
Comparative Genomics of Prophages Sato and Sole Expands the Genetic Diversity Found in the Genus Betatectivirus


Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Strains, Phages and Culture Conditions
2.2. Production of Concentrated Phage Stocks
2.3. Phage DNA Extraction, Sequencing and Assembly
2.4. Phage Genome Annotation and Analysis
2.5. Detection of a Plasmidial Prophage State by Plasmid Gel Electrophoresis
2.6. Phylogenetic Methods
2.7. Nucleotide Sequence Accession Numbers
3. Results and Discussion
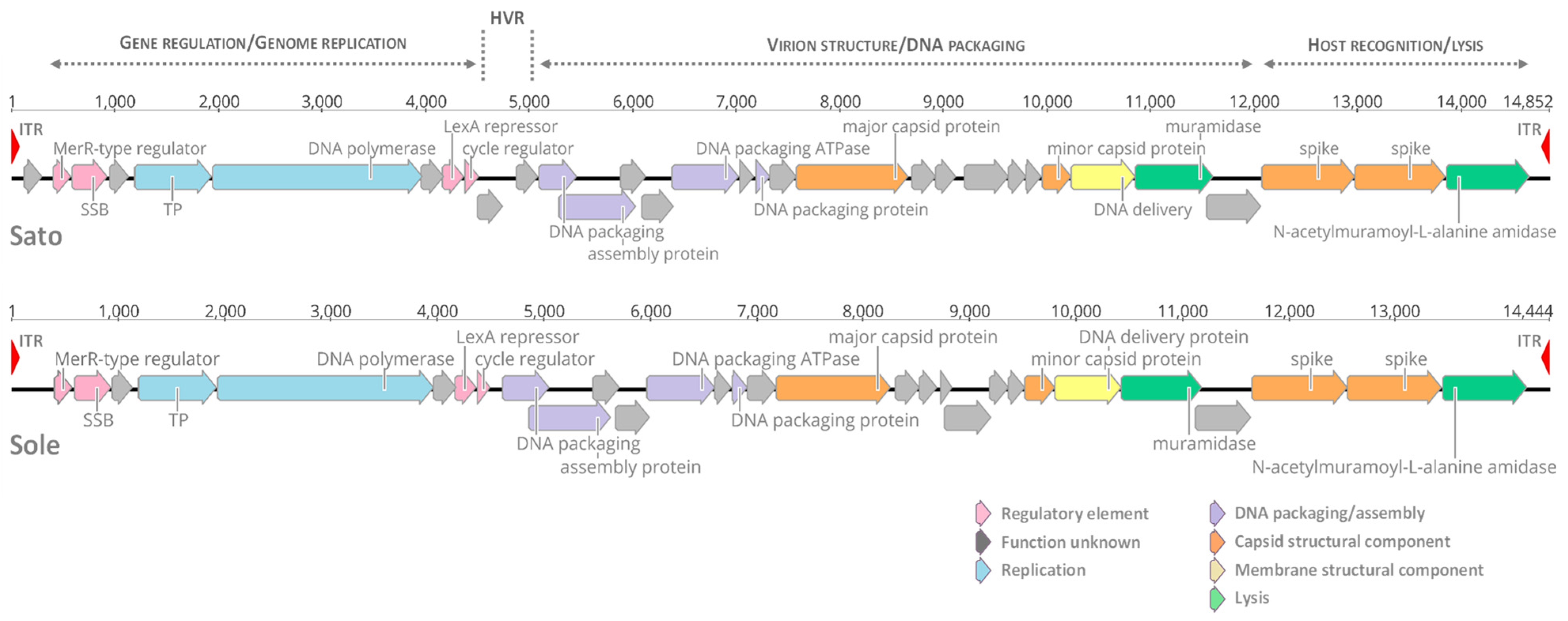
3.1. Genomic Features of Tectiviruses Sato and Sole
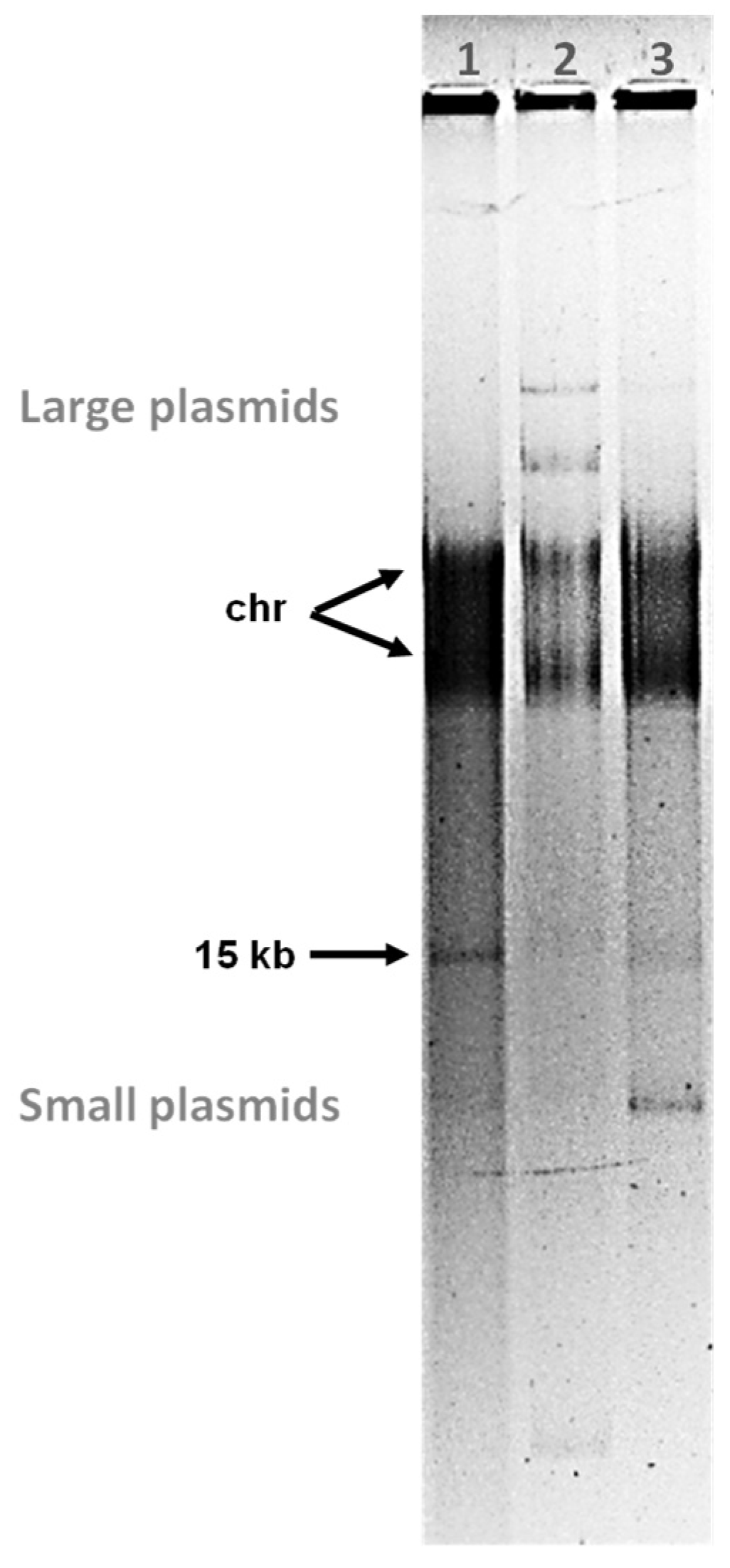
3.2. Tectiviruses Sato and Sole Are Plasmidial Prophages
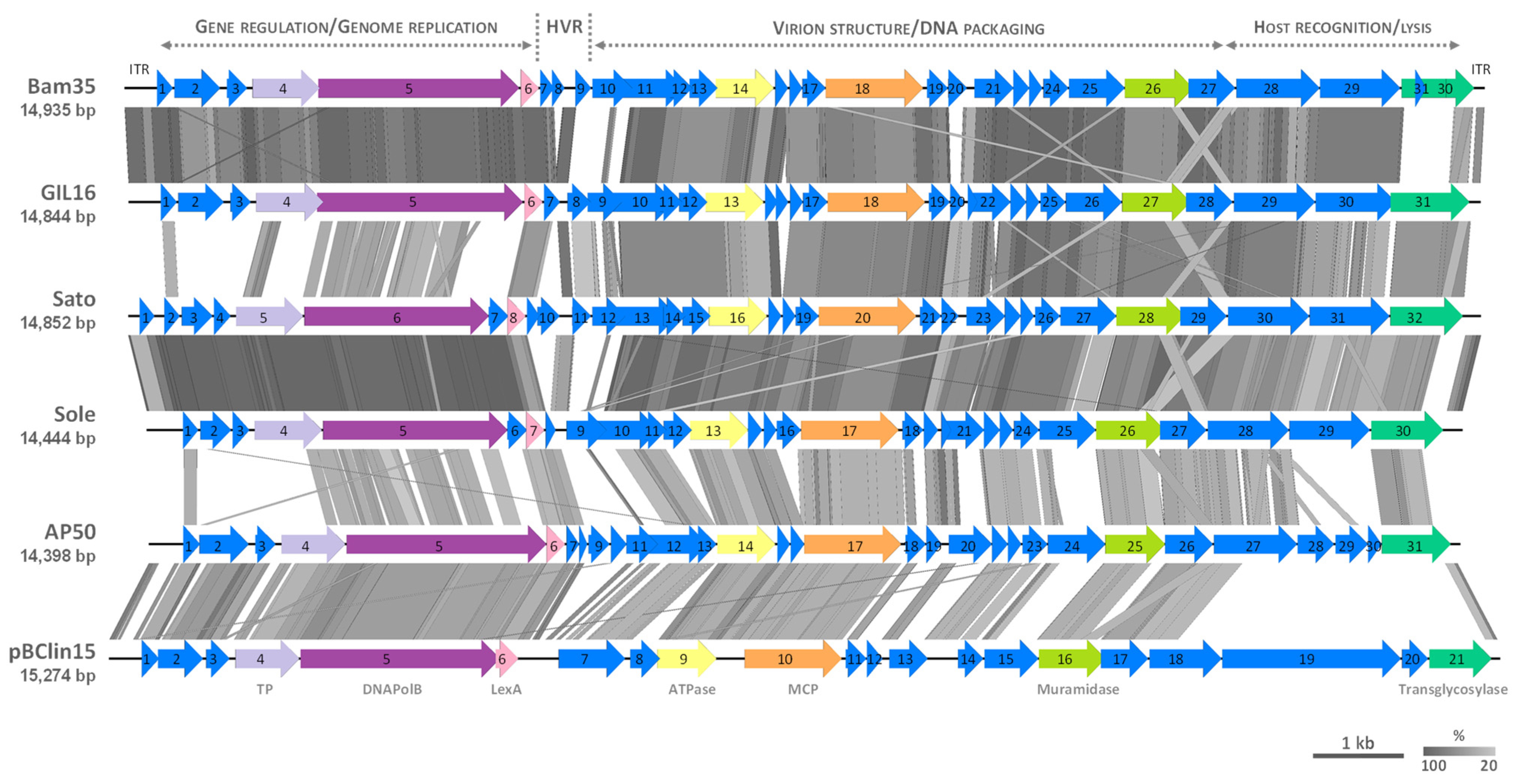
3.3. Comparative Genomics of Tectiviruses Sato and Sole
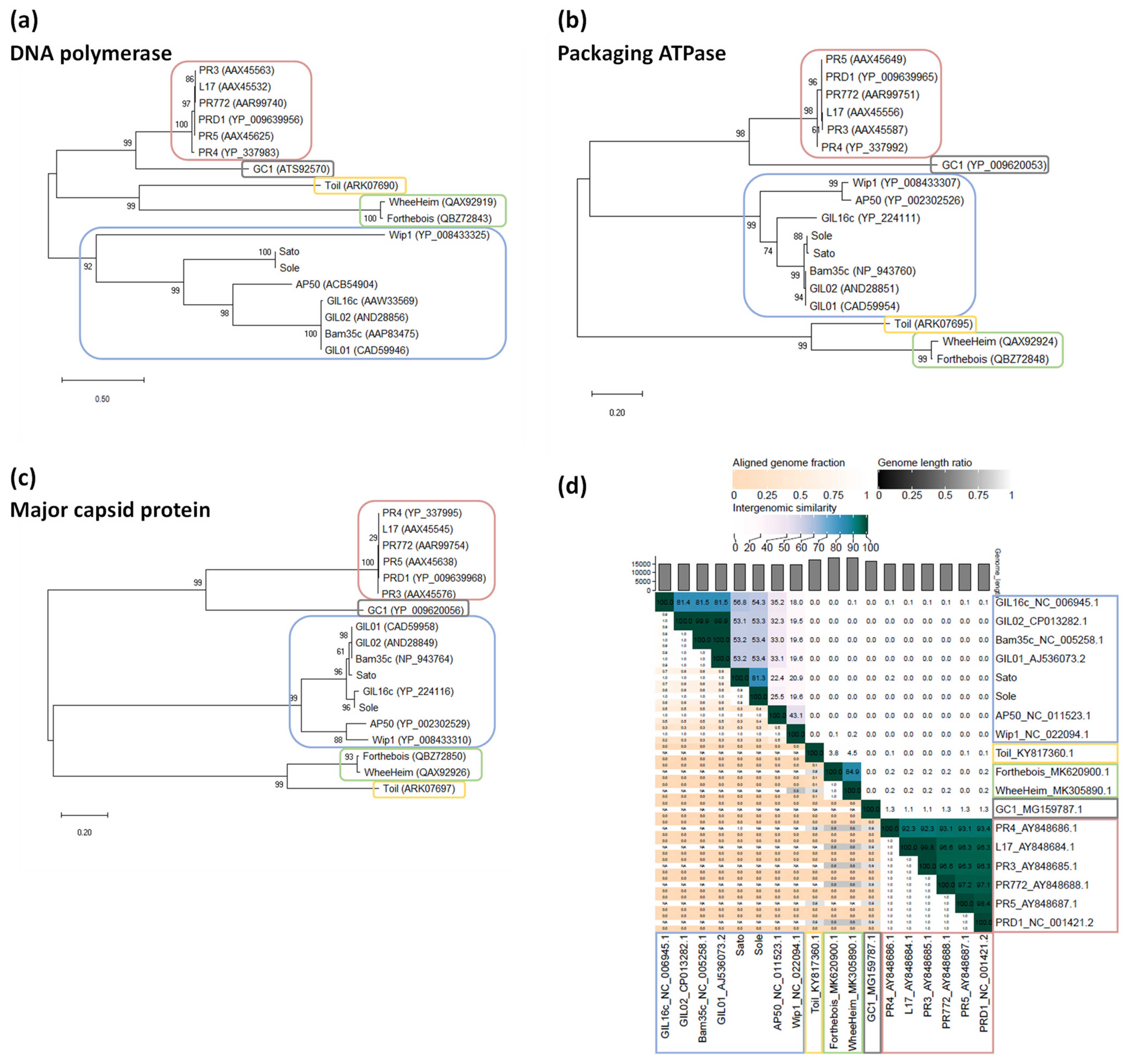
3.4. Phylogenetic Position of Sato and Sole within the Family Tectiviridae


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sato | Sole | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Original Host | GenBank Acc. No. | Genome Length (bp) | Percent Cover | Percent Identity | % nt SequenceIdentity 1 | Percent Cover | Percent Identity | % nt SequenceIdentity 1 | |
| Phage | |||||||||
| Bam35 2 | B. thuringiensis | NC_005258 | 14,935 | 60 | 85.82 | 51.49 | 55 | 88.96 | 48.93 |
| GIL01 | B. thuringiensis | AJ536073 | 14,931 | 60 | 85.84 | 51.50 | 60 | 88.60 | 53.16 |
