
CRISPR-Mediated Gene Activation (CRISPRa) of pp38/pp24 Orchestrates Events Triggering Lytic Infection in Marek’s Disease Virus-Transformed Cell Lines


Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
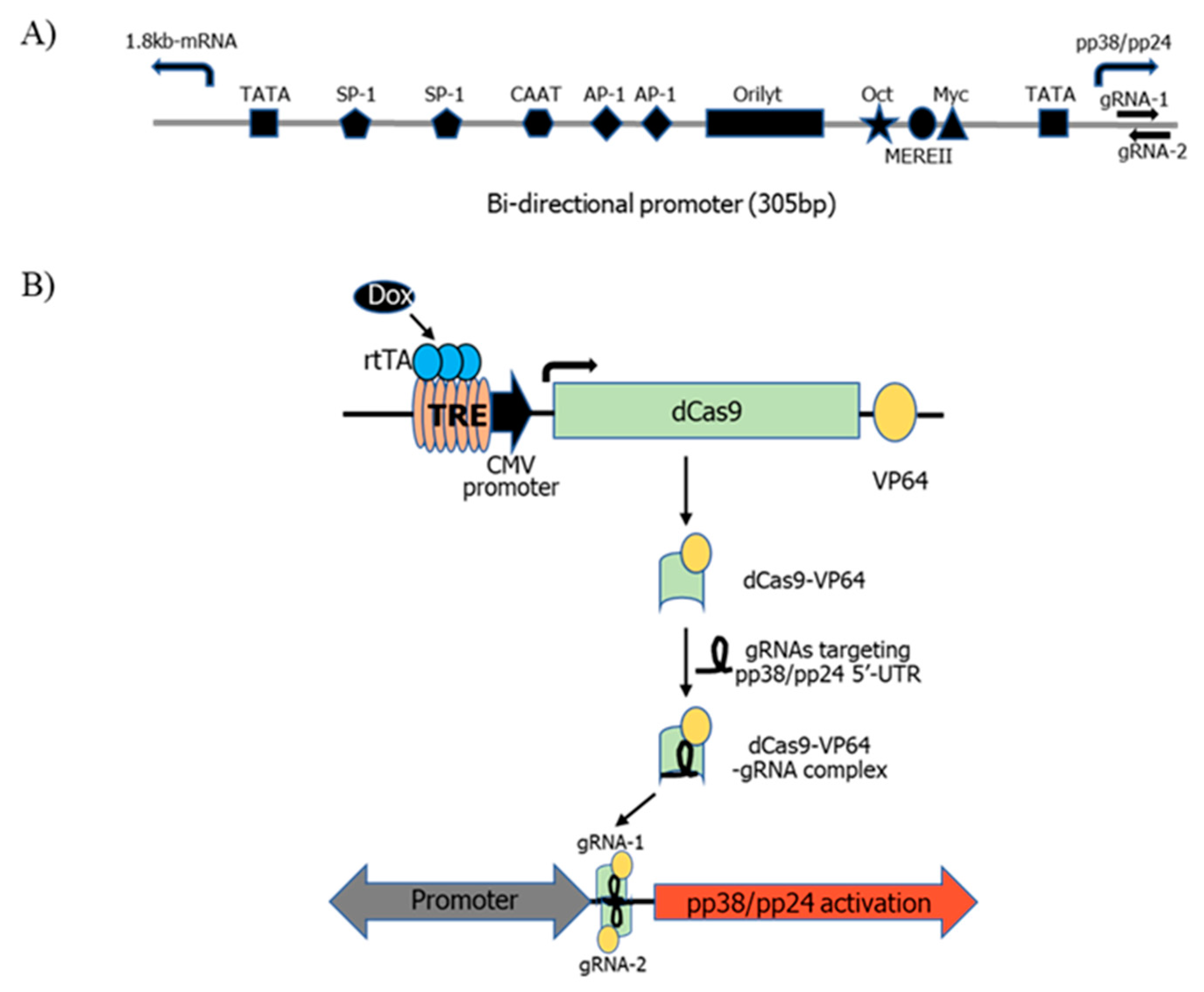
2.2. Generation of dCas9-VP64 Stable Cell Line
2.3. gRNA Design
2.4. Transfection of gRNAs into 4523T-dCas9-VP64 Cells
2.5. FACS Analysis
2.6. RNA Extraction and RT-qPCR
2.7. Reactivation of MDV from pp38/pp24 Activated 4523T Cells
2.8. Immunofluorescence Assay (IFA)
2.9. Caspase Assay Using Caspase-3/7 Green Reagent
2.10. DNA Fragmentation Assay
3. Results
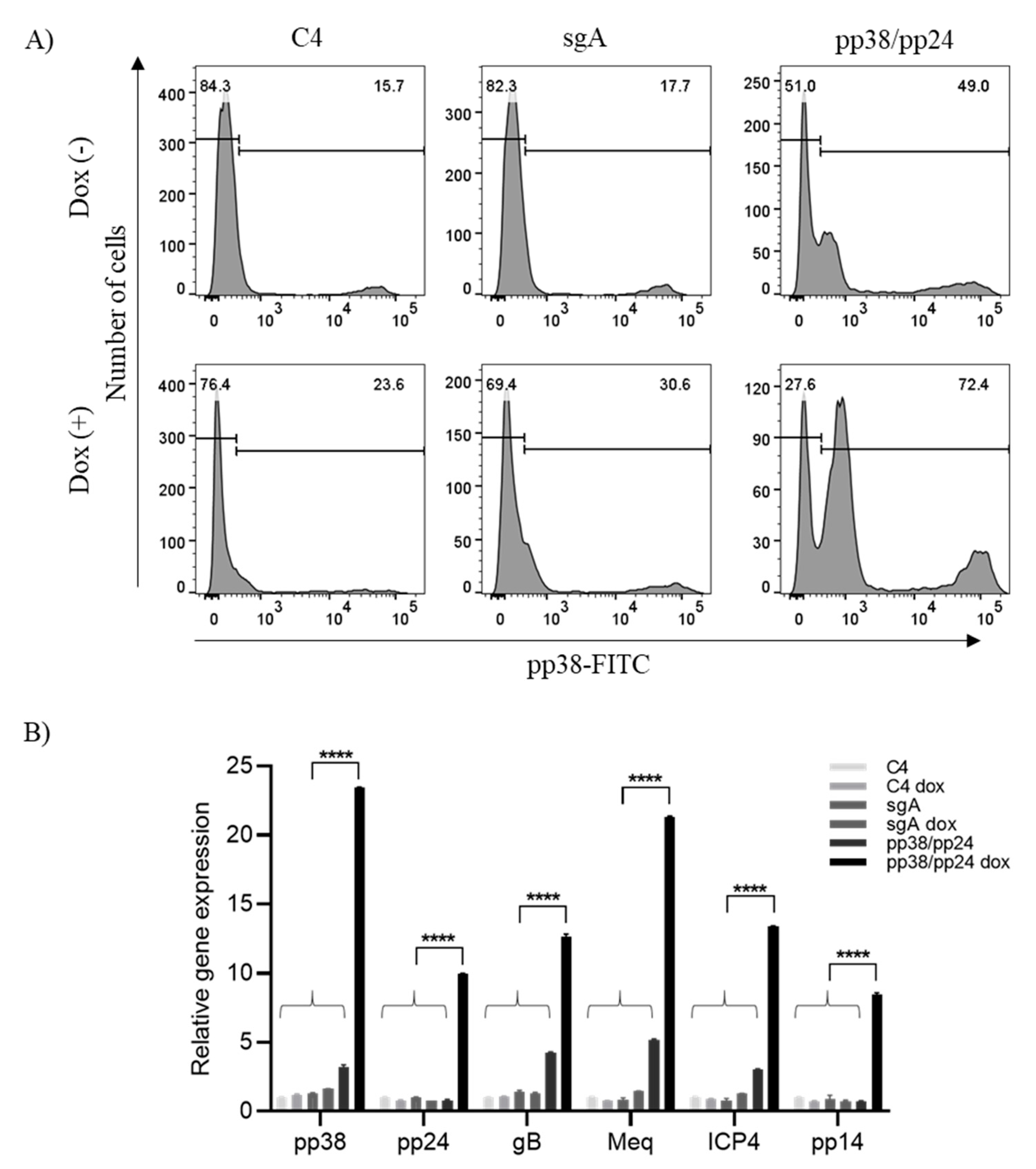
3.1. pp38/pp24 Activation by CRISPRa
3.2. pp38/pp24 Activation Induces Overexpression of Other MDV Lytic Genes
3.3. Differential Expression of Host Genes in Response to pp38/pp24 Overexpression
3.4. Enforced Expression of pp38/pp24 Enhances MDV Reactivation
3.5. Cytolysis Induced by pp38/pp24 Expression
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Davison, A.J. Herpesvirus systematics. Vet. Microbiol. 2010, 143, 52–69. [Google Scholar] [CrossRef] [Green Version]
- Nicoll, M.P.; Proença, J.T.; Efstathiou, S. The molecular basis of herpes simplex virus latency. FEMS Microbiol. Rev. 2012, 36, 684–705. [Google Scholar] [CrossRef]
- Brown, A.C.; Nair, V.; Allday, M.J. Epigenetic regulation of the latency-associated region of marek’s disease virus in tumor-derived t-cell lines and primary lymphoma. J. Virol. 2012, 86, 1683–1695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mwangi, W.N.; Vasoya, D.; Kgosana, L.B.; Watson, M.; Nair, V. Differentially expressed genes during spontaneous lytic switch of marek’s disease virus in lymphoblastoid cell lines determined by global gene expression profiling. J. Gen. Virol. 2017, 98, 779–790. [Google Scholar] [CrossRef]
- Halford, W.P.; Kemp, C.D.; Isler, J.A.; Davido, D.J.; Schaffer, P.A. Icp0, icp4, or vp16 expressed from adenovirus vectors induces reactivation of latent herpes simplex virus type 1 in primary cultures of latently infected trigeminal ganglion cells. J. Virol. 2001, 75, 6143–6153. [Google Scholar] [CrossRef] [Green Version]
