
Nanopore Sequencing Is a Credible Alternative to Recover Complete Genomes of Geminiviruses

 , , and
, , and
Abstract
:1. Introduction
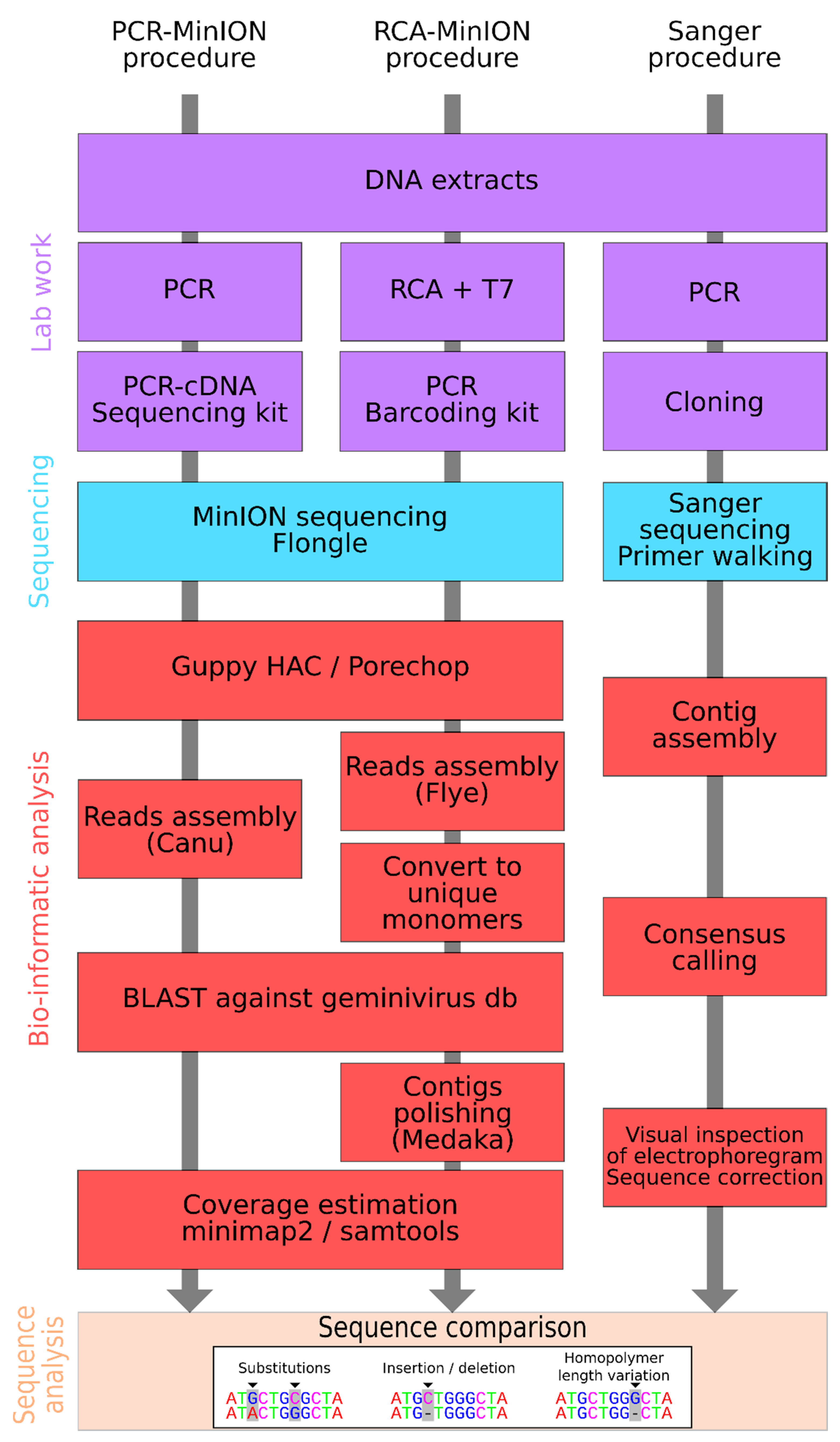
2. Material and Methods
2.1. Sampling and DNA Extraction
2.2. Full Genome Cloning and Sanger Sequencing
2.3. Minion Sequencing
2.4. MinION Sequence Assembly
2.5. Sequence Comparison and Phylogenetic Analysis
3. Results and Discussion
3.1. Sanger Sequences
3.2. Long Read Sequencing and Assembly
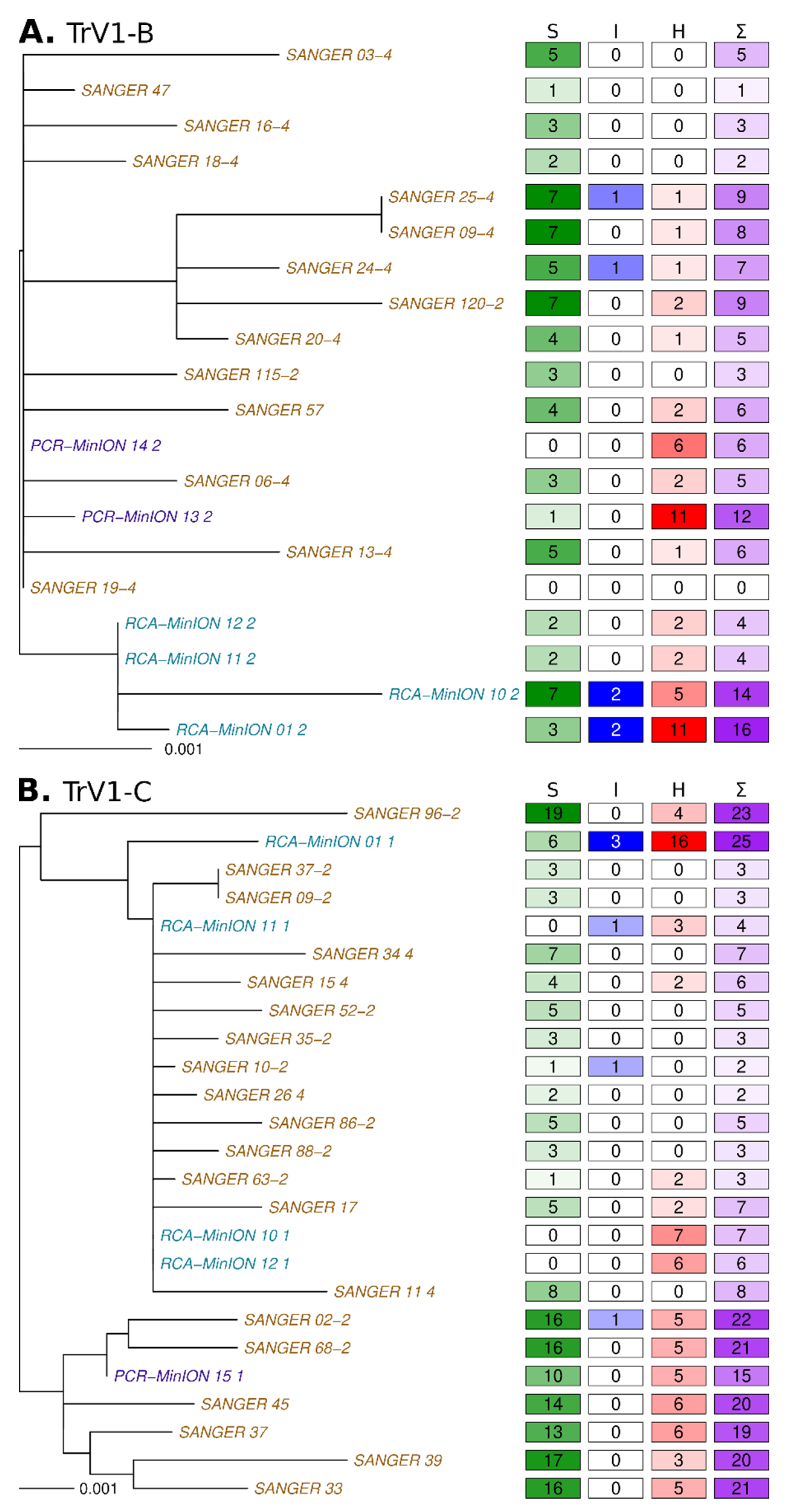
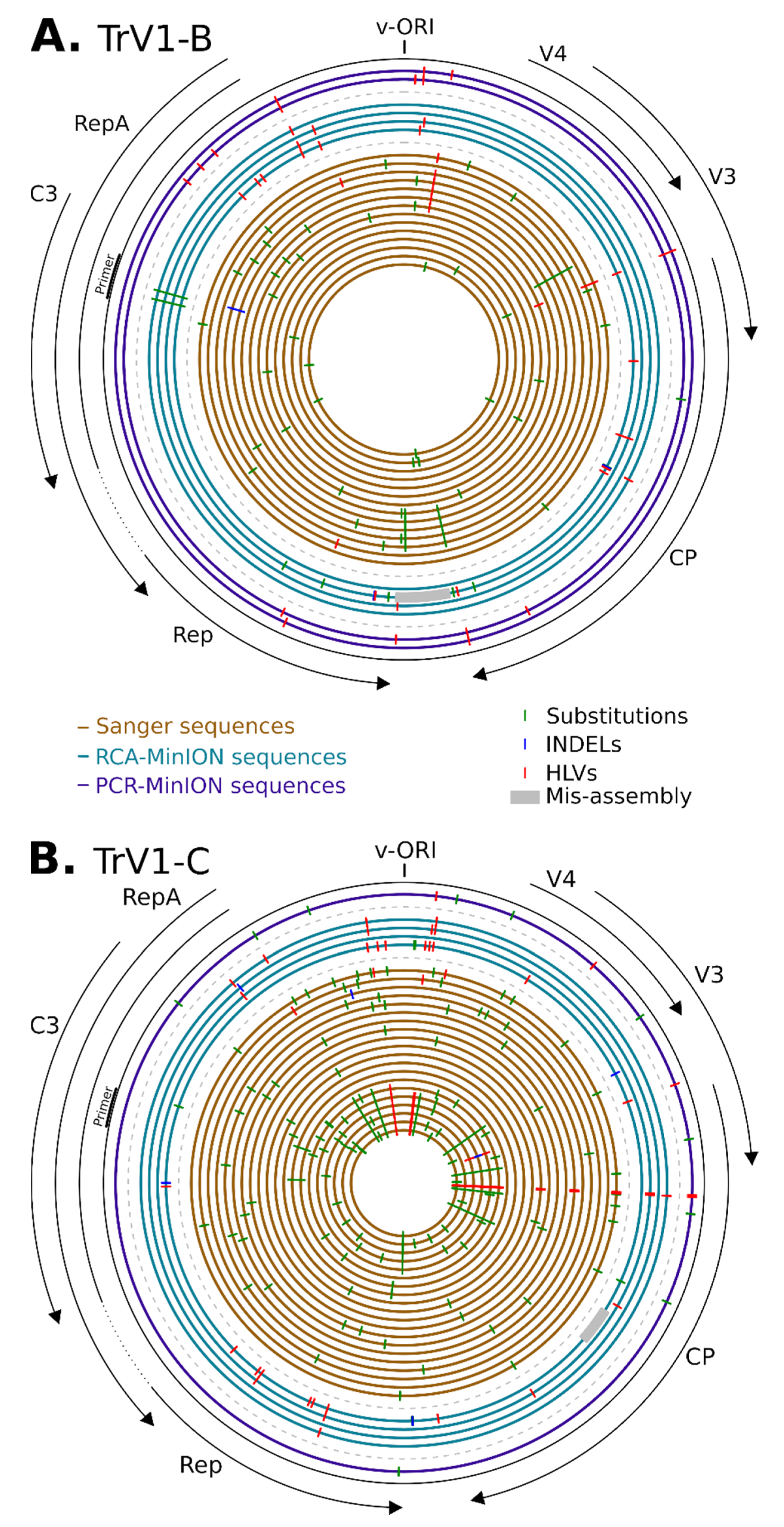
3.3. Sequences Comparison
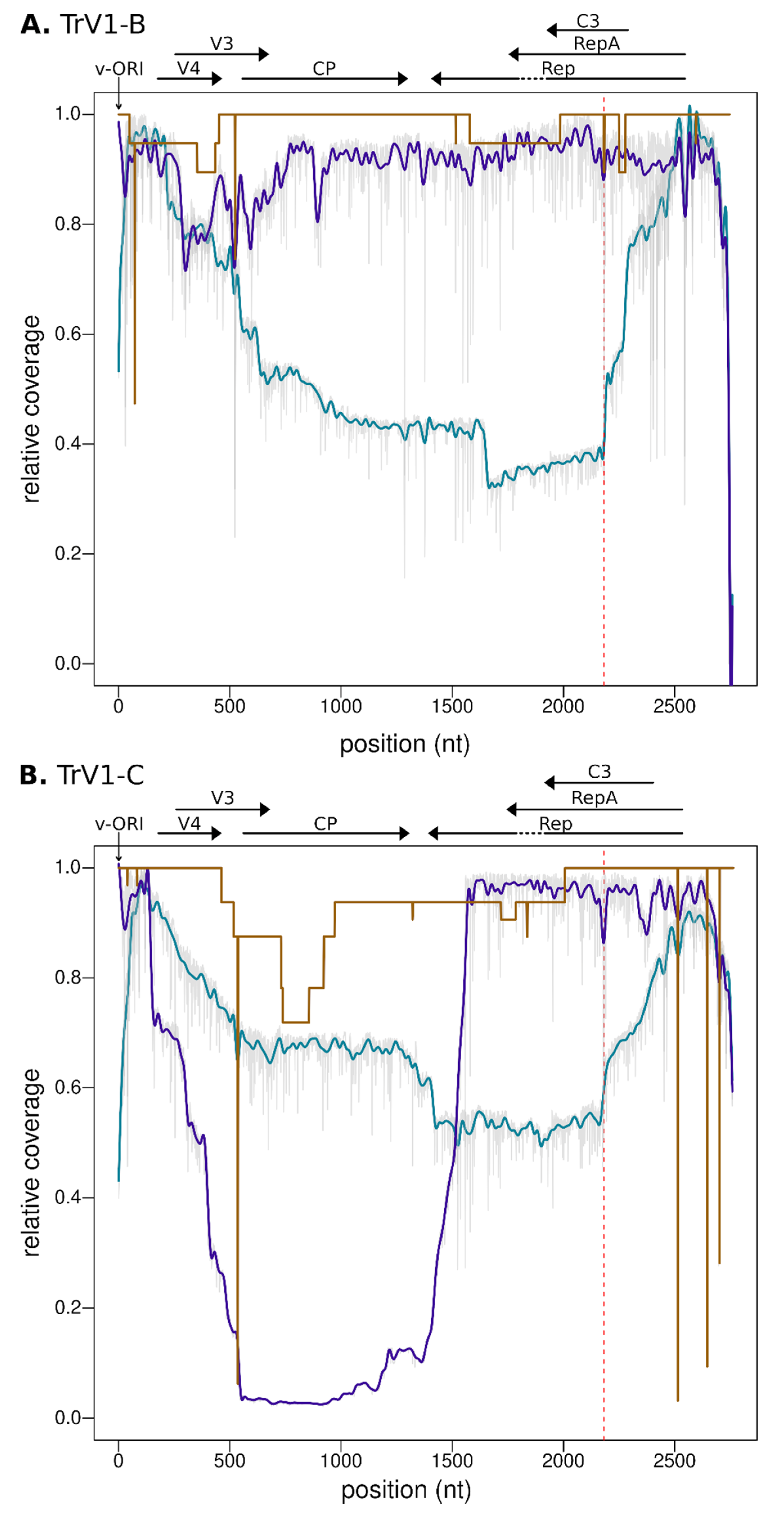
3.4. Defective Genome and Sequence Coverage
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dolja, V.V.; Koonin, E.V. Metagenomics reshapes the concepts of RNA virus evolution by revealing extensive horizontal virus transfer. Virus Res. 2018, 244, 36–52. [Google Scholar] [CrossRef] [PubMed]
- Greninger, A.L. A decade of RNA virus metagenomics is (not) enough. Virus Res. 2018, 244, 218–229. [Google Scholar] [CrossRef]
- Koonin, E.V.; Dolja, V.V. Metaviromics: A tectonic shift in understanding virus evolution. Virus Res. 2018, 246, A1–A3. [Google Scholar] [CrossRef] [PubMed]
- Roux, S.; Matthijnssens, J.; Dutilh, B.E. Metagenomics in Virology. Encycl. Virol. 2021, 1, 133–140. [Google Scholar] [CrossRef]
- Roux, S.; Brum, J.R.; Dutilh, B.E.; Sunagawa, S.; Duhaime, M.B.; Loy, A.; Poulos, B.T.; Solonenko, N.; Lara, E.; Poulain, J.; et al. Ecogenomics and potential biogeochemical impacts of globally abundant ocean viruses. Nat. Cell Biol. 2016, 537, 689–693. [Google Scholar] [CrossRef] [Green Version]
- McMullen, A.; Martinez-Hernandez, F.; Martinez-Garcia, M. Absolute quantification of infecting viral particles by chip-based digital polymerase chain reaction. Environ. Microbiol. Rep. 2019, 11, 855–860. [Google Scholar] [CrossRef]
- Breitbart, M.; Hewson, I.; Felts, B.; Mahaffy, J.M.; Nulton, J.; Salamon, P.; Rohwer, F. Metagenomic Analyses of an Uncultured Viral Community from Human Feces. J. Bacteriol. 2003, 185, 6220–6223. [Google Scholar] [CrossRef] [Green Version]
- Wylie, K.M.; Weinstock, G.M.; Storch, G.A. Emerging view of the human virome. Transl. Res. 2012, 160, 283–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lecuit, M.; Eloit, M. The diagnosis of infectious diseases by whole genome next generation sequencing: A new era is opening. Front. Cell. Infect. Microbiol. 2014, 4, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maclot, F.; Candresse, T.; Filloux, D.; Malmstrom, C.M.; Roumagnac, P.; Van Der Vlugt, R.; Massart, S. Illuminating an Ecological Blackbox: Using High Throughput Sequencing to Characterize the Plant Virome Across Scales. Front. Microbiol. 2020, 11, 578064. [Google Scholar] [CrossRef]