| GIL02 | B. thuringiensis | CP013282 | 14,961 | 60 | 85.84 | 51.50 | 60 | 88.60 | 53.16 |
| GIL16 2 | B. thuringiensis | NC_006945 | 14,844 | 66 | 84.43 | 55.72 | 59 | 92.52 | 54.59 |
| AP50 2 | B. anthracis | NC_011523 | 14,398 | 29 | 66.38 | 19.25 | 31 | 65.81 | 20.40 |
| Wip1 | B. anthracis | NC_022094 | 14,319 | 28 | 65.01 | 18.20 | 26 | 66.70 | 17.34 |
| Sato | B. cereus3 | MZ089978 | 14,852 | - | - | - | 93 | 96.77 | 90.00 |
| Sole | B. cereus s.l. | MZ089979 | 14,444 | 90 | 96.77 | 87.09 | - | - | - |
| Plasmid | |||||||||
| pBClin15 | B. cereus | AE016878 | 15,274 | 37 | 75.30 | 27.86 | 40 | 72.46 | 28.98 |
| pEFR-1-4 | B. cereus | CP064076 | 14,727 | 99 | 99.99 | 98.99 | 92 | 96.94 | 89.18 |
| pEFR-4-5 | B. paranthracis | CP064084 | 14,728 | 99 | 99.99 | 98.99 | 92 | 96.94 | 89.18 |
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liu, Y.; Du, J.; Lai, Q.; Zeng, R.; Ye, D.; Xu, J.; Shao, Z. Proposal of nine novel species of the Bacillus cereus group. Int. J. Syst. Evol. Microbiol. 2017, 67, 2499–2508. [Google Scholar] [CrossRef]
- Carroll, L.M.; Wiedmann, M.; Kovac, J. Proposal of a taxonomic nomenclature for the Bacillus cereus group which reconciles genomic definitions of bacterial species with clinical and industrial phenotypes. mBio 2020, 11, e00034-20. [Google Scholar] [CrossRef] [Green Version]
