
A Primer on the Analysis of High-Throughput Sequencing Data for Detection of Plant Viruses

 , , , , , , , , , , , , add
Show full author list
, , , , , , , , , , , , add
Show full author list
Abstract
:1. Introduction
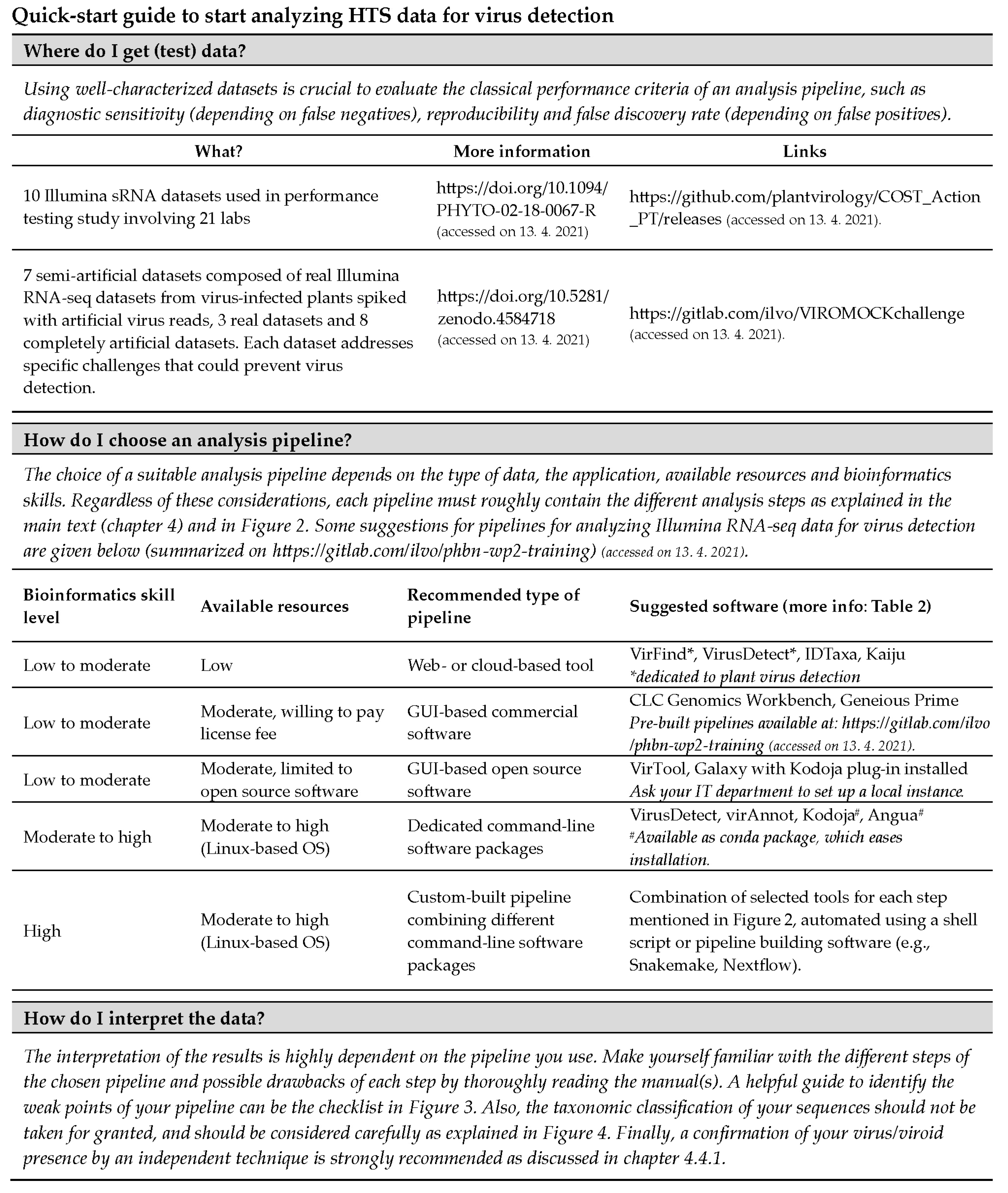
2. What Should I Anticipate and How Should I Prepare?
3. Starting the Project: How Do I Prepare Samples and Sequence Nucleic Acids?
3.1. Input Material and Nucleic Acids Preparation
3.1.1. Total RNA/DNA
3.1.2. Small RNA (sRNA)
3.1.3. Virion-Associated Nucleic Acids (VANA)
3.1.4. Double-Stranded RNA
3.2. Library Preparation and Sequencing
3.3. Contamination
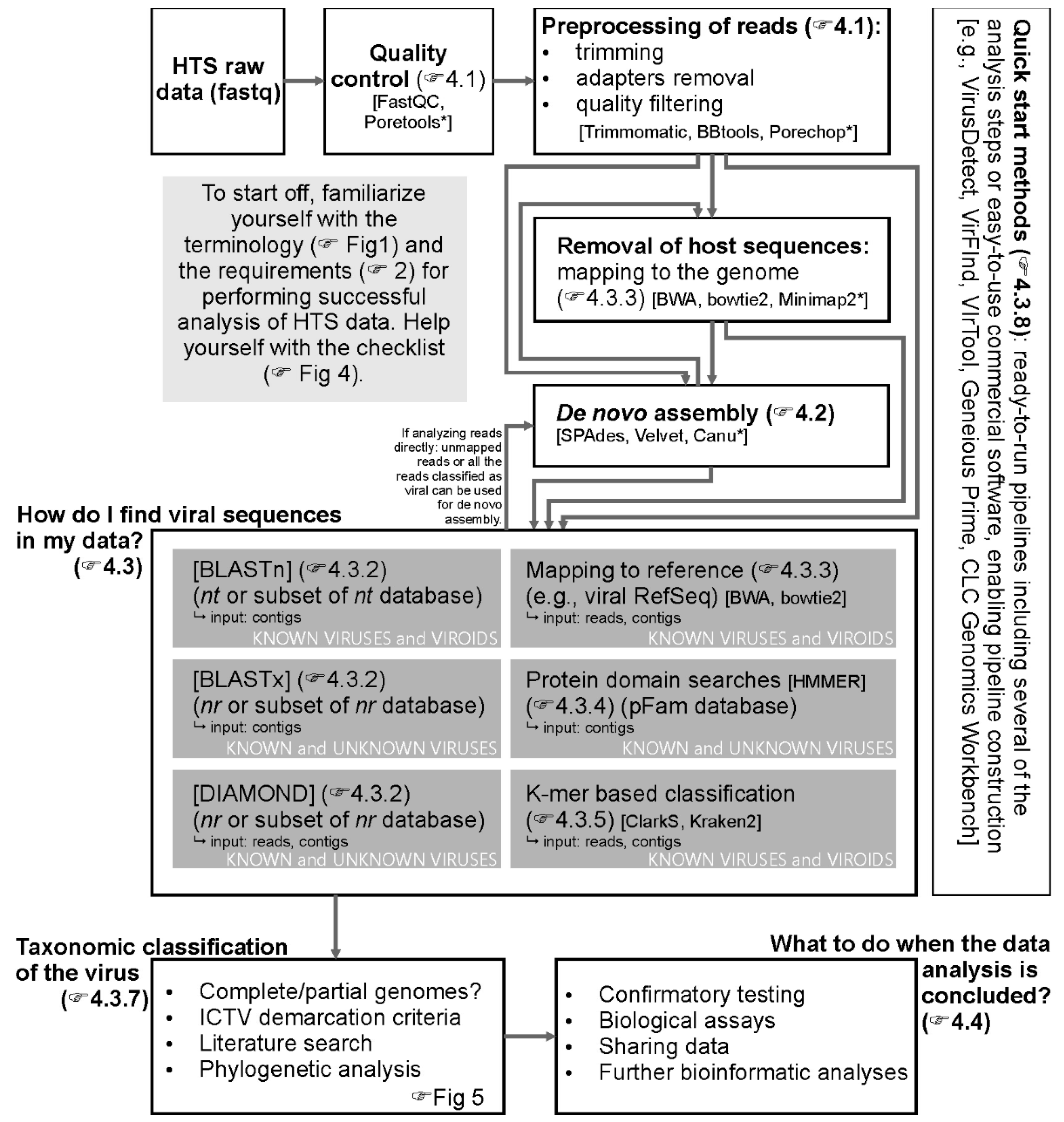
4. How Do I Analyze the Data?
4.1. Demultiplexing, Quality Control, and Trimming
4.2. De Novo Assembly
4.3. How Do I Find and Classify Viral Sequences in My Data?
4.3.1. Databases
4.3.2. BLAST and BLAST-Like Approaches
- BLASTn can be used to compare a nucleotide sequence with a nucleotide database. It is less computationally intensive than BLASTx, but because of the higher divergence rate of nucleotide sequences, it is less efficient for the annotation of novel viruses not represented in the database used.
- BLASTp can be used to compare a protein sequence with a database of protein sequences.
- BLASTx can be used to compare a nucleotide sequence translated in all six reading frames with a database of protein sequences. While computationally intensive, it is the most efficient BLAST program for the annotation of novel viruses.
- tBLASTn can be used to compare a protein sequence with all six possible reading frames of a nucleotide database and is often used to identify proteins in new, unannotated genomes.
- tBLASTx can be used to compare all six reading frames of a nucleotide sequence with all six reading frames of a nucleotide database. It is the costliest in computation time.
- MegaBLAST can be used to compare nucleotide sequences expected to be already present or closely related to those in a nucleotide database. It can be much faster than BLASTn and is able to handle much longer sequences but deals less efficiently with very divergent sequences.
4.3.3. Mapping Reads (or Contigs) to Reference Database
4.3.4. Protein Domain Searches
4.3.5. K-mer Approaches and Machine Learning-Based Approaches
4.3.6. Which Analysis Approach Should I Choose?
4.3.7. Taxonomic Classification
4.3.8. “Quick Start” Methods
4.4. What to Do When The Data Analysis Is Concluded?
4.4.1. Identity Confirmation by an Independent Technique
4.4.2. Biological Characterization Post HTS Detection
4.4.3. Sharing Data to Leverage Knowledge
4.4.4. Recombination Analysis
4.4.5. Additional Bioinformatics Analyses
5. Conclusions and Outlook
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pipeline | Brief Description | Web Link/Publication | Pros | Cons |
|---|---|---|---|---|
| VirusDetect | Virus discovery using sRNA and RNAseq sequences | http://virusdetect.feilab.net, accessed on 13 April 2021 [147] |
|
|
| Virtool | HTS sample manager with virus detection, discovery and analysis workflows | www.virtool.ca, accessed on 13 April 2021 https://github.com/virtool/virtool, accessed on 13 April 2021 [36] |
|
|
| virAnnot | Command-line tool for virus detection and viral diversity estimation | [148] |
|
|
| VirFind | Online virus discovery tool | http://virfind.org, accessed on 13 April 2021 [149] |
|
|
| Angua | Command-line tool for virus detection | https://fred.fera.co.uk/smcgreig/angua3, accessed on 13 April 2021 |
|
|
| Kodoja | k-mer based command-line tool for virus detection | https://github.com/abaizan/kodoja, accessed on 13 April 2021 [107] |
