
A Cell Culture System to Investigate Marek’s Disease Virus Integration into Host Chromosomes

 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells
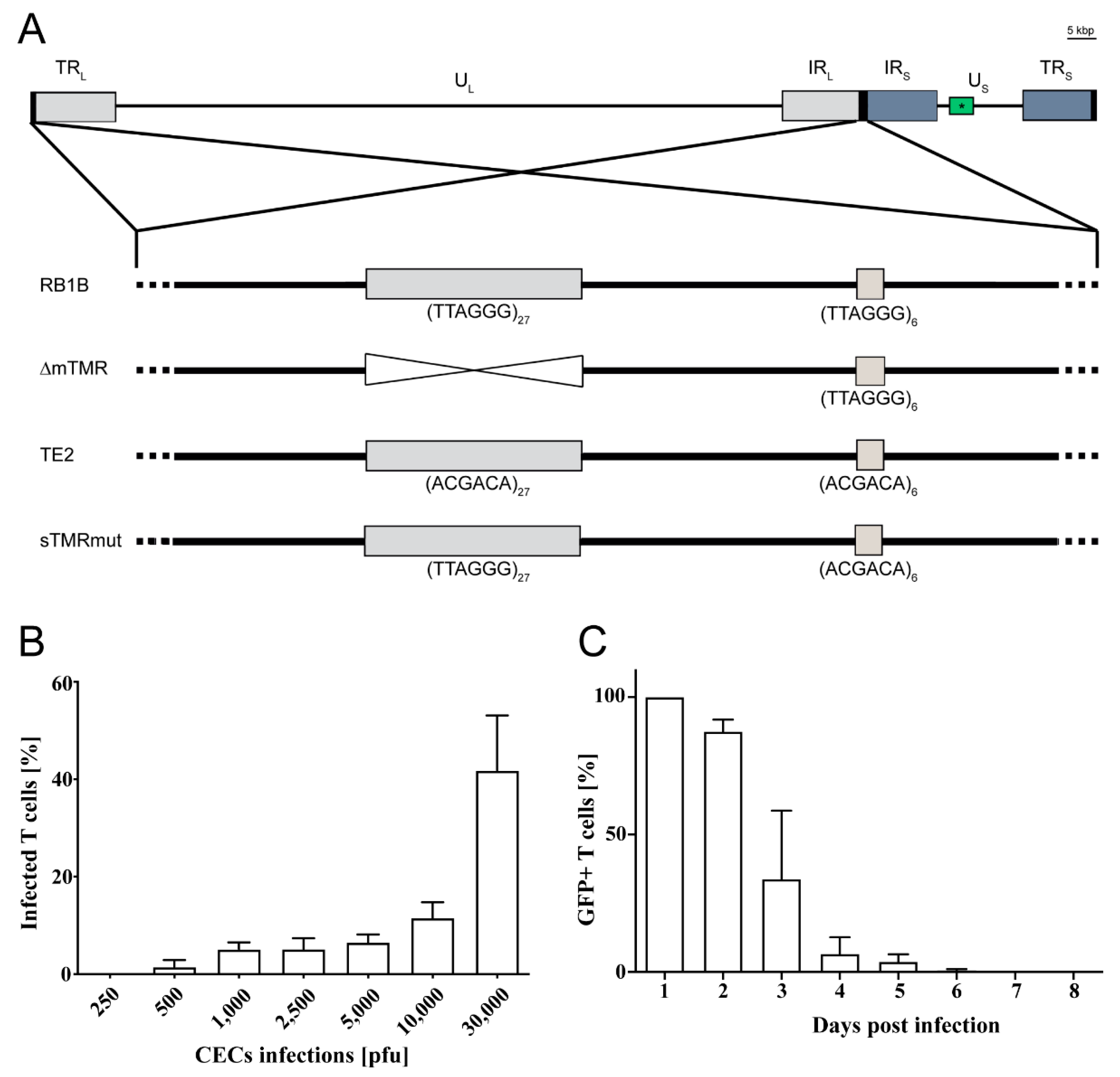
2.2. Generation of Mutant Viruses
2.3. T Cell Infection
2.4. Quantification of MDV Genome Copy Numbers by qPCR
2.5. RT-qPCR
2.6. Flow Cytometry
2.7. Fluorescence In Situ Hybridization
2.8. Reactivation
2.9. Statistical Analyses
3. Results and Discussion
3.1. MDV Efficiently Infects CU91 T Cells
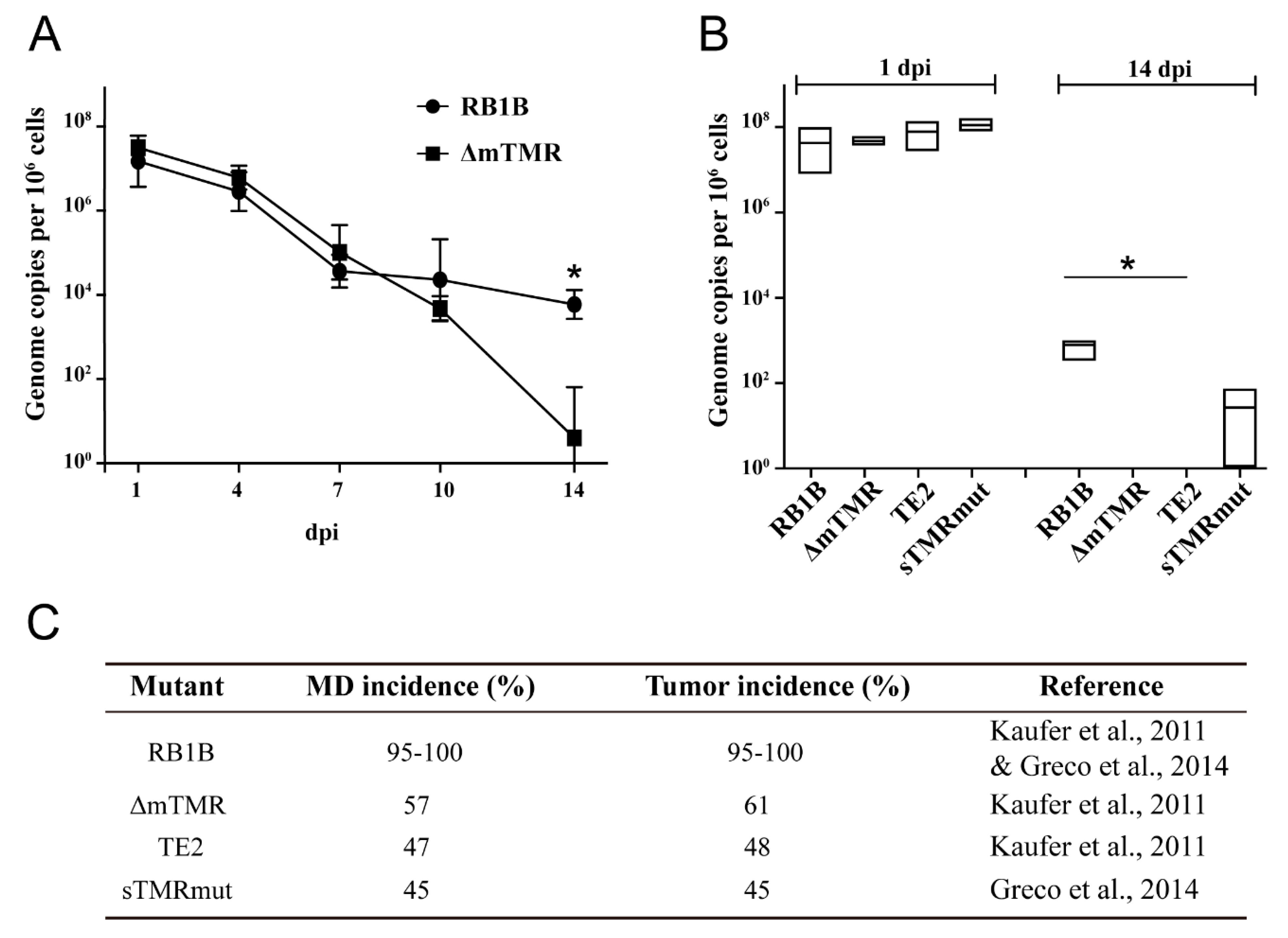
3.2. MDV Genome Maintenance in Infected CU91 T Cells Is Dependent on Viral TMR
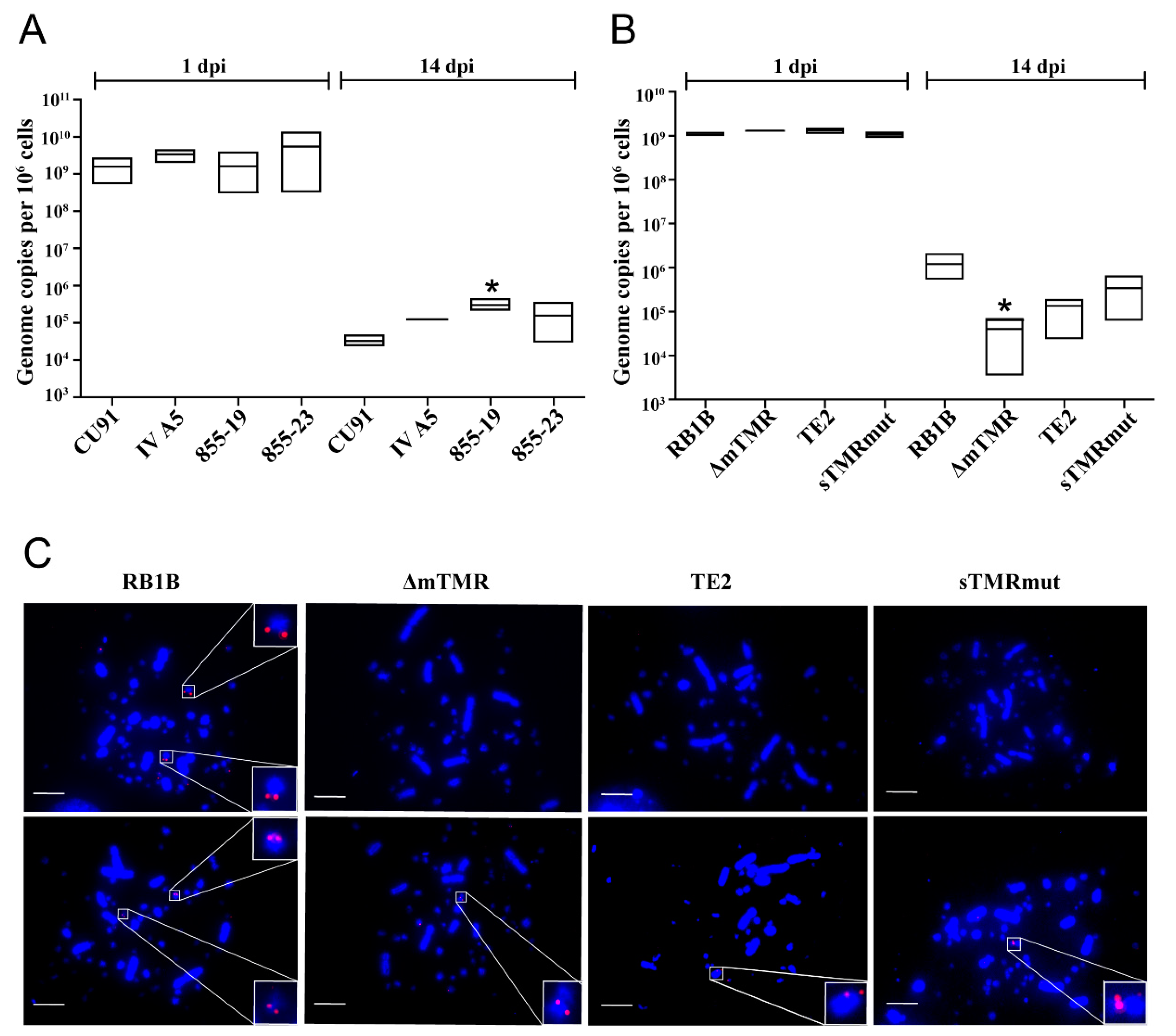
3.3. MDV Genome Maintenance Differs between Chicken T Cell Lines
3.4. Impaired Genome Maintenance and Integration of TMR Mutants in 855-19 T Cells
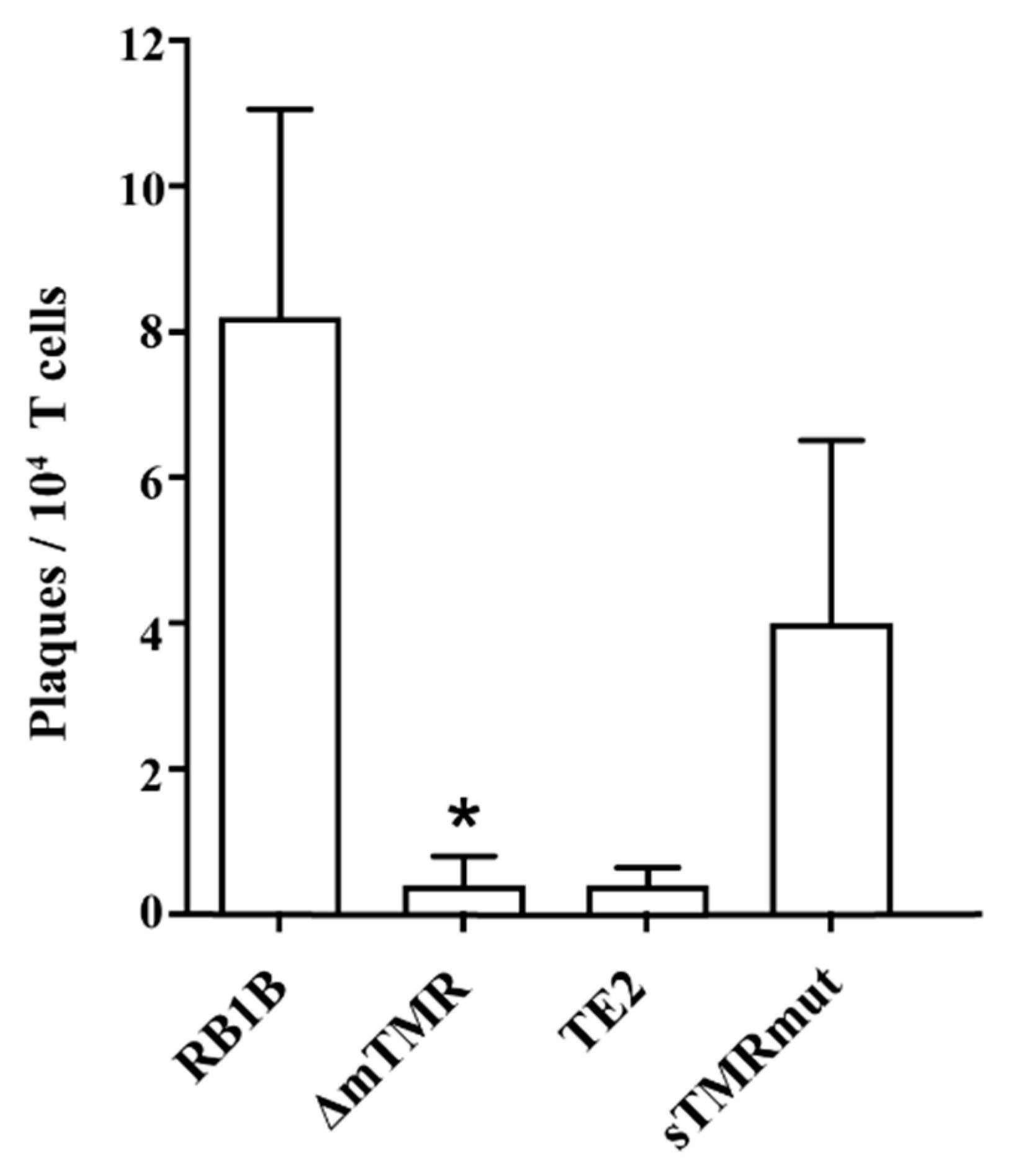
3.5. The MDV Genome Can Reactivate from Latency
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Morrow, C.; Fehler, F. Marek’s Disease: A worldwide problem. In Marek’s Disease; Davison, F., Nair, V., Eds.; Elsevier: Amsterdam, The Netherlands, 2004; pp. 49–61. [Google Scholar]
- Bertzbach, L.D.; Conradie, A.M.; You, Y.; Kaufer, B.B. Latest insights into Marek’s disease virus pathogenesis and tumorigenesis. Cancers 2020, 12, 647. [Google Scholar] [CrossRef] [Green Version]
- Read, A.F.; Baigent, S.J.; Powers, C.; Kgosana, L.B.; Blackwell, L.; Smith, L.P.; Kennedy, D.A.; Walkden-Brown, S.W.; Nair, V.K. Imperfect Vaccination Can Enhance the Transmission of Highly Virulent Pathogens. PLoS Biol. 2015, 13, e1002198. [Google Scholar] [CrossRef]