- Perng, G.C.; Slanina, S.M.; Yukht, A.; Ghiasi, H.; Nesburn, A.B.; Wechsler, S.L. The latency-associated transcript gene enhances establishment of herpes simplex virus type 1 latency in rabbits. J. Virol. 2000, 74, 1885–1891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Q.; Anderson, A.S.; Morgan, R.W. Marek’s disease virus (mdv) icp4, pp38, and meq genes are involved in the maintenance of transformation of mdcc-msb1 mdv-transformed lymphoblastoid cells. J. Virol. 1996, 70, 1125–1131. [Google Scholar] [CrossRef] [Green Version]
- Parcells, M.S.; Dienglewicz, R.L.; Anderson, A.S.; Morgan, R.W. Recombinant marek’s disease virus (mdv)-derived lymphoblastoid cell lines: Regulation of a marker gene within the context of the mdv genome. J. Virol. 1999, 73, 1362–1373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lupiani, B.M.; Liao, Y.; Jin, D.; Izumiya, Y.; Reddy, S.M. Marek’s disease virus. In Avian Virology: Current Research and Future Trends; Samal, S.K., Ed.; Caister Academic Press: Norfolk, UK, 2019; pp. 345–364. [Google Scholar]
- Chen, X.B.; Sondermeijer, P.J.; Velicer, L.F. Identification of a unique marek’s disease virus gene which encodes a 38-kilodalton phosphoprotein and is expressed in both lytically infected cells and latently infected lymphoblastoid tumor cells. J. Virol. 1992, 66, 85–94. [Google Scholar] [CrossRef] [Green Version]
- Cui, Z.Z.; Lee, L.F.; Liu, J.L.; Kung, H.J. Structural analysis and transcriptional mapping of the marek’s disease virus gene encoding pp38, an antigen associated with transformed cells. J. Virol. 1991, 65, 6509–6515. [Google Scholar] [CrossRef] [Green Version]
- Hong, Y.; Coussens, P.M. Identification of an immediate-early gene in the marek’s disease virus long internal repeat region which encodes a unique 14-kilodalton polypeptide. J. Virol. 1994, 68, 3593–3603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, J.; Cui, Z.; Lee, L.F. Marek’s disease virus unique genes pp38 and pp24 are essential for transactivating the bi-directional promoters for the 1.8 kb mrna transcripts. Virus Genes 2007, 35, 643–650. [Google Scholar] [CrossRef]
- Gimeno, I.M.; Witter, R.L.; Hunt, H.D.; Reddy, S.M.; Lee, L.F.; Silva, R.F. The pp38 gene of marek’s disease virus (mdv) is necessary for cytolytic infection of b cells and maintenance of the transformed state but not for cytolytic infection of the feather follicle epithelium and horizontal spread of mdv. J. Virol. 2005, 79, 4545–4549. [Google Scholar] [CrossRef] [Green Version]