- Dávila-Ramos, S.; Castelán-Sánchez, H.G.; Martínez-Ávila, L.; Sánchez-Carbente, M.D.R.; Peralta, R.; Hernández-Mendoza, A.; Dobson, A.D.W.; Gonzalez, R.A.; Pastor, N.; Batista-García, R.A. A Review on Viral Metagenomics in Extreme Environments. Front. Microbiol. 2019, 10, 2403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodwin, S.; McPherson, J.D.; McCombie, W.R. Coming of age: Ten years of next-generation sequencing technologies. Nat. Rev. Genet. 2016, 17, 333–351. [Google Scholar] [CrossRef]
- Shi, M.; Lin, X.-D.; Tian, J.-H.; Chen, L.-J.; Chen, X.; Li, C.-X.; Qin, X.-C.; Li, J.; Cao, J.-P.; Eden, J.-S.; et al. Redefining the invertebrate RNA virosphere. Nat. Cell Biol. 2016, 540, 539–543. [Google Scholar] [CrossRef]
- Shi, M.; Zhang, Y.-Z.; Holmes, E.C. Meta-transcriptomics and the evolutionary biology of RNA viruses. Virus Res. 2018, 243, 83–90. [Google Scholar] [CrossRef]
- Zhang, Y.-Z.; Shi, M.; Holmes, E.C. Using Metagenomics to Characterize an Expanding Virosphere. Cell 2018, 172, 1168–1172. [Google Scholar] [CrossRef] [PubMed]
- Rosario, K.; Duffy, S.; Breitbart, M. A field guide to eukaryotic circular single-stranded DNA viruses: Insights gained from metagenomics. Arch. Virol. 2012, 157, 1851–1871. [Google Scholar] [CrossRef] [PubMed]
- Simmonds, P.; Adams, M.J.; Benkő, M.; Breitbart, M.; Brister, J.R.; Carstens, E.B.; Davison, A.J.; Delwart, E.; Gorbalenya, A.E.; Harrach, M.B.B.; et al. Virus taxonomy in the age of metagenomics. Nat. Rev. Genet. 2017, 15, 161–168. [Google Scholar] [CrossRef]
- Batovska, J.; Lynch, S.E.; Rodoni, B.C.; Sawbridge, T.I.; Cogan, N.O. Metagenomic arbovirus detection using MinION nanopore sequencing. J. Virol. Methods 2017, 249, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Roux, S.; Adriaenssens, E.M.; Dutilh, B.E.; Koonin, E.V.; Kropinski, A.M.; Krupovic, M.; Kuhn, J.H.; Lavigne, R.; Brister, J.R.; Varsani, A.; et al. Minimum Information about an Uncultivated Virus Genome (MIUViG). Nat. Biotechnol. 2019, 37, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Claverie, S.; Ouattara, A.; Hoareau, M.; Filloux, D.; Varsani, A.; Roumagnac, P.; Martin, D.P.; Lett, J.-M.; Lefeuvre, P. Exploring the diversity of Poaceae-infecting mastreviruses on Reunion Island using a viral metagenomics-based approach. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef] [Green Version]