- Osterrieder, N.; Kamil, J.P.; Schumacher, D.; Tischer, B.K.; Trapp, S. Marek’s disease virus: From miasma to model. Nat. Rev. Microbiol. 2006, 4, 283–294. [Google Scholar] [CrossRef]
- Davison, F.; Nair, V. Use of Marek’s disease vaccines: Could they be driving the virus to increasing virulence? Expert Rev. Vaccines 2005, 4, 77–88. [Google Scholar] [CrossRef]
- Baigent, S.J.; Ross, L.J.; Davison, T.F. Differential susceptibility to Marek’s disease is associated with differences in number, but not phenotype or location, of pp38+ lymphocytes. J. Gen. Virol. 1998, 79 Pt 11, 2795–2802. [Google Scholar] [CrossRef] [Green Version]
- Baigent, S.J.; Ross, L.J.; Davison, T.F. A flow cytometric method for identifying Marek’s disease virus pp38 expression in lymphocyte subpopulations. Avian Pathol. 1996, 25, 255–267. [Google Scholar] [CrossRef]
- Bertzbach, L.D.; Laparidou, M.; Härtle, S.; Etches, R.J.; Kaspers, B.; Schusser, B.; Kaufer, B.B. Unraveling the role of B cells in the pathogenesis of an oncogenic avian herpesvirus. Proc. Natl. Acad. Sci. USA 2018, 115, 11603–11607. [Google Scholar] [CrossRef] [Green Version]
- Schat, K.A.; Chen, C.L.; Calnek, B.W.; Char, D. Transformation of T-lymphocyte subsets by Marek’s disease herpesvirus. J. Virol. 1991, 65, 1408–1413. [Google Scholar] [CrossRef] [Green Version]
- Parcells, M.S.; Burnside, J.; Morgan, R.W. Cancer Associated Viruses. Marek’s Disease Virus-Induced T-Cell Lymphomas; Springer: Boston, MA, USA, 2012; pp. 307–335. [Google Scholar] [CrossRef]
- Delecluse, H.J.; Hammerschmidt, W. Status of Marek’s disease virus in established lymphoma cell lines: Herpesvirus integration is common. J. Virol. 1993, 67, 82–92. [Google Scholar] [CrossRef] [Green Version]