- Jensen, G.B.; Hansen, B.M.; Eilenberg, J.; Mahillon, J. The hidden lifestyles of Bacillus cereus and relatives. Environ. Microbiol. 2003, 5, 631–640. [Google Scholar] [CrossRef]
- Gillis, A.; Fayad, N.; Makart, L.; Bolotin, A.; Sorokin, A.; Kallassy, M.; Mahillon, J. Role of plasmid plasticity and mobile genetic elements in the entomopathogen Bacillus thuringiensis serovar israelensis. FEMS Microbiol. Rev. 2018, 42, 829–856. [Google Scholar] [CrossRef]
- Gillis, A.; Mahillon, J. Phages preying on Bacillus anthracis, Bacillus cereus, and Bacillus thuringiensis: Past, present and future. Viruses 2014, 6, 2623–2672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasko, D.A.; Altherr, M.R.; Han, C.S.; Ravel, J. Genomics of the Bacillus cereus group of organisms. FEMS Microbiol. Rev. 2005, 29, 303–329. [Google Scholar] [PubMed]
- Meric, G.; Mageiros, L.; Pascoe, B.; Woodcock, D.J.; Mourkas, E.; Lamble, S.; Bowden, R.; Jolley, K.A.; Raymond, B.; Sheppard, S.K. Lineage-specific plasmid acquisition and the evolution of specialized pathogens in Bacillus thuringiensis and the Bacillus cereus group. Mol. Ecol. 2018, 27, 1524–1540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brüssow, H.; Canchaya, C.; Hardt, W.D. Phages and the evolution of bacterial pathogens: From genomic rearrangements to lysogenic conversion. Microbiol. Mol. Biol. Rev. 2004, 68, 560–602. [Google Scholar] [CrossRef] [Green Version]