- International Committee on Taxonomy of Viruses Executive Committee. The new scope of virus taxonomy: Partitioning the virosphere into 15 hierarchical ranks. Nat. Microbiol. 2020, 5, 668–674. [Google Scholar] [CrossRef]
- Roossinck, M.J.; Martin, D.P.; Roumagnac, P. Plant Virus Metagenomics: Advances in Virus Discovery. Phytopathology 2015, 105, 716–727. [Google Scholar] [CrossRef] [Green Version]
- Quiñones-Mateu, M.E.; Avila, S.; Reyes-Teran, G.; Martinez, M.A. Deep sequencing: Becoming a critical tool in clinical virology. J. Clin. Virol. 2014, 61, 9–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mikheyev, A.S.; Tin, M.M.Y. A first look at the Oxford Nanopore MinION sequencer. Mol. Ecol. Resour. 2014, 14, 1097–1102. [Google Scholar] [CrossRef]
- Sanger, F.; Coulson, A. A rapid method for determining sequences in DNA by primed synthesis with DNA polymerase. J. Mol. Biol. 1975, 94, 441–448. [Google Scholar] [CrossRef]
- Jain, M.; Olsen, H.E.; Paten, B.; Akeson, M. The Oxford Nanopore MinION: Delivery of nanopore sequencing to the genomics community. Genome Biol. 2016, 17, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Pomerantz, A.; Peñafiel, N.; Arteaga, A.; Bustamante, L.; Pichardo, F.; Coloma, L.A.; Barrio-Amorós, C.L.; Salazar-Valenzuela, D.; Prost, S. Real-time DNA barcoding in a rainforest using nanopore sequencing: Opportunities for rapid biodiversity assessments and local capacity building. GigaScience 2018, 7, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Lewandowski, K.; Lumley, S.; Pullan, S.; Vipond, R.; Carroll, M.; Foster, D.; Matthews, P.C.; Peto, T.; Crook, D. Detection of Viral Pathogens with Multiplex Nanopore MinION Sequencing: Be Careful With Cross-Talk. Front. Microbiol. 2018, 9, 2225. [Google Scholar] [CrossRef]
- Filloux, D.; Fernandez, E.; Loire, E.; Claude, L.; Galzi, S.; Candresse, T.; Winter, S.; Jeeva, M.L.; Makeshkumar, T.; Martin, D.P.; et al. Nanopore-based detection and characterization of yam viruses. Sci. Rep. 2018, 8, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ewang, J.; Moore, N.E.; Edeng, Y.-M.; Eccles, D.A.; Hall, R.J. MinION nanopore sequencing of an influenza genome. Front. Microbiol. 2015, 6, 766. [Google Scholar] [CrossRef] [Green Version]
- Quick, J.; Loman, N.J.; Duraffour, S.; Simpson, J.T.; Severi, E.; Cowley, L.; Bore, J.A.; Koundouno, R.; Dudas, G.; Mikhail, A.; et al. Real-time, portable genome sequencing for Ebola surveillance. Nat. Cell Biol. 2016, 530, 228–232. [Google Scholar] [CrossRef] [Green Version]