- Kaufer, B.B.; Jarosinski, K.W.; Osterrieder, N. Herpesvirus telomeric repeats facilitate genomic integration into host telomeres and mobilization of viral DNA during reactivation. J. Exp. Med. 2011, 208, 605–615. [Google Scholar] [CrossRef]
- Robinson, C.M.; Hunt, H.D.; Cheng, H.H.; Delany, M.E. Chromosomal integration of an avian oncogenic herpesvirus reveals telomeric preferences and evidence for lymphoma clonality. Herpesviridae 2010, 1, 5. [Google Scholar] [CrossRef] [Green Version]
- Osterrieder, N.; Wallaschek, N.; Kaufer, B.B. Herpesvirus Genome Integration into Telomeric Repeats of Host Cell Chromosomes. Annu. Rev. Virol. 2014, 1, 215–235. [Google Scholar] [CrossRef]
- Morissette, G.; Flamand, L. Herpesviruses and chromosomal integration. J. Virol. 2010, 84, 12100–12109. [Google Scholar] [CrossRef] [Green Version]
- Wallaschek, N.; Sanyal, A.; Pirzer, F.; Gravel, A.; Mori, Y.; Flamand, L.; Kaufer, B.B. The Telomeric Repeats of Human Herpesvirus 6A (HHV-6A) Are Required for Efficient Virus Integration. PLoS Pathog. 2016, 12, e1005666. [Google Scholar] [CrossRef] [Green Version]
- Meyne, J.; Ratliff, R.L.; Moyzis, R.K. Conservation of the Human Telomere Sequence (Ttaggg)N among Vertebrates. Proc. Natl. Acad. Sci. USA 1989, 86, 7049–7053. [Google Scholar] [CrossRef] [Green Version]
- Pratt, W.D.; Morgan, R.W.; Schat, K.A. Characterization of reticuloendotheliosis virus-transformed avian T-lymphoblastoid cell lines infected with Marek’s disease virus. J. Virol. 1992, 66, 7239–7244. [Google Scholar] [CrossRef] [Green Version]