|
|
| Truffle | Targeted virus detection using e-probes based approach | [150] |
|
|
| Kaiju | Online metagenomic analysis tool | http://kaiju.binf.ku.dk/, accessed on 13 April 2021 [105] |
|
|
| Galaxy | Workflow system for computational analyses | https://usegalaxy.org, accessed on 13 April 2021 [151] |
|
|
| ID-Seq | Online metagenomic analysis tool | https://idseq.net/, accessed on 13 April 2021 [152] |
|
|
| Geneious Prime | Software for molecular biology and sequence analysis | https://www.geneious.com, accessed on 13 April 2021 |
|
|
| CLC Genomics Workbench | Comprehensive software solution of molecular biology analysis tools | https://digitalinsights.qiagen.com/products-overview/discovery-insights-portfolio/analysis-and-visualization/qiagen-clc-genomics-workbench/, accessed on 13 April 2021 |
|
|
References
- Villamor, D.E.V.; Ho, T.; Al Rwahnih, M.; Martin, R.R.; Tzanetakis, I.E. High throughput sequencing for plant virus detection and discovery. Phytopathology 2019, 109, 716–725. [Google Scholar] [CrossRef]
- Kreuze, J.F.; Perez, A.; Untiveros, M.; Quispe, D.; Fuentes, S.; Barker, I.; Simon, R. Complete viral genome sequence and discovery of novel viruses by deep sequencing of small RNAs: A generic method for diagnosis, discovery and sequencing of viruses. Virology 2009, 388, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, I.P.; Glover, R.H.; Monger, W.A.; Mumford, R.; Jackeviciene, E.; Navalinskiene, M.; Samuitiene, M.; Boonham, N. Next-generation sequencing and metagenomic analysis: A universal diagnostic tool in plant virology. Mol. Plant Pathol. 2009, 10, 537–545. [Google Scholar] [CrossRef] [PubMed]
- Al Rwahnih, M.; Daubert, S.; Golino, D.; Rowhani, A. Deep sequencing analysis of RNAs from a grapevine showing syrah decline symptoms reveals a multiple virus infection that includes a novel virus. Virology 2009, 387, 395–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donaire, L.; Wang, Y.; Gonzalez-Ibeas, D.; Mayer, K.F.; Aranda, M.A.; Llave, C. Deep-sequencing of plant viral small RNAs reveals effective and widespread targeting of viral genomes. Virology 2009, 392, 203–214. [Google Scholar] [CrossRef] [Green Version]
- Massart, S.; Chiumenti, M.; De Jonghe, K.; Glover, R.; Haegeman, A.; Koloniuk, I.; Komínek, P.; Kreuze, J.; Kutnjak, D.; Lotos, L.; et al. Virus detection by high-throughput sequencing of small RNAs: Large-scale performance testing of sequence analysis strategies. Phytopathology 2019, 109, 488–497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olmos, A.; Boonham, N.; Candresse, T.; Gentit, P.; Giovani, B.; Kutnjak, D.; Liefting, L.; Maree, H.J.; Minafra, A.; Moreira, A.; et al. High-throughput sequencing technologies for plant pest diagnosis: Challenges and opportunities. EPPO Bull. 2018, 48, 219–224. [Google Scholar] [CrossRef] [Green Version]
- Weymann, D.; Laskin, J.; Roscoe, R.; Schrader, K.A.; Chia, S.; Yip, S.; Cheung, W.Y.; Gelmon, K.A.; Karsan, A.; Renouf, D.J.; et al. The cost and cost trajectory of whole-genome analysis guiding treatment of patients with advanced cancers. Mol. Genet. Genomic Med. 2017, 5, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Valitest EU Project Consortium Guidelines for the Selection, Development, Validation and Routine Use of High-Throughput Sequencing Analysis in Plant Health Diagnostic Laboratories: Grant Agreement N. 773139: Deliverable N° 2.2. (Confidential). 2020. Available online: https://www.valitest.eu/work_packages/ (accessed on 13 April 2021).