- Yamagishi, J.; Runtuwene, L.R.; Hayashida, K.; Mongan, A.E.; Thi, L.A.N.; Thuy, L.N.; Nhat, C.N.; Limkittikul, K.; Sirivichayakul, C.; Sathirapongsasuti, N.; et al. Serotyping dengue virus with isothermal amplification and a portable sequencer. Sci. Rep. 2017, 7, 1–10. [Google Scholar] [CrossRef]
- Quick, J.; Grubaugh, N.D.; Pullan, S.T.; Claro, I.M.; Smith, A.D.; Gangavarapu, K.; Oliveira, G.; Robles-Sikisaka, R.; Rogers, T.F.; Beutler, N.A.; et al. Multiplex PCR method for MinION and Illumina sequencing of Zika and other virus genomes directly from clinical samples. Nat. Protoc. 2017, 12, 1261–1276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Lee, J.-Y.; Yang, J.-S.; Kim, J.W.; Kim, V.N.; Chang, H. The Architecture of SARS-CoV-2 Transcriptome. Cell 2020, 181, 914–921.e10. [Google Scholar] [CrossRef]
- Adams, I.P.; Braidwood, L.A.; Stomeo, F.; Phiri, N.; Uwumukiza, B.; Feyissa, B.; Mahuku, G.; Wangi, A.; Smith, J.; Mumford, R.; et al. Characterising Maize Viruses Associated with Maize Lethal Necrosis Symptoms in Sub Saharan Africa. bioRxiv 2017. preprint. [Google Scholar] [CrossRef] [Green Version]
- Badial, A.B.; Sherman, D.; Stone, A.; Gopakumar, A.; Wilson, V.; Schneider, W.; King, J. Nanopore Sequencing as a Surveillance Tool for Plant Pathogens in Plant and Insect Tissues. Plant Dis. 2018, 102, 1648–1652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoue-Nagata, A.K.; Albuquerque, L.C.; Rocha, W.B.; Nagata, T. A simple method for cloning the complete begomovirus genome using the bacteriophage φ29 DNA polymerase. J. Virol. Methods 2004, 116, 209–211. [Google Scholar] [CrossRef] [PubMed]
- Rosario, K.; Dayaram, A.; Marinov, M.; Ware, J.; Kraberger, S.; Stainton, D.; Breitbart, M.; Varsani, A. Diverse circular ssDNA viruses discovered in dragonflies (Odonata: Epiprocta). J. Gen. Virol. 2012, 93, 2668–2681. [Google Scholar] [CrossRef]
- Rosario, K.; Schenck, R.O.; Harbeitner, R.C.; Lawler, S.N.; Breitbart, M. Novel circular single-stranded DNA viruses identified in marine invertebrates reveal high sequence diversity and consistent predicted intrinsic disorder patterns within putative structural proteins. Front. Microbiol. 2015, 6, 696. [Google Scholar] [CrossRef] [Green Version]
- Theuns, S.; Vanmechelen, B.; Bernaert, Q.; Deboutte, W.; Vandenhole, M.; Beller, L.; Matthijnssens, J.; Maes, P.; Nauwynck, H.J. Nanopore sequencing as a revolutionary diagnostic tool for porcine viral enteric disease complexes identifies porcine kobuvirus as an important enteric virus. Sci. Rep. 2018, 8, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Boykin, L.M.; Sseruwagi, P.; Alicai, T.; Ateka, E.; Mohammed, I.U.; Stanton, J.-A.L.; Kayuki, C.; Mark, D.; Fute, T.; Erasto, J.; et al. Tree Lab: Portable genomics for Early Detection of Plant Viruses and Pests in Sub-Saharan Africa. Genes 2019, 10, 632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, J.; Zhang, Y.; Dai, M.; Xu, J.; Chen, L.; Zhang, F.; Zhao, N.; Wang, J. Profiling of Human Gut Virome with Oxford Nanopore Technology. Med. Microecol. 2020, 4, 100012. [Google Scholar] [CrossRef]
