comparison of amount and speed of deposition between the PARI LC STAR(R) jet nebulizer and an investigational eFlow(R) nebulizer. J. Aerosol. Med. Pulm. Drug Deliv. 2011, 24, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Reychler, G.; Aubriot, A.S.; Depoortere, V.; Jamar, F.; Liistro, G. Effect of drug targeting nebulization on lung deposition: A randomized crossover scintigraphic comparison between central and peripheral delivery. Respir. Care 2014, 59, 1501–1507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dugernier, J.; Reychler, G.; Wittebole, X.; Roeseler, J.; Depoortere, V.; Sottiaux, T.; Michotte, J.B.; Vanbever, R.; Dugernier, T.; Goffette, P.; et al. Aerosol delivery with two ventilation modes during mechanical ventilation: A randomized study. Ann. Intensive Care 2016, 6, 73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bachmann, M.C.; Morais, C.; Bugedo, G.; Bruhn, A.; Morales, A.; Borges, J.B.; Costa, E.; Retamal, J. Electrical impedance tomography in acute respiratory distress syndrome. Crit. Care 2018, 22, 263. [Google Scholar] [CrossRef] [Green Version]
- Palmer, L.B.; Smaldone, G.C. Reduction of bacterial resistance with inhaled antibiotics in the intensive care unit. Am. J. Respir. Crit. Care Med. 2014, 189, 1225–1233. [Google Scholar] [CrossRef]
- Sole-Lleonart, C.; Rouby, J.J.; Blot, S.; Poulakou, G.; Chastre, J.; Palmer, L.B.; Bassetti, M.; Luyt, C.E.; Pereira, J.M.; Riera, J.; et al. Nebulization of Antiinfective Agents in Invasively Mechanically Ventilated Adults: A Systematic Review and Meta-analysis. Anesthesiology 2017, 126, 890–908. [Google Scholar] [CrossRef]
- Xu, F.; He, L.L.; Che, L.Q.; Li, W.; Ying, S.M.; Chen, Z.H.; Shen, H.H. Aerosolized antibiotics for ventilator-associated pneumonia: A pairwise and Bayesian network meta-analysis. Crit. Care 2018, 22, 301. [Google Scholar] [CrossRef] [Green Version]
- Alves, J.; Alp, E.; Koulenti, D.; Zhang, Z.; Ehrmann, S.; Blot, S.; Bassetti, M.; Conway-Morris, A.; Reina, R.; Teran, E.; et al. Nebulization of antimicrobial agents in mechanically ventilated adults in 2017: An international cross-sectional survey. Eur J. Clin. Microbiol. Infect. Dis. 2018, 37, 785–794. [Google Scholar] [CrossRef]
- Kollef, M.H.; Ricard, J.D.; Roux, D.; Francois, B.; Ischaki, E.; Rozgonyi, Z.; Boulain, T.; Ivanyi, Z.; Janos, G.; Garot, D.; et al. A Randomized Trial of the Amikacin Fosfomycin Inhalation System for the Adjunctive Therapy of Gram-Negative Ventilator-Associated Pneumonia: IASIS Trial. Chest 2017, 151, 1239–1246. [Google Scholar] [CrossRef]
- Niederman, M.S.; Alder, J.; Bassetti, M.; Boateng, F.; Cao, B.; Corkery, K.; Dhand, R.; Kaye, K.S.; Lawatscheck, R.; McLeroth, P.; et al. Inhaled amikacin adjunctive to intravenous standard-of-care antibiotics in mechanically ventilated patients with Gram-negative pneumonia (INHALE): A double-blind, randomised, placebo-controlled, phase 3, superiority trial. Lancet Infect. Dis. 2020, 20, 330–340. [Google Scholar] [CrossRef]
- Stokker, J.; Karami, M.; Hoek, R.; Gommers, D.; van der Eerden, M. Effect of adjunctive tobramycin inhalation versus placebo on early clinical response in the treatment of ventilator-associated pneumonia: The VAPORISE randomized-controlled trial. Intensive Care Med. 2020, 46, 546–548. [Google Scholar] [CrossRef]
- Schreiber, M.P.; Shorr, A.F. Inhaled antibiotics for the treatment of pneumonia. Curr. Opin. Pulm. Med. 2019, 25, 289–293. [Google Scholar] [CrossRef]