- Maliogka, V.I.; Minafra, A.; Saldarelli, P.; Ruiz-García, A.B.; Glasa, M.; Katis, N.; Olmos, A. Recent advances on detection and characterization of fruit tree viruses using high-throughput sequencing technologies. Viruses 2018, 10, 436. [Google Scholar] [CrossRef] [Green Version]
- Roossinck, M.J. Deep sequencing for discovery and evolutionary analysis of plant viruses. Virus Res. 2017, 239, 82–86. [Google Scholar] [CrossRef] [PubMed]
- Roossinck, M.J.; Martin, D.P.; Roumagnac, P. Plant virus metagenomics: Advances in virus discovery. Phytopathology 2015, 105, 716–727. [Google Scholar] [CrossRef] [Green Version]
- Marais, A.; Faure, C.; Bergey, B.; Candresse, T. Viral double-stranded RNAs (dsRNAs) from plants: Alternative nucleic acid substrates for high-throughput sequencing. In Viral Metagenomics: Methods and Protocols; Pantaleo, V., Chiumenti, M., Eds.; Humana Press: New York, NY, USA, 2018; pp. 45–53. ISBN 978-1-4939-7682-9. [Google Scholar]
- Massart, S.; Olmos, A.; Jijakli, H.; Candresse, T. Current impact and future directions of high throughput sequencing in plant virus diagnostics. Virus Res. 2014, 188, 90–96. [Google Scholar] [CrossRef]
- Pecman, A.; Kutnjak, D.; Gutiérrez-Aguirre, I.; Adams, I.; Fox, A.; Boonham, N.; Ravnikar, M. Next generation sequencing for detection and discovery of plant viruses and viroids: Comparison of two approaches. Front. Microbiol. 2017, 8. [Google Scholar] [CrossRef] [Green Version]
- Boone, M.; De Koker, A.; Callewaert, N. Survey and summary capturing the “ome”: The expanding molecular toolbox for RNA and DNA library construction. Nucleic Acids Res. 2018, 46, 2701–2721. [Google Scholar] [CrossRef]
- Visser, M.; Bester, R.; Burger, J.T.; Maree, H.J. Next-generation sequencing for virus detection: Covering all the bases. Virol. J. 2016, 13, 4–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Idris, A.; Al-Saleh, M.; Piatek, M.J.; Al-Shahwan, I.; Ali, S.; Brown, J.K. Viral metagenomics: Analysis of begomoviruses by illumina high-throughput sequencing. Viruses 2014, 6, 1219–1236. [Google Scholar] [CrossRef] [Green Version]
- Sukal, A.C.; Kidanemariam, D.B.; Dale, J.L.; Harding, R.M.; James, A.P. Assessment and optimization of rolling circle amplification protocols for the detection and characterization of badnaviruses. Virology 2019, 529, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Wyant, P.S.; Strohmeier, S.; Schäfer, B.; Krenz, B.; Assunção, I.P.; de Andrade Lima, G.S.; Jeske, H. Circular DNA genomics (circomics) exemplified for geminiviruses in bean crops and weeds of northeastern Brazil. Virology 2012, 427, 151–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vivek, A.T.; Zahra, S.; Kumar, S. From current knowledge to best practice: A primer on viral diagnostics using deep sequencing of virus-derived small interfering RNAs (vsiRNAs) in infected plants. Methods 2020, 183, 30–37. [Google Scholar] [CrossRef]
- Kutnjak, D.; Rupar, M.; Gutierrez-Aguirre, I.; Curk, T.; Kreuze, J.F.; Ravnikar, M. Deep sequencing of virus-derived small interfering RNAs and RNA from viral particles shows highly similar mutational landscapes of a plant virus population. J. Virol. 2015, 89, 4760–4769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seguin, J.; Rajeswaran, R.; Malpica-López, N.; Martin, R.R.; Kasschau, K.; Dolja, V.V.; Otten, P.; Farinelli, L.; Pooggin, M.M. De novo reconstruction of consensus master genomes of plant RNA and DNA viruses from siRNAs. PLoS ONE 2014, 9, e88513. [Google Scholar] [CrossRef] [PubMed]
- Smith, O.; Clapham, A.; Rose, P.; Liu, Y.; Wang, J.; Allaby, R.G. A complete ancient RNA genome: Identification, reconstruction and evolutionary history of archaeological Barley Stripe Mosaic Virus. Sci. Rep. 2014, 4, 4003. [Google Scholar] [CrossRef] [Green Version]
- Turco, S.; Golyaev, V.; Seguin, J.; Gilli, C.; Farinelli, L.; Boller, T.; Schumpp, O.; Pooggin, M.M. Small RNA-omics for virome reconstruction and antiviral defense characterization in mixed infections of cultivated solanum plants. Mol. Plant-Microbe Interact. 2018, 31, 707–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melcher, U.; Muthukumar, V.; Wiley, G.B.; Min, B.E.; Palmer, M.W.; Verchot-Lubicz, J.; Ali, A.; Nelson, R.S.; Roe, B.A.; Thapa, V.; et al. Evidence for novel viruses by analysis of nucleic acids in virus-like particle fractions from Ambrosia psilostachya. J. Virol. Methods 2008, 152, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Muthukumar, V.; Melcher, U.; Pierce, M.; Wiley, G.B.; Roe, B.A.; Palmer, M.W.; Thapa, V.; Ali, A.; Ding, T. Non-cultivated plants of the tallgrass prairie preserve of northeastern oklahoma frequently contain virus-like sequences in particulate fractions. Virus Res. 2009, 141, 169–173. [Google Scholar] [CrossRef] [PubMed]
- Bernardo, P.; Charles-Dominique, T.; Barakat, M.; Ortet, P.; Fernandez, E.; Filloux, D.; Hartnady, P.; Rebelo, T.A.; Cousins, S.R.; Mesleard, F.; et al. Geometagenomics illuminates the impact of agriculture on the distribution and prevalence of plant viruses at the ecosystem scale. ISME J. 2018, 12, 173–184. [Google Scholar] [CrossRef] [Green Version]
- Filloux, D.; Dallot, S.; Delaunay, A.; Galzi, S.; Jacquot, E.; Roumagnac, P. Metagenomics approaches based on virion-associated nucleic acids (VANA): An innovative tool for assessing without a priori viral diversity of plants. Methods Mol. Biol. 2015, 1302, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Marais, A.; Lefebvre, M.; Theil, S.; Svanella-Dumas, L.; Faure, C.; Candresse, T. Phytovirome analysis of wild plant populations: Comparison of double-stranded rna and virion-associated nucleic acid metagenomic approaches. J. Virol. 2019, 94. [Google Scholar] [CrossRef] [PubMed]