- Bhaya, D.; Davison, M.; Barrangou, R. Crispr-cas systems in bacteria and archaea: Versatile small rnas for adaptive defense and regulation. Annu. Rev. Genet. 2011, 45, 273–297. [Google Scholar] [CrossRef] [Green Version]
- Knott, G.J.; Doudna, J.A. Crispr-cas guides the future of genetic engineering. Science 2018, 361, 866–869. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F. Development of Crispr-Cas Systems for Genome Editing and Beyond; Published online by Cambridge University Press: Cambridge, UK, 2019. [Google Scholar]
- Dong, C.; Fontana, J.; Patel, A.; Carothers, J.M.; Zalatan, J.G. Synthetic crispr-cas gene activators for transcriptional reprogramming in bacteria. Nat. Commun. 2018, 9, 2489. [Google Scholar] [CrossRef] [Green Version]
- Forstneric, V.; Oven, I.; Ogorevc, J.; Lainscek, D.; Praznik, A.; Lebar, T.; Jerala, R.; Horvat, S. Crispra-mediated foxp3 gene upregulation in mammalian cells. Cell Biosci. 2019, 9, 93. [Google Scholar] [CrossRef] [Green Version]
- Elbasani, E.; Falasco, F.; Gramolelli, S.; Nurminen, V.; Gunther, T.; Weltner, J.; Balboa, D.; Grundhoff, A.; Otonkoski, T.; Ojala, P.M. Kaposi’s sarcoma-associated herpesvirus reactivation by targeting of a dcas9-based transcription activator to the orf50 promoter. Viruses 2020, 12, 952. [Google Scholar] [CrossRef] [PubMed]
- Mwangi, W.N.; Smith, L.P.; Baigent, S.J.; Beal, R.K.; Nair, V.; Smith, A.L. Clonal structure of rapid-onset mdv-driven cd4+ lymphomas and responding cd8+ t cells. PLoS Pathog. 2011, 7, e1001337. [Google Scholar] [CrossRef] [Green Version]
- Baigent, S.J.; Ross, L.J.; Davison, T.F. A flow cytometric method for identifying marek’s disease virus pp38 expression in lymphocyte subpopulations. Avian Pathol. 1996, 25, 255–267. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Tang, N.; Sadigh, Y.; Baigent, S.; Shen, Z.; Nair, V.; Yao, Y. Application of crispr/cas9 gene editing system on mdv-1 genome for the study of gene function. Viruses 2018, 10, 279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, X.; Tan, C.; Wang, F.; Wang, Y.; Zhou, R.; Cui, D.; You, W.; Zhao, H.; Ren, J.; Feng, B. Knock-in of large reporter genes in human cells via crispr/cas9-induced homology-dependent and independent DNA repair. Nucleic Acids Res. 2016, 44, e85. [Google Scholar] [CrossRef] [Green Version]