- Palmer, L.B.; Smaldone, G.C.; Chen, J.J.; Baram, D.; Duan, T.; Monteforte, M.; Varela, M.; Tempone, A.K.; O’Riordan, T.; Daroowalla, F.; et al. Aerosolized antibiotics and ventilator-associated tracheobronchitis in the intensive care unit. Crit. Care Med. 2008, 36, 2008–2013. [Google Scholar] [CrossRef]
- Rello, J.; Sole-Lleonart, C.; Rouby, J.J.; Chastre, J.; Blot, S.; Poulakou, G.; Luyt, C.E.; Riera, J.; Palmer, L.B.; Pereira, J.M.; et al. Use of nebulized antimicrobials for the treatment of respiratory infections in invasively mechanically ventilated adults: A position paper from the European Society of Clinical Microbiology and Infectious Diseases. Clin. Microbiol. Infect. 2017, 23, 629–639. [Google Scholar] [CrossRef] [Green Version]
- Zampieri, F.G.; Nassar, A.P., Jr.; Gusmao-Flores, D.; Taniguchi, L.U.; Torres, A.; Ranzani, O.T. Nebulized antibiotics for ventilator-associated pneumonia: A systematic review and meta-analysis. Crit. Care 2015, 19, 150. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.; Zhang, J.; Liu, H.X.; Zhu, Y.G.; Qu, J.M. Intravenous combined with aerosolised polymyxin versus intravenous polymyxin alone in the treatment of pneumonia caused by multidrug-resistant pathogens: A systematic review and meta-analysis. Int. J. Antimicrob. Agents 2015, 46, 603–609. [Google Scholar] [CrossRef]
- Valachis, A.; Samonis, G.; Kofteridis, D.P. The role of aerosolized colistin in the treatment of ventilator-associated pneumonia: A systematic review and metaanalysis. Crit. Care Med. 2015, 43, 527–533. [Google Scholar] [CrossRef]
- Roberts, J.A. Using PK/PD to optimize antibiotic dosing for critically ill patients. Curr. Pharm Biotechnol. 2011, 12, 2070–2079. [Google Scholar] [CrossRef]
- Roberts, J.A.; Roberts, M.S.; Semark, A.; Udy, A.A.; Kirkpatrick, C.M.; Paterson, D.L.; Roberts, M.J.; Kruger, P.; Lipman, J. Antibiotic dosing in the ’at risk’ critically ill patient: Linking pathophysiology with pharmacokinetics/pharmacodynamics in sepsis and trauma patients. BMC Anesthesiol. 2011, 11, 3. [Google Scholar] [CrossRef] [Green Version]
- Shekar, K.; Fraser, J.F.; Smith, M.T.; Roberts, J.A. Pharmacokinetic changes in patients receiving extracorporeal membrane oxygenation. J. Crit. Care 2012, 27, 741. [Google Scholar] [CrossRef]
- Abdulla, A.; Dijkstra, A.; Hunfeld, N.G.M.; Endeman, H.; Bahmany, S.; Ewoldt, T.M.J.; Muller, A.E.; van Gelder, T.; Gommers, D.; Koch, B.C.P. Failure of target attainment of beta-lactam antibiotics in critically ill patients and associated risk factors: A two-center prospective study (EXPAT). Crit. Care 2020, 24, 558. [Google Scholar] [CrossRef]
- Scheetz, M.H.; Lodise, T.P.; Downes, K.J.; Drusano, G.; Neely, M. The case for precision dosing: Medical conservatism does not justify inaction. J. Antimicrob. Chemother. 2021. [Google Scholar] [CrossRef]
- Carlier, M.; Carrette, S.; Stove, V.; Verstraete, A.G.; De Waele, J.J. Does consistent piperacillin dosing result in consistent therapeutic concentrations in critically ill patients? A longitudinal study over an entire antibiotic course. Int. J. Antimicrob. Agents 2014, 43, 470–473. [Google Scholar] [CrossRef]
- Lodise, T.P., Jr.; Lomaestro, B.; Drusano, G.L. Piperacillin-tazobactam for Pseudomonas aeruginosa infection: Clinical implications of an extended-infusion dosing strategy. Clin. Infect. Dis. 2007, 44, 357–363. [Google Scholar] [CrossRef]