- Hatfull, G.F.; Hendrix, R.W. Bacteriophages and their genomes. Curr. Opin. Virol. 2011, 1, 298–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gillis, A.; Mahillon, J. Prevalence, genetic diversity and host range of tectiviruses among members of the Bacillus cereus group. Appl. Environ. Microbiol. 2014, 80, 4138–4152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jalasvuori, M.; Koskinen, K. Extending the hosts of Tectiviridae into four additional genera of gram-positive bacteria and more diverse Bacillus species. Virology 2018, 518, 136–142. [Google Scholar] [CrossRef]
- Adriaenssens, E.M.; Wittmann, J.; Kuhn, J.H.; Turner, D.; Sullivan, M.B.; Dutilh, B.E.; Jang, H.B.; van Zyl, L.J.; Klumpp, J.; Lobocka, M.; et al. Taxonomy of prokaryotic viruses: 2017 update from the ICTV Bacterial and Archaeal Viruses Subcommittee. Arch. Virol. 2018, 163, 1125–1129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strömsten, N.J.; Benson, S.D.; Burnett, R.M.; Bamford, D.H.; Bamford, J.K. The Bacillus thuringiensis linear double-stranded DNA phage Bam35, which is highly similar to the Bacillus cereus linear plasmid pBClin15, has a prophage state. J. Bacteriol. 2003, 185, 6985–6989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gillis, A.; Mahillon, J. Influence of lysogeny of tectiviruses GIL01 and GIL16 on Bacillus thuringiensis growth, biofilm formation, and swarming motility. Appl. Environ. Microbiol. 2014, 80, 7620–7630. [Google Scholar] [CrossRef] [Green Version]