- Roossinck, M.J. Plants, viruses and the environment: Ecology and mutualism. Virology 2015, 479–480, 271–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hull, R. Origins and evolution of plant viruses. In Plant Virology; Elsevier: London, UK, 2014; pp. 423–476. [Google Scholar]
- Al Rwahnih, M.; Daubert, S.; Golino, D.; Islas, C.; Rowhani, A. Comparison of next-generation sequencing versus biological indexing for the optimal detection of viral pathogens in grapevine. Phytopathology 2015, 105, 758–763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kesanakurti, P.; Belton, M.; Saeed, H.; Rast, H.; Boyes, I.; Rott, M. Screening for plant viruses by next generation sequencing using a modified double strand RNA extraction protocol with an internal amplification control. J. Virol. Methods 2016, 236, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Loconsole, G.; Saldarelli, P.; Doddapaneni, H.; Savino, V.; Martelli, G.P.; Saponari, M. Identification of a single-stranded DNA virus associated with citrus chlorotic dwarf disease, a new member in the family geminiviridae. Virology 2012, 432, 162–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rott, M.; Xiang, Y.; Boyes, I.; Belton, M.; Saeed, H.; Kesanakurti, P.; Hayes, S.; Lawrence, T.; Birch, C.; Bhagwat, B.; et al. Application of next generation sequencing for diagnostic testing of tree fruit viruses and viroids. Plant Dis. 2017, 101, 1489–1499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, F.; Wagner, V.; Rasmussen, S.B.; Hartmann, R.; Paludan, S.R. Double-stranded RNA is produced by positive-strand RNA viruses and DNA viruses but not in detectable amounts by negative-strand RNA viruses. J. Virol. 2006, 80, 5059–5064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaafar, Y.Z.A.; Ziebell, H. Comparative study on three viral enrichment approaches based on RNA extraction for plant virus/viroid detection using high-throughput sequencing. PLoS ONE 2020, 15, e0237951. [Google Scholar] [CrossRef] [PubMed]
- Thapa, V.; McGlinn, D.J.; Melcher, U.; Palmer, M.W.; Roossinck, M.J. Determinants of taxonomic composition of plant viruses at the nature conservancy’s tallgrass prairie preserve, Oklahoma. Virus Evol. 2015, 1, vev007. [Google Scholar] [CrossRef] [PubMed]
- Blouin, A.G.; Ross, H.A.; Hobson-Peters, J.; O’Brien, C.A.; Warren, B.; MacDiarmid, R. A new virus discovered by immunocapture of double-stranded RNA, a rapid method for virus enrichment in metagenomic studies. Mol. Ecol. Resour. 2016, 16, 1255–1263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, K.; Tomita, R.; Sakamoto, M. Recombinant plant dsRNA-binding protein as an effective tool for the isolation of viral replicative form dsRNA and universal detection of RNA viruses. J. Gen. Plant Pathol. 2009, 75, 87–91. [Google Scholar] [CrossRef]
- Roossinck, M.J.; Saha, P.; Wiley, G.B.; Quan, J.; White, J.D.; Lai, H.; Chavarría, F.; Shen, G.; Roe, B.A. Ecogenomics: Using massively parallel pyrosequencing to understand virus ecology. Mol. Ecol. 2010, 19, 81–88. [Google Scholar] [CrossRef]
- Chalupowicz, L.; Dombrovsky, A.; Gaba, V.; Luria, N.; Reuven, M.; Beerman, A.; Lachman, O.; Dror, O.; Nissan, G.; Manulis-Sasson, S. Diagnosis of plant diseases using the nanopore sequencing platform. Plant Pathol. 2019, 68, 229–238. [Google Scholar] [CrossRef]
- Lusk, R.W. Diverse and widespread contamination evident in the unmapped depths of high throughput sequencing data. PLoS ONE 2014, 9, e110808. [Google Scholar] [CrossRef] [Green Version]
- Laurence, M.; Hatzis, C.; Brash, D.E. Common contaminants in next-generation sequencing that hinder discovery of low-abundance microbes. PLoS ONE 2014, 9, e97876. [Google Scholar] [CrossRef] [PubMed]
- Schmieder, R.; Edwards, R. Fast identification and removal of sequence contamination from genomic and metagenomic datasets. PLoS ONE 2011, 6, e17288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naccache, S.N.; Greninger, A.L.; Lee, D.; Coffey, L.L.; Phan, T.; Rein-Weston, A.; Aronsohn, A.; Hackett, J.; Delwart, E.L.; Chiu, C.Y. The perils of pathogen discovery: Origin of a novel parvovirus-like hybrid genome traced to nucleic acid extraction spin columns. J. Virol. 2013, 87, 11966–11977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10. [Google Scholar] [CrossRef]
- Illumina bcl2fastq and bcl2fastq2 Conversion Software; v.2.20; Illumina: San Diego, CA, USA, 2019; Available online: https://emea.support.illumina.com/sequencing/sequencing_software/bcl2fastq-conversion-software.html (accessed on 13 April 2021).
- Oxford Nanopore Technologies Guppy: Local Accelerated Basecalling for Nanopore Data. Available online: https://community.nanoporetech.com/downloads (accessed on 13 April 2021).
- Illumina Effects of Index Misassignment on Multiplexing and Downstream Analysis (770-2017-004-D). Available online: https://www.illumina.com/content/dam/illumina-marketing/documents/products/whitepapers/index-hopping-white-paper-770-2017-004.pdf (accessed on 13 April 2021).
- van der Valk, T.; Vezzi, F.; Ormestad, M.; Dalén, L.; Guschanski, K. Index hopping on the Illumina HiseqX platform and its consequences for ancient DNA studies. Mol. Ecol. Resour. 2020, 20, 1171–1181. [Google Scholar] [CrossRef]
- MacConaill, L.E.; Burns, R.T.; Nag, A.; Coleman, H.A.; Slevin, M.K.; Giorda, K.; Light, M.; Lai, K.; Jarosz, M.; McNeill, M.S.; et al. Unique, dual-indexed sequencing adapters with UMIs effectively eliminate index cross-talk and significantly improve sensitivity of massively parallel sequencing. BMC Genom. 2018, 19, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wick, B. Porechop. Available online: https://github.com/rrwick/Porechop (accessed on 13 April 2021).