- Baigent, S.J.; Petherbridge, L.J.; Howes, K.; Smith, L.P.; Currie, R.J.; Nair, V.K. Absolute quantitation of marek’s disease virus genome copy number in chicken feather and lymphocyte samples using real-time pcr. J. Virol. Methods 2005, 123, 53–64. [Google Scholar] [CrossRef]
- Zhang, Y.; Luo, J.; Tang, N.; Teng, M.; Reddy, V.; Moffat, K.; Shen, Z.; Nair, V.; Yao, Y. Targeted editing of the pp38 gene in marek’s disease virus-transformed cell lines using crispr/cas9 system. Viruses 2019, 11, 391. [Google Scholar] [CrossRef] [Green Version]
- Barrow, A.D.; Burgess, S.C.; Howes, K.; Nair, V.K. Monocytosis is associated with the onset of leukocyte and viral infiltration of the brain in chickens infected with the very virulent marek’s disease virus strain c12/130. Avian Pathol. 2003, 32, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, L.A.; Horlbeck, M.A.; Adamson, B.; Villalta, J.E.; Chen, Y.; Whitehead, E.H.; Guimaraes, C.; Panning, B.; Ploegh, H.L.; Bassik, M.C.; et al. Genome-scale crispr-mediated control of gene repression and activation. Cell 2014, 159, 647–661. [Google Scholar] [CrossRef] [Green Version]
- Sanson, K.R.; Hanna, R.E.; Hegde, M.; Donovan, K.F.; Strand, C.; Sullender, M.E.; Vaimberg, E.W.; Goodale, A.; Root, D.E.; Piccioni, F.; et al. Optimized libraries for crispr-cas9 genetic screens with multiple modalities. Nat. Commun. 2018, 9, 5416. [Google Scholar] [CrossRef] [Green Version]
- Cho, K.O.; Ohashi, K.; Onuma, M. Electron microscopic and immunohistochemical localization of marek’s disease (md) herpesvirus particles in md skin lymphomas. Vet. Pathol. 1999, 36, 314–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kennedy, P.G.; Rovnak, J.; Badani, H.; Cohrs, R.J. A comparison of herpes simplex virus type 1 and varicella-zoster virus latency and reactivation. J. Gen. Virol. 2015, 96, 1581–1602. [Google Scholar] [CrossRef]
- Bradley, G.; Hayashi, M.; Lancz, G.; Tanaka, A.; Nonoyama, M. Structure of the marek’s disease virus bamhi-h gene family: Genes of putative importance for tumor induction. J. Virol. 1989, 63, 2534–2542. [Google Scholar] [CrossRef] [Green Version]
- Zhu, G.S.; Iwata, A.; Gong, M.; Ueda, S.; Hirai, K. Marek’s disease virus type 1-specific phosphorylated proteins pp38 and pp24 with common amino acid termini are encoded from the opposite junction regions between the long unique and inverted repeat sequences of viral genome. Virology 1994, 200, 816–820. [Google Scholar] [CrossRef]