- Mäntynen, S.; Sundberg, L.-R.; Oksanen, H.M.; Poranen, M.M. Half a century of research on membrane-containing bacteriophages: Bringing new concepts to modern virology. Viruses 2019, 11, 76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caldentey, J.; Tuma, R.; Bamford, D.H. Assembly of bacteriophage PRD1 spike complex: Role of the multidomain protein p5. Biochemistry 2000, 39, 10566–10573. [Google Scholar] [CrossRef] [PubMed]
- Peralta, B.; Gil-Carton, D.; Castaño-Díez, D.; Bertin, A.; Boulogne, C.; Oksanen, H.M.; Bamford, D.H.; Abrescia, N.G.A. Mechanism of membranous tunnelling nanotube formation in viral genome delivery. PLoS Biol. 2013, 11, e1001667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berjón-Otero, M.; Villar, L.; Salas, M.; Redrejo-Rodríguez, M. Disclosing early steps of protein-primed genome replication of the gram-positive tectivirus Bam35. Nucleic Acids Res. 2016, 44, 9733–9744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gillis, A.; Jang, H.B.; Kuhn, J.H.; Kropinski, A.M.; Adriaenssens, E.; Lavigne, R. To Rename the Existing Genus Tectivirus (Proposed New Name Alphatectivirus) in the Family Tectiviridae, Create a New Genus, Betatectivirus, and Include Three (3) New Species; ICTV Taxonomic Proposal, 2017.013B. Available online: https://talk.ictvonline.org/files/ictv_official_taxonomy_updates_since_the_8th_report/m/prokaryote-official/7109 (accessed on 30 April 2021).