- De Coster, W.; D’Hert, S.; Schultz, D.T.; Cruts, M.; Van Broeckhoven, C. NanoPack: Visualizing and processing long-read sequencing data. Bioinformatics 2018, 34, 2666–2669. [Google Scholar] [CrossRef] [PubMed]
- Cock, P.J.A.; Fields, C.J.; Goto, N.; Heuer, M.L.; Rice, P.M. The Sanger FASTQ file format for sequences with quality scores, and the Solexa/Illumina FASTQ variants. Nucleic Acids Res. 2009, 38, 1767–1771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrews, S. FastQC. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 13 April 2021).
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef] [Green Version]
- Loman, N.J.; Quinlan, A.R. Poretools: A toolkit for analyzing nanopore sequence data. Bioinformatics 2014, 30, 3399–3401. [Google Scholar] [CrossRef] [PubMed]
- Najoshi Sickle—A Windowed Adaptive Trimming Tool for FASTQ Files Using Quality. Available online: https://github.com/najoshi/sickle (accessed on 13 April 2021).
- Andino, R.; Domingo, E. Viral quasispecies. Virology 2015, 479–480, 46–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paszkiewicz, K.; Studholme, D.J. De novo assembly of short sequence reads. Brief. Bioinform. 2010, 11, 457–472. [Google Scholar] [CrossRef] [Green Version]
- Sohn, J.-I.; Nam, J.-W. The present and future of de novo whole-genome assembly. Brief. Bioinform. 2018, 19, 23–40. [Google Scholar] [CrossRef] [Green Version]
- Luo, R.; Liu, B.; Xie, Y.; Li, Z.; Huang, W.; Yuan, J.; He, G.; Chen, Y.; Pan, Q.; Liu, Y.; et al. SOAPdenovo2: An empirically improved memory-efficient short-read de novo assembler. Gigascience 2012, 1, 2047-217X-1-18. [Google Scholar] [CrossRef] [PubMed]
- Gnerre, S.; MacCallum, I.; Przybylski, D.; Ribeiro, F.J.; Burton, J.N.; Walker, B.J.; Sharpe, T.; Hall, G.; Shea, T.P.; Sykes, S.; et al. High-quality draft assemblies of mammalian genomes from massively parallel sequence data. Proc. Natl. Acad. Sci. USA 2011, 108, 1513–1518. [Google Scholar] [CrossRef] [Green Version]
- Simpson, J.T.; Wong, K.; Jackman, S.D.; Schein, J.E.; Jones, S.J.M.; Birol, I. ABySS: A parallel assembler for short read sequence data. Genome Res. 2009, 19, 1117–1123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zerbino, D.R.; Birney, E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008, 18, 821–829. [Google Scholar] [CrossRef] [Green Version]
- Peng, Y.; Leung, H.C.M.; Yiu, S.M.; Chin, F.Y.L. IDBA-UD: A de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 2012, 28, 1420–1428. [Google Scholar] [CrossRef] [Green Version]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nurk, S.; Bankevich, A.; Antipov, D.; Gurevich, A.A.; Korobeynikov, A.; Lapidus, A.; Prjibelski, A.D.; Pyshkin, A.; Sirotkin, A.; Sirotkin, Y.; et al. Assembling single-cell genomes and mini-metagenomes from chimeric MDA products. J. Comput. Biol. 2013, 20, 714–737. [Google Scholar] [CrossRef] [Green Version]
- Bushmanova, E.; Antipov, D.; Lapidus, A.; Prjibelski, A.D. RnaSPAdes: A de novo transcriptome assembler and its application to RNA-Seq data. Gigascience 2019, 8, giz100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edwards, D.J.; Holt, K.E. Beginner’s guide to comparative bacterial genome analysis using next-generation sequence data. Microb. Inform. Exp. 2013, 3, 2. [Google Scholar] [CrossRef] [Green Version]
- Rang, F.J.; Kloosterman, W.P.; de Ridder, J. From squiggle to basepair: Computational approaches for improving nanopore sequencing read accuracy. Genome Biol. 2018, 19, 90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koren, S.; Schatz, M.C.; Walenz, B.P.; Martin, J.; Howard, J.T.; Ganapathy, G.; Wang, Z.; Rasko, D.A.; McCombie, W.R.; Jarvis, E.D.; et al. Hybrid error correction and de novo assembly of single-molecule sequencing reads. Nat. Biotechnol. 2012, 30, 693–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and accurate long-read assembly via adaptive κ-mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chin, C.S.; Peluso, P.; Sedlazeck, F.J.; Nattestad, M.; Concepcion, G.T.; Clum, A.; Dunn, C.; O’Malley, R.; Figueroa-Balderas, R.; Morales-Cruz, A.; et al. Phased diploid genome assembly with single-molecule real-time sequencing. Nat. Methods 2016, 13, 1050–1054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oxford Nanopore Technologies Pomoxis—Bioinformatics Tools for Nanopore Research. Available online: https://github.com/nanoporetech/pomoxis (accessed on 13 April 2021).