- Goncalves-Pereira, J.; Oliveira, B.S.; Janeiro, S.; Estilita, J.; Monteiro, C.; Salgueiro, A.; Vieira, A.; Gouveia, J.; Paulino, C.; Bento, L.; et al. Continuous infusion of piperacillin/tazobactam in septic critically ill patients--a multicenter propensity matched analysis. PLoS ONE 2012, 7, e49845. [Google Scholar] [CrossRef]
- Shiu, J.; Wang, E.; Tejani, A.M.; Wasdell, M. Continuous versus intermittent infusions of antibiotics for the treatment of severe acute infections. Cochrane Database Syst. Rev. 2013. [Google Scholar] [CrossRef]
- Roberts, J.A.; Paul, S.K.; Akova, M.; Bassetti, M.; De Waele, J.J.; Dimopoulos, G.; Kaukonen, K.M.; Koulenti, D.; Martin, C.; Montravers, P.; et al. DALI: Defining antibiotic levels in intensive care unit patients: Are current beta-lactam antibiotic doses sufficient for critically ill patients? Clin. Infect. Dis. 2014, 58, 1072–1083. [Google Scholar] [CrossRef]
- Abdul-Aziz, M.H.; Sulaiman, H.; Mat-Nor, M.B.; Rai, V.; Wong, K.K.; Hasan, M.S.; Abd Rahman, A.N.; Jamal, J.A.; Wallis, S.C.; Lipman, J.; et al. Beta-Lactam Infusion in Severe Sepsis (BLISS): A prospective, two-centre, open-labelled randomised controlled trial of continuous versus intermittent beta-lactam infusion in critically ill patients with severe sepsis. Intensive Care Med. 2016, 42, 1535–1545. [Google Scholar] [CrossRef]
- Dulhunty, J.M.; Roberts, J.A.; Davis, J.S.; Webb, S.A.; Bellomo, R.; Gomersall, C.; Shirwadkar, C.; Eastwood, G.M.; Myburgh, J.; Paterson, D.L.; et al. Continuous infusion of beta-lactam antibiotics in severe sepsis: A multicenter double-blind, randomized controlled trial. Clin. Infect. Dis. 2013, 56, 236–244. [Google Scholar] [CrossRef] [Green Version]
- Roberts, J.A.; Abdul-Aziz, M.H.; Davis, J.S.; Dulhunty, J.M.; Cotta, M.O.; Myburgh, J.; Bellomo, R.; Lipman, J. Continuous versus Intermittent beta-Lactam Infusion in Severe Sepsis. A Meta-analysis of Individual Patient Data from Randomized Trials. Am. J. Respir. Crit. Care Med. 2016, 194, 681–691. [Google Scholar] [CrossRef]
- Lipman, J.; Brett, S.J.; De Waele, J.J.; Cotta, M.O.; Davis, J.S.; Finfer, S.; Glass, P.; Knowles, S.; McGuinness, S.; Myburgh, J.; et al. A protocol for a phase 3 multicentre randomised controlled trial of continuous versus intermittent beta-lactam antibiotic infusion in critically ill patients with sepsis: BLING III. Crit. Care Resusc. 2019, 21, 63–68. [Google Scholar] [PubMed]
- Udy, A.A.; Dulhunty, J.M.; Roberts, J.A.; Davis, J.S.; Webb, S.A.R.; Bellomo, R.; Gomersall, C.; Shirwadkar, C.; Eastwood, G.M.; Myburgh, J.; et al. Association between augmented renal clearance and clinical outcomes in patients receiving beta-lactam antibiotic therapy by continuous or intermittent infusion: A nested cohort study of the BLING-II randomised, placebo-controlled, clinical trial. Int. J. Antimicrob. Agents 2017, 49, 624–630. [Google Scholar] [CrossRef] [PubMed]
- Craig, W.A. Optimizing aminoglycoside use. Crit. Care Clin. 2011, 27, 107–121. [Google Scholar] [CrossRef] [PubMed]
- Taccone, F.S.; Laterre, P.F.; Spapen, H.; Dugernier, T.; Delattre, I.; Layeux, B.; De Backer, D.; Wittebole, X.; Wallemacq, P.; Vincent, J.L.; et al. Revisiting the loading dose of amikacin for patients with severe sepsis and septic shock. Crit. Care 2010, 14, R53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barclay, M.L.; Kirkpatrick, C.M.; Begg, E.J. Once daily aminoglycoside therapy. Is it less toxic than multiple daily doses and how should it be monitored? Clin. Pharmacokinet. 1999, 36, 89–98. [Google Scholar] [CrossRef] [PubMed]