- Verheust, C.; Jensen, G.; Mahillon, J. pGIL01, a linear tectiviral plasmid prophage originating from Bacillus thuringiensis serovar israelensis. Microbiology 2003, 149, 2083–2092. [Google Scholar] [CrossRef] [Green Version]
- Bolotin, A.; Gillis, A.; Sanchis, V.; Nielsen-LeRoux, C.; Mahillon, J.; Lereclus, D.; Sorokin, A. Comparative genomics of extrachromosomal elements in Bacillus thuringiensis subsp. israelensis. Res. Microbiol. 2017, 168, 331–344. [Google Scholar] [CrossRef] [Green Version]
- Jalasvuori, M.; Palmu, S.; Gillis, A.; Kokko, H.; Mahillon, J.; Bamford, J.K.H.; Fornelos, N. Identification of five novel tectiviruses in Bacillus strains: Analysis of a highly variable region generating genetic diversity. Res. Microbiol. 2013, 164, 118–126. [Google Scholar] [CrossRef]
- Sozhamannan, S.; McKinstry, M.; Lentz, S.M.; Jalasvuori, M.; McAfee, F.; Smith, A.; Dabbs, J.; Ackermann, H.-W.; Bamford, J.K.H.; Mateczun, A.; et al. Molecular characterization of a variant of Bacillus anthracis-specific phage AP50 with improved bacteriolytic activity. Appl. Environ. Microbiol. 2008, 74, 6792–6796. [Google Scholar] [CrossRef] [Green Version]
- Caruso, S.M.; deCarvalho, T.N.; Huynh, A.; Morcos, G.; Kuo, N.; Parsa, S.; Erill, I. A novel genus of actinobacterial Tectiviridae. Viruses 2019, 11, 1134. [Google Scholar] [CrossRef] [Green Version]
- Sambrook, J.; Russell, D.W. Molecular Cloning: A Laboratory Manual, 3rd ed.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2001. [Google Scholar]
- Hock, L.; Gillis, A.; Mahillon, J. Complete genome sequence of bacteriophage Deep-Purple, a novel member of the family Siphoviridae infecting Bacillus cereus. Arch. Virol. 2018, 163, 2555–2559. [Google Scholar] [CrossRef] [PubMed]
- Brettin, T.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Olsen, G.J.; Olson, R.; Overbeek, R.; Parrello, B.; Pusch, G.D.; et al. RASTtk: A modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes. Sci. Rep. 2015, 5, 8365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delcher, A.L.; Bratke, K.A.; Powers, E.C.; Salzberg, S.L. Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics 2007, 23, 673–679. [Google Scholar] [CrossRef] [PubMed]
- Besemer, J.; Lomsadze, A.; Borodovsky, M. GeneMarkS: A self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res. 2001, 29, 2607–2618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef] [Green Version]
- Chan, P.P.; Lin, B.Y.; Mak, A.J.; Lowe, T.M. tRNAscan-SE 2.0: Improved detection and functional classification of transfer RNA genes. bioRxiv 2019. [Google Scholar] [CrossRef] [Green Version]
- Krogh, A.; Larsson, B.; von Heijne, G.; Sonnhammer, E.L.L. Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. J. Mol. Biol. 2001, 305, 567–580. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, M.J.; Petty, N.K.; Beatson, S.A. Easyfig: A genome comparison visualizer. Bioinformatics 2011, 27, 1009–1010. [Google Scholar] [CrossRef]
- Edgar, R.C. Muscle: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- Muhire, B.M.; Varsani, A.; Martin, D.P. SDT: A virus classification tool based on pairwise sequence alignment and identity calculation. PLoS ONE 2014, 9, e108277. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, L.; Stephens, A.; Nam, S.Z.; Rau, D.; Kubler, J.; Lozajic, M.; Gabler, F.; Soding, J.; Lupas, A.N.; Alva, V. A completely reimplemented MPI bioinformatics toolkit with a new HHpred server at its core. J. Mol. Biol. 2018, 430, 2237–2243. [Google Scholar] [CrossRef] [PubMed]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J.E. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef] [Green Version]