- Chalupowicz, L.; Dombrovsky, A.; Gaba, V.; Luria, N.; Reuven, M.; Beerman, A.; Lachman, O.; Dror, O.; Nissan, G.; Manulis-Sasson, S. Diagnosis of plant diseases using the Nanopore sequencing platform. Plant Pathol. 2018, 68, 229–238. [Google Scholar] [CrossRef]
- Naito, F.Y.B.; Melo, F.L.; Fonseca, M.E.N.; Santos, C.A.F.; Chanes, C.R.; Ribeiro, B.M.; Gilbertson, R.L.; Boiteux, L.S.; Pereira-Carvalho, R.D.C. Nanopore sequencing of a novel bipartite New World begomovirus infecting cowpea. Arch. Virol. 2019, 164, 1907–1910. [Google Scholar] [CrossRef]
- Leiva, A.M.; Siriwan, W.; Alvarez, D.L.; Barrantes, I.; Hemniam, N.; Saokham, K.; Cuellar, W.J. Nanopore-Based Complete Genome Sequence of a Sri Lankan Cassava Mosaic Virus (Geminivirus) Strain from Thailand. Microbiol. Resour. Announc. 2020, 9, 9. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Svanella-Dumas, L.; Julian, C.; Galzi, S.; Fernandez, E.; Yvon, M.; Pirolles, E.; Lefebvre, M.; Filloux, D.; Roumagnac, P.; et al. Genome characterization and diversity of trifolium virus 1: Identification of a novel legume-infective capulavirus. Arch. Virol. 2021, in press. [Google Scholar]
- Varsani, A.; Roumagnac, P.; Fuchs, M.; Navas-Castillo, J.; Moriones, E.; Idris, A.; Briddon, R.W.; Rivera-Bustamante, R.; Zerbini, F.M.; Martin, D.P. Capulavirus and Grablovirus: Two new genera in the family Geminiviridae. Arch. Virol. 2017, 162, 1819–1831. [Google Scholar] [CrossRef] [Green Version]
- Fauquet, C.M.; Briddon, R.W.; Brown, J.K.; Moriones, E.; Stanley, J.; Zerbini, M.; Zhou, X. Geminivirus strain demarcation and nomenclature. Arch. Virol. 2008, 153, 783–821. [Google Scholar] [CrossRef] [Green Version]
- Roumagnac, P.; Granier, M.; Bernardo, P.; Deshoux, M.; Ferdinand, R.; Galzi, S.; Fernandez, E.; Julian, C.; Abt, I.; Filloux, D.; et al. Alfalfa Leaf Curl Virus: An Aphid-Transmitted Geminivirus. J. Virol. 2015, 89, 9683–9688. [Google Scholar] [CrossRef] [Green Version]
- Varsani, A.; Navas-Castillo, J.; Moriones, E.; Hernández-Zepeda, C.; Idris, A.; Brown, J.K.; Zerbini, F.M.; Martin, D.P. Establishment of three new genera in the family Geminiviridae: Becurtovirus, Eragrovirus and Turncurtovirus. Arch. Virol. 2014, 159, 2193–2203. [Google Scholar] [CrossRef]