- Shigekane, H.; Kawaguchi, Y.; Shirakata, M.; Sakaguchi, M.; Hirai, K. The bi-directional transcriptional promoters for the latency-relating transcripts of the pp38/pp24 mrnas and the 1.8 kb-mrna in the long inverted repeats of marek’s disease virus serotype 1 DNA are regulated by common promoter-specific enhancers. Arch. Virol. 1999, 144, 1893–1907. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Cui, Z.; Lee, L.F.; Cui, X.; Reddy, S.M. The role of pp38 in regulation of marek’s disease virus bi-directional promoter between pp38 and 1.8-kb mrna. Virus Genes 2006, 32, 193–201. [Google Scholar] [CrossRef]
- Jiabo, D.; Zhizhong, C.; Shijin, J.; Sanjay, R. The enhancement effect of pp38 gene product on the activity of its upstream bi-directional promoter in marek’s disease virus. Sci. China C Life Sci. 2006, 49, 53–62. [Google Scholar]
- Ding, J.; Cui, Z.; Jiang, S.; Li, Y. Study on the structure of heteropolymer pp38/pp24 and its enhancement on the bi-directional promoter upstream of pp38 gene in marek’s disease virus. Sci. China C Life Sci. 2008, 51, 821–826. [Google Scholar] [CrossRef]
- Fujimoto, Y.; Tedder, T.F. Cd83: A regulatory molecule of the immune system with great potential for therapeutic application. J. Med. Dent. Sci. 2006, 53, 85–91. [Google Scholar] [PubMed]
- Kaiser, P.; Underwood, G.; Davison, F. Differential cytokine responses following marek’s disease virus infection of chickens differing in resistance to marek’s disease. J. Virol. 2003, 77, 762–768. [Google Scholar] [CrossRef] [Green Version]
- Xing, Z.; Schat, K.A. Expression of cytokine genes in marek’s disease virus-infected chickens and chicken embryo fibroblast cultures. Immunology 2000, 100, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Degterev, A.; Huang, Z.; Boyce, M.; Li, Y.; Jagtap, P.; Mizushima, N.; Cuny, G.D.; Mitchison, T.J.; Moskowitz, M.A.; Yuan, J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat. Chem. Biol. 2005, 1, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Dhuriya, Y.K.; Sharma, D. Necroptosis: A regulated inflammatory mode of cell death. J. Neuroinflam. 2018, 15, 199. [Google Scholar] [CrossRef] [Green Version]