- Viertlboeck, B.C.; Crooijmans, R.P.M.A.; Groenen, M.A.M.; Göbel, T.W.F. Chicken Ig-like receptor B2, a member of a multigene family, is mainly expressed on B lymphocytes, recruits both Src homology 2 domain containing protein tyrosine phosphatase (SHP)-1 and SHP-2, and inhibits proliferation. J. Immunol. 2004, 173, 7385–7393. [Google Scholar] [CrossRef] [Green Version]
- Schat, K.; Purchase, H. Cell-Culture Methods. In A Laboratory Manual for the Isolation and Identification of Avian Pathogens, 4th ed.; American Association of Avian Pathologists: Kennett Square, PA, USA, 1998. [Google Scholar]
- Conradie, A.M.; Bertzbach, L.D.; Trimpert, J.; Patria, J.N.; Murata, S.; Parcells, M.S.; Kaufer, B.B. Distinct polymorphisms in a single herpesvirus gene are capable of enhancing virulence and mediating vaccinal resistance. PLoS Pathog. 2020, 16, e1009104. [Google Scholar] [CrossRef]
- Tischer, B.K.; Smith, G.A.; Osterrieder, N. En passant mutagenesis: A two step markerless red recombination system. Methods Mol. Biol. 2010, 634, 421–430. [Google Scholar] [CrossRef]
- Tischer, B.K.; von Einem, J.; Kaufer, B.; Osterrieder, N. Two-step red-mediated recombination for versatile high-efficiency markerless DNA manipulation in Escherichia coli. Biotechniques 2006, 40, 191–197. [Google Scholar]
- Greco, A.; Fester, N.; Engel, A.T.; Kaufer, B.B. Role of the short telomeric repeat region in Marek’s disease virus replication, genomic integration, and lymphomagenesis. J. Virol. 2014, 88, 14138–14147. [Google Scholar] [CrossRef] [Green Version]
- Bertzbach, L.D.; van Haarlem, D.A.; Härtle, S.; Kaufer, B.B.; Jansen, C.A. Marek’s Disease Virus Infection of Natural Killer Cells. Microorganisms 2019, 7, 588. [Google Scholar] [CrossRef] [Green Version]