- Zerbini, F.M.; Briddon, R.W.; Idris, A.; Martin, D.P.; Moriones, E.; Navas-Castillo, J.; Rivera-Bustamante, R.; Roumagnac, P.; Varsani, A. ICTV Report Consortium ICTV Virus Taxonomy Profile: Geminiviridae. J. Gen. Virol. 2017, 98, 131–133. [Google Scholar] [CrossRef] [PubMed]
- Claverie, S.; Bernardo, P.; Kraberger, S.; Hartnady, P.; Lefeuvre, P.; Lett, J.-M.; Galzi, S.; Filloux, D.; Harkins, G.W.; Varsani, A.; et al. From Spatial Metagenomics to Molecular Characterization of Plant Viruses: A Geminivirus Case Study. In Advances in Clinical Chemistry; Elsevier: Amsterdam, The Netherlands, 2018; Volume 101, pp. 55–83. [Google Scholar]
- Bernardo, P.; Golden, M.; Akram, M.; Naimuddin; Nadarajan, N.; Fernandez, E.; Granier, M.; Rebelo, A.G.; Peterschmitt, M.; Martin, D.P.; et al. Identification and characterisation of a highly divergent geminivirus: Evolutionary and taxonomic implications. Virus Res. 2013, 177, 35–45. [Google Scholar] [CrossRef]
- Susi, H.; Filloux, D.; Frilander, M.J.; Roumagnac, P.; Laine, A.-L. Diverse and variable virus communities in wild plant populations revealed by metagenomic tools. PeerJ 2019, 7, e6140. [Google Scholar] [CrossRef] [PubMed]
- Oxford Nanopore Technologies. Available online: https://nanoporetech.com/ (accessed on 18 March 2021).
- Porechop. Available online: https://github.com/rrwick/Porechop (accessed on 18 March 2021).
- De Coster, W.; D’Hert, S.; Schultz, D.T.; Cruts, M.; Van Broeckhoven, C. NanoPack: Visualizing and processing long-read sequencing data. Bioinformatics 2018, 34, 2666–2669. [Google Scholar] [CrossRef] [PubMed]
- Kolmogorov, M.; Yuan, J.; Lin, Y.; Pevzner, P.A. Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 2019, 37, 540–546. [Google Scholar] [CrossRef]
- Medaka. Available online: https://github.com/nanoporetech/medaka (accessed on 18 March 2021).
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and accurate long-read assembly via adaptivek-mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef] [PubMed]
- Rozewicki, J.; Li, S.; Amada, K.M.; Standley, D.M.; Katoh, K. MAFFT-DASH: Integrated protein sequence and structural alignment. Nucleic Acids Res. 2019, 47, W5–W10. [Google Scholar] [CrossRef]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately Maximum-Likelihood Trees for Large Alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef] [PubMed]
- Paradis, E.; Schliep, K. ape 5.0: An environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 2018, 35, 526–528. [Google Scholar] [CrossRef]