- Roy, A.; Kucukural, A.; Zhang, Y. I-TASSER: A unified platform for automated protein structure and function prediction. Nat. Protoc. 2010, 5, 725–738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gillis, A.; Guo, S.; Bolotin, A.; Makart, L.; Sorokin, A.; Mahillon, J. Detection of the cryptic prophage-like molecule pBtic235 in Bacillus thuringiensis subsp. israelensis. Res. Microbiol. 2017, 168, 319–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Moraru, C.; Varsani, A.; Kropinski, A.M. VIRIDIC—A novel tool to calculate the intergenomic similarities of prokaryote-infecting viruses. Viruses 2020, 12, 1268. [Google Scholar] [CrossRef]
- Turner, D.; Kropinski, A.M.; Adriaenssens, E.M. A roadmap for genome-based phage taxonomy. Viruses 2021, 13, 506. [Google Scholar] [CrossRef] [PubMed]
- Fornelos, N.; Bamford, J.K.H.; Mahillon, J. Phage-borne factors and host LexA regulate the lytic switch in phage GIL01. J. Bacteriol. 2011, 193, 6008–6019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berjón-Otero, M.; Lechuga, A.; Mehla, J.; Uetz, P.; Salas, M.; Redrejo-Rodríguez, M. Bam35 tectivirus intraviral interaction map unveils new function and localization of phage orfan proteins. J. Virol. 2017, 91, e00870-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lechuga, A.; Kazlauskas, D.; Salas, M.; Redrejo-Rodríguez, M. Unlimited cooperativity of Betatectivirus SSB, a novel DNA binding protein related to an atypical group of SSBs from protein-primed replicating bacterial viruses. bioRxiv 2021. [Google Scholar] [CrossRef]
- Van der Auwera, G.A.; Feldgarden, M.; Kolter, R.; Mahillon, J. Whole-genome sequences of 94 environmental isolates of Bacillus cereus sensu lato. Genome Announc. 2013, 1, e00380-13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stabell, F.B.; Egge-Jacobsen, W.; Risoen, P.A.; Kolstø, A.B.; Økstad, O.A. Orf 2 from the Bacillus cereus linear plasmid pBClin15 encodes a DNA binding protein. Lett. Appl. Microbiol. 2009, 48, 51–57. [Google Scholar] [CrossRef]
- Berjón-Otero, M.; Villar, L.; de Vega, M.; Salas, M.; Redrejo-Rodríguez, M. DNA polymerase from temperate phage Bam35 is endowed with processive polymerization and abasic sites translesion synthesis capacity. Proc. Nat. Acad. Sci. USA. 2015, 112, E3476–E3484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravantti, J.J.; Gaidelyte, A.; Bamford, D.H.; Bamford, J.K.H. Comparative analysis of bacterial viruses Bam35, infecting a gram-positive host, and PRD1, infecting gram-negative hosts, demonstrates a viral lineage. Virology 2003, 313, 401–414. [Google Scholar] [CrossRef] [Green Version]
- Fornelos, N.; Browning, D.F.; Pavlin, A.; Podlesek, Z.; Hodnik, V.; Salas, M.; Butala, M. Lytic gene expression in the temperate bacteriophage GIL01 is activated by a phage-encoded lexa homologue. Nucleic Acids Res. 2018, 46, 9432–9443. [Google Scholar] [CrossRef]
- Marceau, A.H. Functions of single-strand DNA-binding proteins in DNA replication, recombination, and repair. In Single-Stranded DNA Binding Proteins: Methods and Protocols; Keck, J.L., Ed.; Humana Press: Totowa, NJ, USA, 2012; pp. 1–21. [Google Scholar]
- Theobald, D.L.; Mitton-Fry, R.M.; Wuttke, D.S. Nucleic acid recognition by OB-fold proteins. Annu. Rev. Biophys. Biomol. Struct. 2003, 32, 115–133. [Google Scholar] [CrossRef] [Green Version]
- Salas, M. Protein-priming of DNA replication. Annu. Rev. Biochem. 1991, 60, 39–71. [Google Scholar] [CrossRef] [PubMed]