- Schumacher, D.; Tischer, B.K.; Fuchs, W.; Osterrieder, N. Reconstitution of Marek’s disease virus serotype 1 (MDV-1) from DNA cloned as a bacterial artificial chromosome and characterization of a glycoprotein B-negative MDV-1 mutant. J. Virol. 2000, 74, 11088–11098. [Google Scholar] [CrossRef] [Green Version]
- Bertzbach, L.D.; Pfaff, F.; Pauker, V.I.; Kheimar, A.M.; Höper, D.; Härtle, S.; Karger, A.; Kaufer, B.B. The Transcriptional Landscape of Marek’s Disease Virus in Primary Chicken B Cells Reveals Novel Splice Variants and Genes. Viruses 2019, 11, 264. [Google Scholar] [CrossRef] [Green Version]
- Jarosinski, K.; Kattenhorn, L.; Kaufer, B.; Ploegh, H.; Osterrieder, N. A herpesvirus ubiquitin-specific protease is critical for efficient T cell lymphoma formation. Proc. Natl. Acad. Sci. USA 2007, 104, 20025–20030. [Google Scholar] [CrossRef] [Green Version]
- Vychodil, T.; Conradie, A.M.; Trimpert, J.; Aswad, A.; Bertzbach, L.D.; Kaufer, B.B. Marek’s Disease Virus Requires Both Copies of the Inverted Repeat Regions for Efficient In Vivo Replication and Pathogenesis. J. Virol. 2021, 95, e01256-20. [Google Scholar] [CrossRef]
- Kheimar, A.; Trimpert, J.; Groenke, N.; Kaufer, B.B. Overexpression of cellular telomerase RNA enhances virus-induced cancer formation. Oncogene 2019, 38, 1778–1786. [Google Scholar] [CrossRef]
- Kaufer, B.B. Detection of integrated herpesvirus genomes by fluorescence in situ hybridization (FISH). Methods Mol. Biol. 2013, 1064, 141–152. [Google Scholar] [CrossRef]