- Muhire, B.M.; Varsani, A.; Martin, D.P. SDT: A Virus Classification Tool Based on Pairwise Sequence Alignment and Identity Calculation. PLoS ONE 2014, 9, e108277. [Google Scholar] [CrossRef]
- Yilmaz, S.; Allgaier, M.; Hugenholtz, P. Multiple displacement amplification compromises quantitative analysis of metagenomes. Nat. Methods 2010, 7, 943–944. [Google Scholar] [CrossRef]
- Kim, K.-H.; Bae, J.-W. Amplification Methods Bias Metagenomic Libraries of Uncultured Single-Stranded and Double-Stranded DNA Viruses. Appl. Environ. Microbiol. 2011, 77, 7663–7668. [Google Scholar] [CrossRef] [Green Version]
- Gallet, R.; Fabre, F.; Michalakis, Y.; Blanc, S. The Number of Target Molecules of the Amplification Step Limits Accuracy and Sensitivity in Ultradeep-Sequencing Viral Population Studies. J. Virol. 2017, 91, 91. [Google Scholar] [CrossRef] [Green Version]
- Laehnemann, D.; Borkhardt, A.; McHardy, A.C. Denoising DNA deep sequencing data—high-throughput sequencing errors and their correction. Brief. Bioinform. 2016, 17, 154–179. [Google Scholar] [CrossRef] [PubMed]
- Wick, R.R.; Judd, L.M.; Holt, K.E. Performance of neural network basecalling tools for Oxford Nanopore sequencing. Genome Biol. 2019, 20, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gargis, A.S.; Cherney, B.; Conley, A.B.; McLaughlin, H.P.; Sue, D. Rapid Detection of Genetic Engineering, Structural Variation, and Antimicrobial Resistance Markers in Bacterial Biothreat Pathogens by Nanopore Sequencing. Sci. Rep. 2019, 9, 13501–13514. [Google Scholar] [CrossRef]
- Seah, A.; Lim, M.C.; McAloose, D.; Prost, S.; Seimon, T.A. MinION-Based DNA Barcoding of Preserved and Non-Invasively Collected Wildlife Samples. Genes 2020, 11, 445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shendure, J.; Ji, H. Next-generation DNA sequencing. Nat. Biotechnol. 2008, 26, 1135–1145. [Google Scholar] [CrossRef]
- Ge, L.; Zhang, J.; Zhou, X.; Li, H. Genetic Structure and Population Variability of Tomato Yellow Leaf Curl China Virus. J. Virol. 2007, 81, 5902–5907. [Google Scholar] [CrossRef] [Green Version]
- Harkins, G.W.; Delport, W.; Duffy, S.; Wood, N.; Monjane, A.L.; Owor, B.E.; Donaldson, L.; Saumtally, S.; Triton, G.; Briddon, R.W.; et al. Experimental evidence indicating that mastreviruses probably did not co-diverge with their hosts. Virol. J. 2009, 6, 104. [Google Scholar] [CrossRef] [Green Version]
- Domingo, E.; Sheldon, J.; Perales, C. Viral Quasispecies Evolution. Microbiol. Mol. Biol. Rev. 2012, 76, 159–216. [Google Scholar] [CrossRef] [Green Version]