- Filloux, D.; Fernandez, E.; Loire, E.; Claude, L.; Galzi, S.; Candresse, T.; Winter, S.; Jeeva, M.L.; Makeshkumar, T.; Martin, D.P.; et al. Nanopore-based detection and characterization of yam viruses. Sci. Rep. 2018, 8, 17879. [Google Scholar] [CrossRef] [Green Version]
- Boykin, L.M.; Sseruwagi, P.; Alicai, T.; Ateka, E.; Mohammed, I.U.; Stanton, J.A.L.; Kayuki, C.; Mark, D.; Fute, T.; Erasto, J.; et al. Tree lab: Portable genomics for early detection of plant viruses and pests in sub-saharan africa. Genes 2019, 10, 632. [Google Scholar] [CrossRef] [Green Version]
- Naito, F.Y.B.; Melo, F.L.; Fonseca, M.E.N.; Santos, C.A.F.; Chanes, C.R.; Ribeiro, B.M.; Gilbertson, R.L.; Boiteux, L.S.; de Cássia Pereira-Carvalho, R. Nanopore sequencing of a novel bipartite new world begomovirus infecting cowpea. Arch. Virol. 2019, 164, 1907–1910. [Google Scholar] [CrossRef]
- Leiva, A.M.; Siriwan, W.; Lopez-Alvarez, D.; Barrantes, I.; Hemniam, N.; Saokham, K.; Cuellar, W.J. Nanopore-based complete genome sequence of a sri lankan cassava mosaic virus (geminivirus) strain from Thailand. Microbiol. Resour. Announc. 2020, 9. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Finn, R.D.; Clements, J.; Eddy, S.R. HMMER web server: Interactive sequence similarity searching. Nucleic Acids Res. 2011, 39, W29–W37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stobbe, A.H.; Daniels, J.; Espindola, A.S.; Verma, R.; Melcher, U.; Ochoa-Corona, F.; Garzon, C.; Fletcher, J.; Schneider, W. E-probe diagnostic nucleic acid analysis (EDNA): A theoretical approach for handling of next generation sequencing data for diagnostics. J. Microbiol. Methods 2013, 94, 356–366. [Google Scholar] [CrossRef] [PubMed]
- Punta, M.; Coggill, P.C.; Eberhardt, R.Y.; Mistry, J.; Tate, J.; Boursnell, C.; Pang, N.; Forslund, K.; Ceric, G.; Clements, J.; et al. The Pfam protein families database. Nucleic Acids Res. 2012, 40, 290–301. [Google Scholar] [CrossRef] [PubMed]
- Marchler-Bauer, A.; Panchenko, A.R.; Shoemarker, B.A.; Thiessen, P.A.; Geer, L.Y.; Bryant, S.H. CDD: A database of conserved domain alignments with links to domain three-dimensional structure. Nucleic Acids Res. 2002, 30, 281–283. [Google Scholar] [CrossRef] [PubMed]
- Agranovsky, A.A.; Boyko, V.P.; Karasev, A.V.; Koonin, E.V.; Dolja, V.V. Putative 65 kDa protein of beet yellows closterovirus is a homologue of HSP70 heat shock proteins. J. Mol. Biol. 1991, 217, 603–610. [Google Scholar] [CrossRef]
- Amselem, J.; Cornut, G.; Choisne, N.; Alaux, M.; Alfama-Depauw, F.; Jamilloux, V.; Maumus, F.; Letellier, T.; Luyten, I.; Pommier, C.; et al. RepetDB: A unified resource for transposable element references. Mob. DNA 2019, 10, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geering, A.D.W.; Maumus, F.; Copetti, D.; Choisne, N.; Zwickl, D.J.; Zytnicki, M.; McTaggart, A.R.; Scalabrin, S.; Vezzulli, S.; Wing, R.A.; et al. Endogenous florendoviruses are major components of plant genomes and hallmarks of virus evolution. Nat. Commun. 2014, 5, 5269. [Google Scholar] [CrossRef] [Green Version]
- Diop, S.I.; Geering, A.D.W.; Alfama-Depauw, F.; Loaec, M.; Teycheney, P.-Y.; Maumus, F. Tracheophyte genomes keep track of the deep evolution of the caulimoviridae. Sci. Rep. 2018, 8, 572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, V.; Lefeuvre, P.; Roumagnac, P.; Filloux, D.; Teycheney, P.-Y.; Martin, D.P.; Maumus, F. Large-scale survey reveals pervasiveness and potential function of endogenous geminiviral sequences in plants. Virus Evol. 2020, 6, veaa071. [Google Scholar] [CrossRef] [PubMed]
- Tangherlini, M.; Dell’Anno, A.; Zeigler Allen, L.; Riccioni, G.; Corinaldesi, C. Assessing viral taxonomic composition in benthic marine ecosystems: Reliability and efficiency of different bioinformatic tools for viral metagenomic analyses. Sci. Rep. 2016, 6, 28428. [Google Scholar] [CrossRef] [Green Version]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kent, W.J. BLAT—The BLAST-like alignment tool. Genome Res. 2002, 12, 656–664. [Google Scholar] [CrossRef] [Green Version]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using diamond. Nat. Methods 2014, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [Green Version]
- Hong, C.; Manimaran, S.; Shen, Y.; Perez-Rogers, J.F.; Byrd, A.L.; Castro-Nallar, E.; Crandall, K.A.; Johnson, W.E. PathoScope 2.0: A complete computational framework for strain identification in environmental or clinical sequencing samples. Microbiome 2014, 2, 33. [Google Scholar] [CrossRef]
- Mistry, J.; Finn, R.D.; Eddy, S.R.; Bateman, A.; Punta, M. Challenges in homology search: HMMER3 and convergent evolution of coiled-coil regions. Nucleic Acids Res. 2013, 41, e121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skewes-Cox, P.; Sharpton, T.J.; Pollard, K.S.; DeRisi, J.L. Profile hidden Markov models for the detection of viruses within metagenomic sequence data. PLoS ONE 2014, 9, e105067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bzhalava, Z.; Hultin, E.; Dillner, J. Extension of the viral ecology in humans using viral profile hidden Markov models. PLoS ONE 2018, 13, e0190938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood, D.E.; Salzberg, S.L. Kraken: Ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 2014, 15, R46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood, D.E.; Lu, J.; Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019, 20, 257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menzel, P.; Ng, K.L.; Krogh, A. Fast and sensitive taxonomic classification for metagenomics with Kaiju. Nat. Commun. 2016, 7, 11257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flygare, S.; Simmon, K.; Miller, C.; Qiao, Y.; Kennedy, B.; Di Sera, T.; Graf, E.H.; Tardif, K.D.; Kapusta, A.; Rynearson, S.; et al. Taxonomer: An interactive metagenomics analysis portal for universal pathogen detection and host mRNA expression profiling. Genome Biol. 2016, 17, 111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baizan-Edge, A.; Cock, P.; MacFarlane, S.; McGavin, W.; Torrance, L.; Jones, S. Kodoja: A workflow for virus detection in plants using k-mer analysis of RNA-sequencing data. J. Gen. Virol. 2019, 100, 533–542. [Google Scholar] [CrossRef] [PubMed]
- Tampuu, A.; Bzhalava, Z.; Dillner, J.; Vicente, R. ViraMiner: Deep learning on raw DNA sequences for identifying viral genomes in human samples. PLoS ONE 2019, 14, e0222271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, J.; Song, K.; Deng, C.; Ahlgren, N.A.; Fuhrman, J.A.; Li, Y.; Xie, X.; Poplin, R.; Sun, F. Identifying viruses from metagenomic data using deep learning. Quant. Biol. 2020, 8, 64–77. [Google Scholar] [CrossRef] [Green Version]
- Abdelkareem, A.O.; Khalil, M.I.; Elaraby, M.; Abbas, H.; Elbehery, A.H.A. VirNet: Deep attention model for viral reads identification. In Proceedings of the 2018 13th International Conference on Computer Engineering and Systems (ICCES), Cairo, Egypt, 18–19 December 2018; pp. 623–626. [Google Scholar]