- De Waele, J.J.; De Neve, N. Aminoglycosides for life-threatening infections: A plea for an individualized approach using intensive therapeutic drug monitoring. Minerva Anestesiol. 2014, 80, 1135–1142. [Google Scholar]
- Cotta, M.O.; Roberts, J.A.; Lipman, J. Antibiotic dose optimization in critically ill patients. Med. Intensiva 2015, 39, 563–572. [Google Scholar] [CrossRef]
- Sawchuk, R.J.; Zaske, D.E.; Cipolle, R.J.; Wargin, W.A.; Strate, R.G. Kinetic model for gentamicin dosing with the use of individual patient parameters. Clin. Pharmacol. Ther. 1977, 21, 362–369. [Google Scholar] [CrossRef]
- Jeffres, M.N.; Isakow, W.; Doherty, J.A.; McKinnon, P.S.; Ritchie, D.J.; Micek, S.T.; Kollef, M.H. Predictors of mortality for methicillin-resistant Staphylococcus aureus health-care-associated pneumonia: Specific evaluation of vancomycin pharmacokinetic indices. Chest 2006, 130, 947–955. [Google Scholar] [CrossRef]
- Kitzis, M.D.; Goldstein, F.W. Monitoring of vancomycin serum levels for the treatment of staphylococcal infections. Clin. Microbiol. Infect. 2006, 12, 92–95. [Google Scholar] [CrossRef] [Green Version]
- Lodise, T.P.; Drusano, G. Vancomycin Area Under the Curve-Guided Dosing and Monitoring for Adult and Pediatric Patients with Suspected or Documented Serious Methicillin-Resistant Staphylococcus aureus Infections: Putting the Safety of Our Patients First. Clin. Infect. Dis. 2021, 72, 1497–1501. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Antibiotics that Stay in Extracellular Fluid (Vd < 0.3 L/kg) | Drugs that Distribute into Total Body Water (Vd 0.7–1 L/kg) | Drug with High Distribution to Tissues (Vd > 1 L/kg) |
|---|---|---|
|
|
|
| Antimicrobial Class | Monitoring/ Sampling | PK/PD Target | Toxicity Threshold |
|---|---|---|---|
| Therapeutic Drug Monitoring Recommended | |||
Beta-lactams
| Cmin/One sample 1 Css (continuous infusion)/One sample 2 | 100% fT> MIC Css > MIC 50–100% fT > MIC 45–100% fT > MIC 50–100% fT > MIC | Nephrotoxicity/Neurotoxicity Cmin > 361 mg/L (Piperacillin nephro-/neurotoxicity) Cmin > 20 mg/L (Cefepime neurotoxicity) Cmin > 44.5 mg/L (Meropenem nephro-/neurotoxicity) |
Aminoglycosides
| AUC-based/Two samples 3 Cmax/MIC/One sample 4 Cmin/One sample 1 | AUC 80–120 mg h/L Cmax/MIC ≥ 8–10 Cmin <0.5 mg/L Cmin <2.5 mg/L | Nephrotoxicity/Ototoxicity Cmin> 1 mg/L Cmin> 5 mg/L |
Glycopeptides
| AUC/MIC/Two samples 5 Cmin/One sample 1 Css/One sample 2 | AUC (0–24)/MIC ≥ 400 Cmin ≥ 15–20 mg/L 6 Css 20–25 mg/L | Nephrotoxicity Cmin > 20 mg/L |
| Therapeutic Drug Monitoring Neither Recommended nor Discouraged | |||
| Colistin | Cmin/One sample 1 AUC (0–24)/MIC | Cmin 2 mg/L Not defined | Nephrotoxicity Cmin > 2.4 mg/L |
| Fluoroquinolones | AUC/MIC/Two samples 7 Cmax/MIC/One sample 4 | fAUC0–24/MIC ≥ 80 Cmax/MIC ≥ 8–12 | Not defined |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Póvoa, P.; Moniz, P.; Pereira, J.G.; Coelho, L. Optimizing Antimicrobial Drug Dosing in Critically Ill Patients. Microorganisms 2021, 9, 1401. https://doi.org/10.3390/microorganisms9071401
Póvoa P, Moniz P, Pereira JG, Coelho L. Optimizing Antimicrobial Drug Dosing in Critically Ill Patients. Microorganisms. 2021; 9(7):1401. https://doi.org/10.3390/microorganisms9071401
Chicago/Turabian StylePóvoa, Pedro, Patrícia Moniz, João Gonçalves Pereira, and Luís Coelho. 2021. "Optimizing Antimicrobial Drug Dosing in Critically Ill Patients" Microorganisms 9, no. 7: 1401. https://doi.org/10.3390/microorganisms9071401