- Schermuly, J.; Greco, A.; Härtle, S.; Osterrieder, N.; Kaufer, B.B.; Kaspers, B. In vitro model for lytic replication, latency, and transformation of an oncogenic alphaherpesvirus. Proc. Natl. Acad. Sci. USA 2015, 112, 7279–7284. [Google Scholar] [CrossRef] [Green Version]
- Arumugaswami, V.; Kumar, P.M.; Konjufca, V.; Dienglewicz, R.L.; Reddy, S.M.; Parcells, M.S. Latency of Marek’s disease virus (MDV) in a reticuloendotheliosis virus-transformed T-cell line. I: Uptake and structure of the latent MDV genome. Avian Dis. 2009, 53, 149–155. [Google Scholar] [CrossRef]
- Arumugaswami, V.; Kumar, P.M.; Konjufca, V.; Dienglewicz, R.L.; Reddy, S.M.; Parcells, M.S. Latency of Marek’s disease virus (MDV) in a reticuloendotheliosis virus-transformed T-cell line. II: Expression of the latent MDV genome. Avian Dis. 2009, 53, 156–165. [Google Scholar] [CrossRef]
- Home, M.; Ohashi, K.; Kodama, H.; Mikami, T. Analysis of marek’s disease tumor-associated surface antigen on MDCC-MSB1-CLO. 18 cells. Int. J. Cancer 1991, 47, 238–243. [Google Scholar] [CrossRef]
- Delecluse, H.-J.; Schüller, S.; Hammerschmidt, W. Latent Marek’s disease virus can be activated from its chromosomally integrated state in herpesvirus-transformed lymphoma cells. EMBO J. 1993, 12, 3277–3286. [Google Scholar] [CrossRef]
- Bencherit, D.; Remy, S.; Le Vern, Y.; Vychodil, T.; Bertzbach, L.D.; Kaufer, B.B.; Denesvre, C.; Trapp-Fragnet, L. Induction of DNA damages upon Marek’s disease virus infection: Implication in viral replication and pathogenesis. J. Virol. 2017, 91, e01658-17. [Google Scholar] [CrossRef] [Green Version]