- Ren, Y.; Xu, Y.; Lee, W.M.; Di Bisceglie, A.M.; Fan, X. In-depth serum virome analysis in patients with acute liver failure with indeterminate etiology. Arch. Virol. 2020, 165, 127–135. [Google Scholar] [CrossRef]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef] [PubMed]
- Warwick-Dugdale, J.; Solonenko, N.; Moore, K.; Chittick, L.; Gregory, A.C.; Allen, M.J.; Sullivan, M.B.; Temperton, B. Long-read viral metagenomics captures abundant and microdiverse viral populations and their niche-defining genomic islands. PeerJ 2019, 7, e6800. [Google Scholar] [CrossRef] [Green Version]
- Lefkowitz, E.J.; Dempsey, D.M.; Hendrickson, R.C.; Orton, R.J.; Siddell, S.G.; Smith, D.B. Virus taxonomy: The database of the international committee on taxonomy of viruses (ICTV). Nucleic Acids Res. 2018, 46, D708–D717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davison, A.J. Journal of general virology—Introduction to ‘ICTV virus taxonomy profiles’. J. Gen. Virol. 2017, 98, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bao, Y.; Chetvernin, V.; Tatusova, T. Improvements to pairwise sequence comparison (PASC): A genome-based web tool for virus classification. Arch. Virol. 2014, 159, 3293–3304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibbs, A.J.; Hajizadeh, M.; Ohshima, K.; Jones, R.A.C. The potyviruses: An evolutionary synthesis is emerging. Viruses 2020, 12, 132. [Google Scholar] [CrossRef] [Green Version]
- Jones, S.; Baizan-Edge, A.; MacFarlane, S.; Torrance, L. Viral diagnostics in plants using next generation sequencing: Computational analysis in practice. Front. Plant Sci. 2017, 8, 1770. [Google Scholar] [CrossRef]
- Blawid, R.; Silva, J.M.F.; Nagata, T. Discovering and sequencing new plant viral genomes by next-generation sequencing: Description of a practical pipeline. Ann. Appl. Biol. 2017, 170, 301–314. [Google Scholar] [CrossRef]
- Roenhorst, J.W.; de Krom, C.; Fox, A.; Mehle, N.; Ravnikar, M.; Werkman, A.W. Ensuring validation in diagnostic testing is fit for purpose: A view from the plant virology laboratory. EPPO Bull. 2018, 48, 105–115. [Google Scholar] [CrossRef]
- Simmonds, P.; Adams, M.J.; Benk, M.; Breitbart, M.; Brister, J.R.; Carstens, E.B.; Davison, A.J.; Delwart, E.; Gorbalenya, A.E.; Harrach, B.; et al. Consensus statement: Virus taxonomy in the age of metagenomics. Nat. Rev. Microbiol. 2017, 15, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Rwahnih, M.A.; Daubert, S.; Úrbez-Torres, J.R.; Cordero, F.; Rowhani, A. Deep sequencing evidence from single grapevine plants reveals a virome dominated by mycoviruses. Arch. Virol. 2011, 156, 397–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marzano, S.Y.L.; Domier, L.L. Novel mycoviruses discovered from metatranscriptomics survey of soybean phyllosphere phytobiomes. Virus Res. 2016, 213, 332–342. [Google Scholar] [CrossRef] [Green Version]
- Kreuze, J. siRNA deep sequencing and assembly: Piecing together viral infections. In Detection and Diagnostics of Plant Pathogens; Gullino, M.L., Bonants, P.J.M., Eds.; Springer: Dordrecht, The Netherlands, 2014; pp. 21–38. ISBN 978-94-017-9020-8. [Google Scholar]
- Massart, S.; Candresse, T.; Gil, J.; Lacomme, C.; Predajna, L.; Ravnikar, M.; Reynard, J.S.; Rumbou, A.; Saldarelli, P.; Škoric, D.; et al. A framework for the evaluation of biosecurity, commercial, regulatory, and scientific impacts of plant viruses and viroids identified by NGS technologies. Front. Microbiol. 2017, 8, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kreuze, J.F.; Perez, A.; Gargurevich, M.G.; Cuellar, W.J. Badnaviruses of sweet potato: Symptomless coinhabitants on a global scale. Front. Plant Sci. 2020, 11, 313. [Google Scholar] [CrossRef] [PubMed]
- Koloniuk, I.; Thekke-Veetil, T.; Reynard, J.S.; Pleško, I.M.; Přibylová, J.; Brodard, J.; Kellenberger, I.; Sarkisova, T.; Špak, J.; Lamovšek, J.; et al. Molecular characterization of divergent closterovirus isolates infecting Ribes species. Viruses 2018, 10, 369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sõmera, M.; Kvarnheden, A.; Desbiez, C.; Blystad, D.R.; Sooväli, P.; Kundu, J.K.; Gantsovski, M.; Nygren, J.; Lecoq, H.; Verdin, E.; et al. Sixty years after the first description: Genome sequence and biological characterization of European wheat striate mosaic virus infecting cereal crops. Phytopathology 2020, 110, 68–79. [Google Scholar] [CrossRef]
- Hammond, J.; Adams, I.; Fowkes, A.R.; McGreig, S.; Botermans, M.; van Oorspronk, J.J.A.; Westenberg, M.; Verbeek, M.; Dullemans, A.M.; Stijger, C.C.M.M.; et al. Sequence analysis of 43-year old samples of plantago lanceolata show that plantain virus x is synonymous with actinidia virus X and is widely distributed. Plant Pathol. 2020, 249–258. [Google Scholar] [CrossRef]
- Tamisier, L.; Haegeman, A.; Foucart, Y.; Fouillien, N.; Rwahnih, M.A.; Buzkan, N.; Candresse, T.; Chiumenti, M.; De Jonghe, K.; Lefebvre, M.; et al. Semi-artificial datasets as a resource for validation of bioinformatics pipelines for plant virus detection. Zenodo 2020, 4273791, 1–15. [Google Scholar] [CrossRef]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, vev003. [Google Scholar] [CrossRef] [Green Version]
- Lole, K.S.; Bollinger, R.C.; Paranjape, R.S.; Gadkari, D.; Kulkarni, S.S.; Novak, N.G.; Ingersoll, R.; Sheppard, H.W.; Ray, S.C. Full-length human immunodeficiency virus type 1 genomes from subtype c-infected seroconverters in india, with evidence of intersubtype recombination. J. Virol. 1999, 73, 152–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simmonds, P.; Midgley, S. Recombination in the genesis and evolution of hepatitis B virus genotypes. J. Virol. 2005, 79, 15467–15476. [Google Scholar] [CrossRef] [Green Version]
- Routh, A.; Johnson, J.E. Discovery of functional genomic motifs in viruses with ViReMa-a virus recombination mapper-for analysis of next-generation sequencing data. Nucleic Acids Res. 2014, 42, e11. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.; Sun, X.; Taylor, A.; Jiao, C.; Xu, Y.; Cai, X.; Wang, X.; Ge, C.; Pan, G.; Wang, Q.; et al. Diversity, distribution, and evolution of tomato viruses in china uncovered by small RNA sequencing. J. Virol. 2017, 91, e00173-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertran, A.; Ciuffo, M.; Margaria, P.; Rosa, C.; Resende, R.O.; Turina, M. Host-specific accumulation and temperature effects on the generation of dimeric viral RNA species derived from the S-RNA of members of the Tospovirus genus. J. Gen. Virol. 2016, 97, 3051–3062. [Google Scholar] [CrossRef] [PubMed]