- Giampietri, C.; Starace, D.; Petrungaro, S.; Filippini, A.; Ziparo, E. Necroptosis: Molecular signalling and translational implications. Int. J. Cell Biol. 2014, 2014, 490275. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Gene | Primer/Probe | Sequence (5′-3′) |
|---|---|---|
| pp38 [25] | pp38-F | GAAAACAGAAGCGGAATGCG |
| pp38-R | CGATCCAAAGCGCTCATCTC | |
| pp38-probe | CCCCGCATTTCTCGCCGTCCTC | |
| Meq [25] | Meq-F | AGCCGGAGAGGC TTTATGC |
| Meq-R | GGCCCGAATACA AGGAATCC | |
| Meq-probe | TTACCGAGGAT CCCGAAC | |
| ICP4 [25] | ICP4-F | CGCCACACGAGAACACAATG |
| ICP4-R | GGTTGGAGTAGAGCTGCAACTGT | |
| ICP4-probe | CGGCCCAGTACAGCCTGCGG | |
| gB [25] | gB-F | GGTTCAACCGTGATCCGTCTA |
| gB-R | CGATTCCTTCACCCCACT | |
| gB-probe | ACCGCCGCGAAAATGTC | |
| pp24 | pp24-F | TTCCCGAAAACGCATAGAAG |
| pp24-R | TCAGAAATCCCGCAAGAAAG | |
| pp14 | pp14-F | TGGCTAAATGCGACTTCGGT |
| pp14-R | CGTGCAACAACGCCCAAATA | |
| RIPK3 | RIPK3-F | AAGGACGCCTCAACCACATC |
| RIPK3-R | GCTGGTCACTGTGGGGTAAG | |
| IFN-γ | IFN-γ-F | AAGGACGCCTCAACCACATC |
| IFN-γ-R | GCTGGTCACTGTGGGGTAAG | |
| CD1c [4] | CD1c-F | AGCAGGCTGGTGCAGATGTA |
| CD1c-R | TGGCCCTCGTAAGCAATGTC | |
| CD83 [4] | CD83-F | CACCCTGTGCAATGTTTGGA |
| CD83-R | CAAAGCATGTCACAGCAACATCT | |
| CCR5 [4] | CCR5-F | CGGTTTAGCGTTACTCTTGATGTAATAA |
| CCR5-R | TGGACGTGTTCAGCTGATGAC | |
| TP63 [4] | TP63-F | CCTCTCCATGCCTTCAACGT |
| TP63-R | CGTGAAATAATCCACACAGGATGA | |
| ICOS [4] | ICOS-F | ACACTGCTGATTCTTATTCCTTAAGTGA |
| ICOS-R | TACATTGCCACTTGAAAGAAACCTA | |
| GAPDH | GAPDH-F | AGAACATCATCCCAGCGTCC |
| GAPDH-R | CGGCAGGTCAGGTCAACA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roy, P.; Moffat, K.; Nair, V.; Yao, Y. CRISPR-Mediated Gene Activation (CRISPRa) of pp38/pp24 Orchestrates Events Triggering Lytic Infection in Marek’s Disease Virus-Transformed Cell Lines. Microorganisms 2021, 9, 1681. https://doi.org/10.3390/microorganisms9081681
Roy P, Moffat K, Nair V, Yao Y. CRISPR-Mediated Gene Activation (CRISPRa) of pp38/pp24 Orchestrates Events Triggering Lytic Infection in Marek’s Disease Virus-Transformed Cell Lines. Microorganisms. 2021; 9(8):1681. https://doi.org/10.3390/microorganisms9081681
Chicago/Turabian StyleRoy, Poornima, Katy Moffat, Venugopal Nair, and Yongxiu Yao. 2021. "CRISPR-Mediated Gene Activation (CRISPRa) of pp38/pp24 Orchestrates Events Triggering Lytic Infection in Marek’s Disease Virus-Transformed Cell Lines" Microorganisms 9, no. 8: 1681. https://doi.org/10.3390/microorganisms9081681