- Wallaschek, N.; Gravel, A.; Flamand, L.; Kaufer, B.B. The putative U94 integrase is dispensable for human herpesvirus 6 (HHV-6) chromosomal integration. J. Gen. Virol. 2016, 97, 1899–1903. [Google Scholar] [CrossRef] [Green Version]
- Wight, D.J.; Wallaschek, N.; Sanyal, A.; Weller, S.K.; Flamand, L.; Kaufer, B.B. Viral Proteins U41 and U70 of Human Herpesvirus 6A Are Dispensable for Telomere Integration. Viruses 2018, 10, 656. [Google Scholar] [CrossRef] [Green Version]
- Kheimar, A.; Previdelli, R.L.; Wight, D.J.; Kaufer, B.B. Telomeres and Telomerase: Role in Marek’s Disease Virus Pathogenesis, Integration and Tumorigenesis. Viruses 2017, 9, 173. [Google Scholar] [CrossRef] [Green Version]
- Previdelli, R.L.; Bertzbach, L.D.; Wight, D.J.; Vychodil, T.; You, Y.; Arndt, S.; Kaufer, B.B. The Role of Marek’s Disease Virus UL12 and UL29 in DNA Recombination and the Virus Lifecycle. Viruses 2019, 11, 111. [Google Scholar] [CrossRef] [Green Version]
- Fragnet, L.; Blasco, M.A.; Klapper, W.; Rasschaert, D. The RNA subunit of telomerase is encoded by Marek’s disease virus. J. Virol. 2003, 77, 5985–5996. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Construct Name | Sequence (5′ → 3′) | |
|---|---|---|
| eGFP in mini-F | for | GGTGACACGCGCGGCCTCGAACACAGCTG CAGGCCATGGTGAGCAAGGGCGAGG |
| rev | CGTCGACCCGGGTACCTCTAGATCCGCTAGC GCTTTACTTGTACAGCTCGTCCATGCC | |
| PCR-based probe 1 | for | ATTACCTGGGGACAGCATGA |
| rev | CACATCGTTTTGCCATGTTG | |
| PCR-based probe 2 | for | CCGCTTCCTATCTCAGCAGA |
| rev | TCAAGCGCTTTCTCATAGGG | |
| PCR-based probe 3 | for | GAGCCAACAAATCCCCTGA |
| rev | GAGGTTGGTGCTGGAATGTT | |
| PCR-based probe 4 | for | CTGTTCATGTCGGAGGTCTG |
| rev | GAGGGAAGCTACGGTTCAAG | |
| PCR-based probe 5 | for | CCGACAATTATTGCCCCGTA |