- Maliogka, V.I.; Salvador, B.; Carbonell, A.; Sáenz, P.; León, D.S.; Oliveros, J.C.; Delgadillo, M.O.; García, J.A.; Simón-Mateo, C.; Progenika Biopharma, S.A. Virus variants with differences in the p1 protein coexist in a plum pox virus population and display particular host-dependent pathogenicity features. Mol. Plant Pathol. 2012, 13, 877–886. [Google Scholar] [CrossRef]
- da Silva, W.; Kutnjak, D.; Xu, Y.; Xu, Y.; Giovannoni, J.; Elena, S.F.; Gray, S. Transmission modes affect the population structure of potato virus Y in potato. PLoS Pathog. 2020, 16, e1008608. [Google Scholar] [CrossRef]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [CrossRef]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Teslenko, M.; Van Der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. Mrbayes 3.2: Efficient bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huson, D.H.; Beier, S.; Flade, I.; Górska, A.; El-Hadidi, M.; Mitra, S.; Ruscheweyh, H.J.; Tappu, R. MEGAN Community edition—Interactive exploration and analysis of large-scale microbiome sequencing data. PLoS Comput. Biol. 2016, 12, e1004957. [Google Scholar] [CrossRef] [Green Version]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018, 4, vey016. [Google Scholar] [CrossRef] [Green Version]
- Fuentes, S.; Gibbs, A.J.; Adams, I.P.; Wilson, C.; Botermans, M.; Fox, A.; Kreuze, J.; Kehoe, M.A.; Jones, R.A.C. Potato virus A isolates from three continents: Their biological properties, phylogenetics, and prehistory. Phytopathology 2021, 111, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Hardy, O.J.; Vekemans, X. SPAGeDI: A versatile computer program to analyse spatial genetic structure at the individual or population levels. Mol. Ecol. Notes 2002, 2, 618–620. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Gao, S.; Padmanabhan, C.; Li, R.; Galvez, M.; Gutierrez, D.; Fuentes, S.; Ling, K.S.; Kreuze, J.; Fei, Z. VirusDetect: An automated pipeline for efficient virus discovery using deep sequencing of small RNAs. Virology 2017, 500, 130–138. [Google Scholar] [CrossRef]
- Lefebvre, M.; Theil, S.; Ma, Y.; Candresse, T. The virannot pipeline: A resource for automated viral diversity estimation and operational taxonomy units assignation for virome sequencing data. Phytobiomes J. 2019, 3, 256–259. [Google Scholar] [CrossRef] [Green Version]
- Ho, T.; Tzanetakis, I.E. Development of a virus detection and discovery pipeline using next generation sequencing. Virology 2014, 471–473, 54–60. [Google Scholar] [CrossRef] [Green Version]
- Visser, M.; Burger, J.T.; Maree, H.J. Targeted virus detection in next-generation sequencing data using an automated e-probe based approach. Virology 2016, 495, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Afgan, E.; Baker, D.; Batut, B.; Van Den Beek, M.; Bouvier, D.; Ech, M.; Chilton, J.; Clements, D.; Coraor, N.; Grüning, B.A.; et al. The galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 2018, 46, W537–W544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalantar, K.L.; Carvalho, T.; de Bourcy, C.F.A.; Dimitrov, B.; Dingle, G.; Egger, R.; Han, J.; Holmes, O.B.; Juan, Y.F.; King, R.; et al. IDseq-An open source cloud-based pipeline and analysis service for metagenomic pathogen detection and monitoring. Gigascience 2020, 9, giaa111. [Google Scholar] [CrossRef] [PubMed]





| Tool Name | Advantages | Limits and Considerations | Important Thresholds |
|---|---|---|---|
| BLASTx or BLASTn | High sensitivity | Slow, intensive use of computing power if a large database is used, BLASTx needed for the detection of divergent novel viruses, BLASTn needed for the detection of viroids and noncoding regions of viral genomes or satellites; performance improved by prior assembly of contigs. | Minimum percentage of identity; length of identified region of similarity; minimal e-value, bit-score. |
| MegaBLAST | Faster than BLASTn, handles longer sequences | Less sensitive than BLASTn, only useful for detection of nucleotide sequences very similar to the ones in the used database; performance improved by prior assembly of contigs. | Minimum percentage of identity; length of identified region of similarity; minimal e-value, bit-score. |
| BLASTp | High sensitivity | Slow, need to translate nucleotide sequences to proteins first; performance improved by prior assembly of contigs; not applicable for viroids or noncoding regions of viral genomes or satellites. | Minimum percentage of identity; length of identified region of similarity; minimal e-value, bit-score. |
| DIAMOND | Faster than BLASTx | Less sensitive, annotation less accurate than BLAST; performance improved by prior assembly of contigs; only available for searches against protein databases; not applicable for viroids or noncoding regions of viral genomes or satellites. | Minimum percentage of identity; length of identified region of similarity; minimal e-value, bit-score; use sensitive mode. |
| Burrows-Wheeler transform-based mapping algorithms (e.g., BWA or Bowtie2) | Does not require prior assembly of contigs, high sensitivity for short sequences | Only allows detection of known agents. Difficult to adjust mapping stringency to (1) allow detection of divergent isolates while (2) avoiding cross-mapping between related agents; prior assembly of contigs reduces cross-mapping between related agents. | Mapping stringency (e.g., mismatch penalties, gap open/extension penalties, percent of read length matching reference, minimum percentage of identity) |
| HMMER or HMMScan | High efficiency for detection of distant homologs | Annotation more complex for protein families shared between cellular organisms and viruses; not applicable for viroids or noncoding regions of viral genomes or satellites. | Minimal e-value. |
| K-mer based classification algorithms (Kraken or Taxonomer) | Fast | Requires large computer memory; accuracy may be limited for the shorter genomes of plant viruses; the confidence scoring of the results is not straight forward. | C/Q ratio for Kraken (advise the manual). |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kutnjak, D.; Tamisier, L.; Adams, I.; Boonham, N.; Candresse, T.; Chiumenti, M.; De Jonghe, K.; Kreuze, J.F.; Lefebvre, M.; Silva, G.; et al. A Primer on the Analysis of High-Throughput Sequencing Data for Detection of Plant Viruses. Microorganisms 2021, 9, 841. https://doi.org/10.3390/microorganisms9040841
Kutnjak D, Tamisier L, Adams I, Boonham N, Candresse T, Chiumenti M, De Jonghe K, Kreuze JF, Lefebvre M, Silva G, et al. A Primer on the Analysis of High-Throughput Sequencing Data for Detection of Plant Viruses. Microorganisms. 2021; 9(4):841. https://doi.org/10.3390/microorganisms9040841
Chicago/Turabian StyleKutnjak, Denis, Lucie Tamisier, Ian Adams, Neil Boonham, Thierry Candresse, Michela Chiumenti, Kris De Jonghe, Jan F. Kreuze, Marie Lefebvre, Gonçalo Silva, and et al. 2021. "A Primer on the Analysis of High-Throughput Sequencing Data for Detection of Plant Viruses" Microorganisms 9, no. 4: 841. https://doi.org/10.3390/microorganisms9040841