- Jeske, H. Barcoding of Plant Viruses with Circular Single-Stranded DNA Based on Rolling Circle Amplification. Viruses 2018, 10, 469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McNaughton, A.L.; Roberts, H.E.; Bonsall, D.; De Cesare, M.; Mokaya, J.; Lumley, S.F.; Golubchik, T.; Piazza, P.; Martin, J.B.; De Lara, C.; et al. Illumina and Nanopore methods for whole genome sequencing of hepatitis B virus (HBV). Sci. Rep. 2019, 9, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viehweger, A.; Krautwurst, S.; Lamkiewicz, K.; Madhugiri, R.; Ziebuhr, J.; Hölzer, M.; Marz, M. Direct RNA nanopore sequencing of full-length coronavirus genomes provides novel insights into structural variants and enables modification analysis. Genome Res. 2019, 29, 1545–1554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, J.J.M.; Ip, Y.C.A.; Ng, C.S.L.; Huang, D. Takeaways from Mobile DNA Barcoding with BentoLab and MinION. Genes 2020, 11, 1121. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Flongle Id | Raw Reads | Passed Reads | Barcode ID | Trimmed Reads | Capulavirus Reads | TrV1-B Reads | TrV1-C Reads | Length TrV1-B Assembly | Length TrV1-C Assembly |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 188,123 | 162,263 | 1 | 110,830 | 27 | 14 | 13 | 2745 | 2771 |
| 2 | 273,088 | 215,143 | 10 | 65,413 | 4809 | 3230 | 1579 | 2769 | 2764 |
| 11 | 40,242 | 3423 | 2244 | 1179 | 2748 | 2763 | |||
| 12 | 46,421 | 2803 | 1585 | 1218 | 2746 | 2765 | |||
| 13 | 492,922 | 386,099 | - | 385,029 | 380,933 | 380,933 | - | 2731 | - |
| 14 | 414,665 | 371,700 | - | 370,755 | 367,381 | 367,381 | - | 2739 | - |
| 15 | 768,144 | 714,730 | - | 602,579 | 337,338 | - | 337,338 | - | 2754 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ben Chehida, S.; Filloux, D.; Fernandez, E.; Moubset, O.; Hoareau, M.; Julian, C.; Blondin, L.; Lett, J.-M.; Roumagnac, P.; Lefeuvre, P. Nanopore Sequencing Is a Credible Alternative to Recover Complete Genomes of Geminiviruses. Microorganisms 2021, 9, 903. https://doi.org/10.3390/microorganisms9050903
Ben Chehida S, Filloux D, Fernandez E, Moubset O, Hoareau M, Julian C, Blondin L, Lett J-M, Roumagnac P, Lefeuvre P. Nanopore Sequencing Is a Credible Alternative to Recover Complete Genomes of Geminiviruses. Microorganisms. 2021; 9(5):903. https://doi.org/10.3390/microorganisms9050903
Chicago/Turabian StyleBen Chehida, Selim, Denis Filloux, Emmanuel Fernandez, Oumaima Moubset, Murielle Hoareau, Charlotte Julian, Laurence Blondin, Jean-Michel Lett, Philippe Roumagnac, and Pierre Lefeuvre. 2021. "Nanopore Sequencing Is a Credible Alternative to Recover Complete Genomes of Geminiviruses" Microorganisms 9, no. 5: 903. https://doi.org/10.3390/microorganisms9050903