| rev | ATCTGGAAACATGTCCGACG | |
| ICP4 | for | CGTGTTTTCCGGCATGTG |
| rev | TCCCATACCAATCCTCATCCA | |
| probe | FAM-CCCCCACCAGGTGCAGGCA-TAM | |
| iNOS | for | GAGTGGTTTAAGGAGTTGGATCTGA |
| rev | TTCCAGACCTCCCACCTCAA | |
| probe | FAM-CTCTGCCTGCTGTTGCCAACATGC-TAM | |
| UL36 | for | GACAAGCTACTACAAATTGCA |
| rev | GACGTCGATTTATCTCTTAACA | |
| probe | FAM-AAGAACTACATCGAACGCACCCATGCTAGC-TAMRA | |
| pp38 | for | GAGCTAACCGGAGAGGGAGA |
| rev | CGCATACCGACTTTCGTCAA | |
| probe | FAM-CTCCCACTGTGACAGCC-TAMRA | |
| vTR | for | CCTAATCGGAGGTATTGATGGTACTG |
| rev | CCCTAGCCCGCTGAAAGTC | |
| probe | FAM-CCCTCCGCCCGCTGTTTACTCG-TAMRA | |
| GAPDH | for | GGTGCTAAGCGTGTTATCATCTCA |
| rev | CATGGTTGACACCCATCACAA | |
| probe | FAM-TGTGCCAACCCCCAAT-TAMRA | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
You, Y.; Vychodil, T.; Aimola, G.; Previdelli, R.L.; Göbel, T.W.; Bertzbach, L.D.; Kaufer, B.B. A Cell Culture System to Investigate Marek’s Disease Virus Integration into Host Chromosomes. Microorganisms 2021, 9, 2489. https://doi.org/10.3390/microorganisms9122489
You Y, Vychodil T, Aimola G, Previdelli RL, Göbel TW, Bertzbach LD, Kaufer BB. A Cell Culture System to Investigate Marek’s Disease Virus Integration into Host Chromosomes. Microorganisms. 2021; 9(12):2489. https://doi.org/10.3390/microorganisms9122489
Chicago/Turabian StyleYou, Yu, Tereza Vychodil, Giulia Aimola, Renato L. Previdelli, Thomas W. Göbel, Luca D. Bertzbach, and Benedikt B. Kaufer. 2021. "A Cell Culture System to Investigate Marek’s Disease Virus Integration into Host Chromosomes" Microorganisms 9, no. 12: 2489. https://doi.org/10.3390/microorganisms9122489