- Salas, M.; Holguera, I.; Redrejo-Rodríguez, M.; de Vega, M. DNA-binding proteins essential for protein-primed bacteriophage φ29 DNA replication. Front. Mol. Biosci. 2016, 3, 37. [Google Scholar] [CrossRef] [Green Version]
- Caldentey, J.; Blanco, L.; Bamford, D.H.; Salas, M. In vitro replication of bacteriophage PRD1 DNA. Characterization of the protein-primed initiation site. Nucleic Acids Res. 1993, 21, 3725–3730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Philippe, C.; Krupovic, M.; Jaomanjaka, F.; Claisse, O.; Petrel, M.; le Marrec, C. Bacteriophage GC1, a novel tectivirus infecting Gluconobacter cerinus, an acetic acid bacterium associated with wine-making. Viruses 2018, 10, 39. [Google Scholar] [CrossRef] [Green Version]
- Gill, J.J.; Wang, B.; Sestak, E.; Young, R.; Chu, K.-H. Characterization of a novel tectivirus phage Toil and its potential as an agent for biolipid extraction. Sci. Rep. 2018, 8, 1062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verheust, C.; Fornelos, N.; Mahillon, J. GIL16, a new gram-positive tectiviral phage related to the Bacillus thuringiensis GIL01 and the Bacillus cereus pBClin15 elements. J. Bacteriol. 2005, 187, 1966–1973. [Google Scholar] [CrossRef] [PubMed] [Green Version]



| Phage CDSs (No. of Amino Acids Encoded) | Protein Function | Reference | ||||
|---|---|---|---|---|---|---|
| Sato | Sole | Bam35 | GIL16 | AP50 | ||
| 1 (54) | - | - | - | - | Hypothetical protein | This work |
| 2 (56) | 1 (56) | 1 (58) | 1 (58) | 1 (61) | MerR-type regulator | [44] |
| 3 (112) | 2 (112) | 2 (167) | 2 (166) | 2 (180) | ssDNA binding protein | [46,48] |
| 4 (61) | 3 (61) | 3 (74) | 3 (74) | 3 (74) | Hypothetical protein | [4,13,45] |
| 5 (245) | 4 (245) | 4 (245) | 4 (245) | 4 (233) | Terminal protein | [18,45] |
| 6 (674) | 5 (674) | 5 (735) | 5 (753) | 5 (731) | DNA polymerase | [45,49,50] |
| 7 (72) | 6 (72) | - | - | - | Hypothetical protein | This work |
| 8 (66) | 7 (66) | 6 (66) | 6 (66) | 6 (67) | LexA-type repressor | [44,51] |
| Protein | Sato vs. Sole | Sato vs. | Sole vs. | ||||
|---|---|---|---|---|---|---|---|
| Bam35 | GIL16 | AP50 | Bam35 | GIL16 | AP50 | ||
| MerR-type regulator | 100 (100) | 36.73 (87) | 36.73 (87) | 40.43 (83) | 36.73 (87) | 36.73 (87) | 40.43 (83) |
| ssDNA binding protein (SSB) | 97.32 (100) | ND 2 | ND | ND | ND | ND | ND |
| Unknown | 95.08 (100) | ND | 33.33 (39) | ND | ND | 25.00 (39) | ND |
| Terminal protein | 99.18 (100) | 36.13 (93) | 37.07 (91) | 34.43 (37) | 36.13 (93) | 37.07 (91) | 34.43 (37) |
| DNA polymerase (DNAPolB) | 99.70 (100) | 39.02 (93) | 39.57 (93) | 38.77 (93) | 38.86 (93) | 39.42 (93) | 38.92 (93) |
| LexA-type repressor | 78.46 (98) | 77.27 (100) | 77.27 (100) | 63.08 (98) | 63.08 (98) | 63.08 (98) | 60.61 (100) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gillis, A.; Hock, L.; Mahillon, J. Comparative Genomics of Prophages Sato and Sole Expands the Genetic Diversity Found in the Genus Betatectivirus. Microorganisms 2021, 9, 1335. https://doi.org/10.3390/microorganisms9061335
Gillis A, Hock L, Mahillon J. Comparative Genomics of Prophages Sato and Sole Expands the Genetic Diversity Found in the Genus Betatectivirus. Microorganisms. 2021; 9(6):1335. https://doi.org/10.3390/microorganisms9061335
Chicago/Turabian StyleGillis, Annika, Louise Hock, and Jacques Mahillon. 2021. "Comparative Genomics of Prophages Sato and Sole Expands the Genetic Diversity Found in the Genus Betatectivirus" Microorganisms 9, no. 6: 1335. https://doi.org/10.3390/microorganisms9061335