Differential Genotyping of Mycobacterium avium Complex and Its Implications in Clinical and Environmental Epidemiology


Abstract
:1. Introduction
2. Significance of Molecular Genotyping Methods Applied to MAC
3. PFGE for MAC and Their Implications in Epidemiological Studies
3.1. Investigation of MAC for Co-Infection and Relapse
3.2. Relationship among Environment, Animal, and Human Isolates
3.3. Comparison of PFGE with Other Genotyping Methods
4. VNTR for MAC and Their Implications in Epidemiological Studies
4.1. Identification of VNTR Loci and Development of VNTR Techniques
4.2. Application of VNTR Method for Clinical and Epidemiological Investigations
4.3. Geographical Relationship between Bacterial Strains Belonging to MAC
4.4. Comparison of VNTR Using Other Genetic Typing Methods

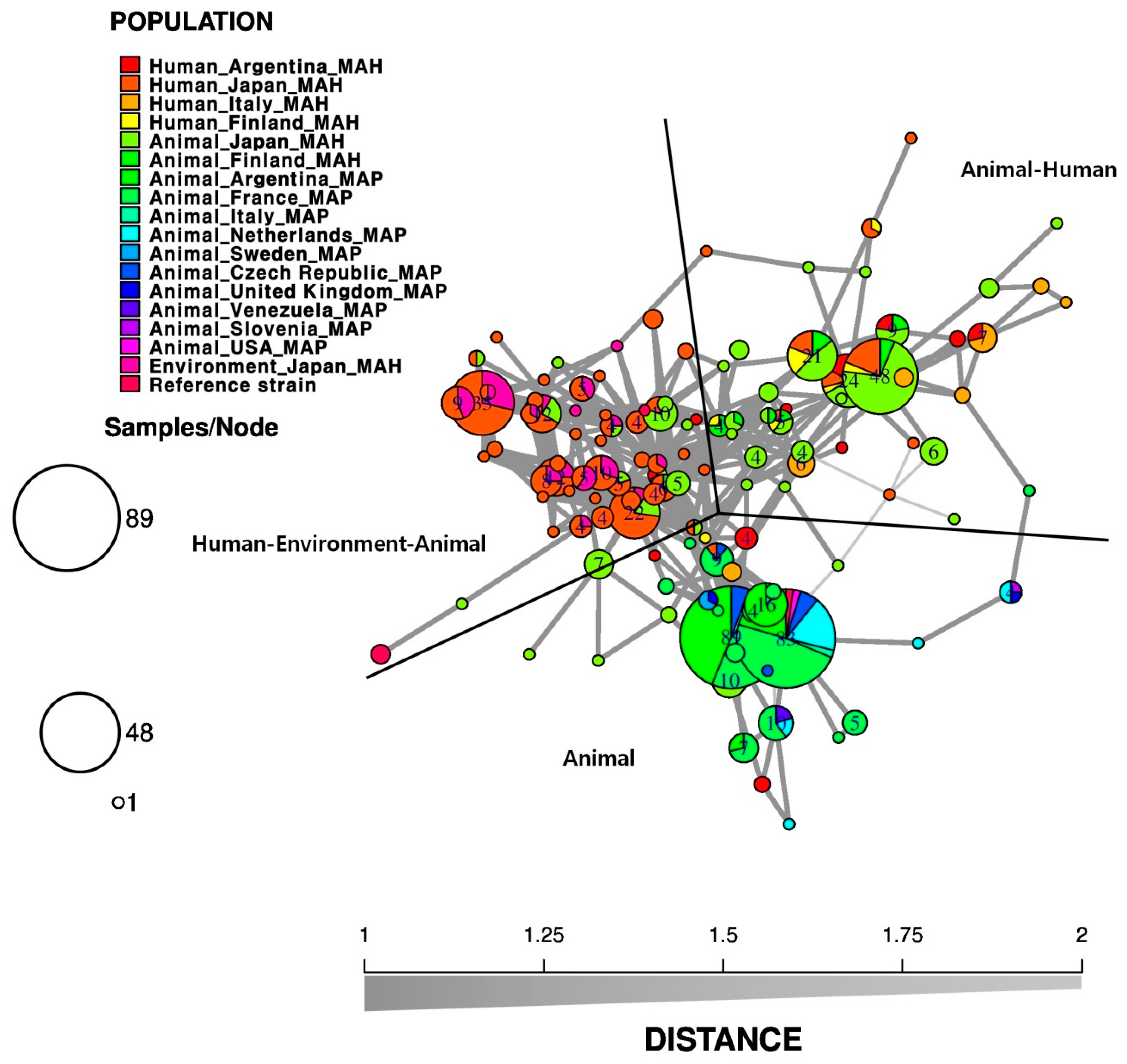{kind=link}
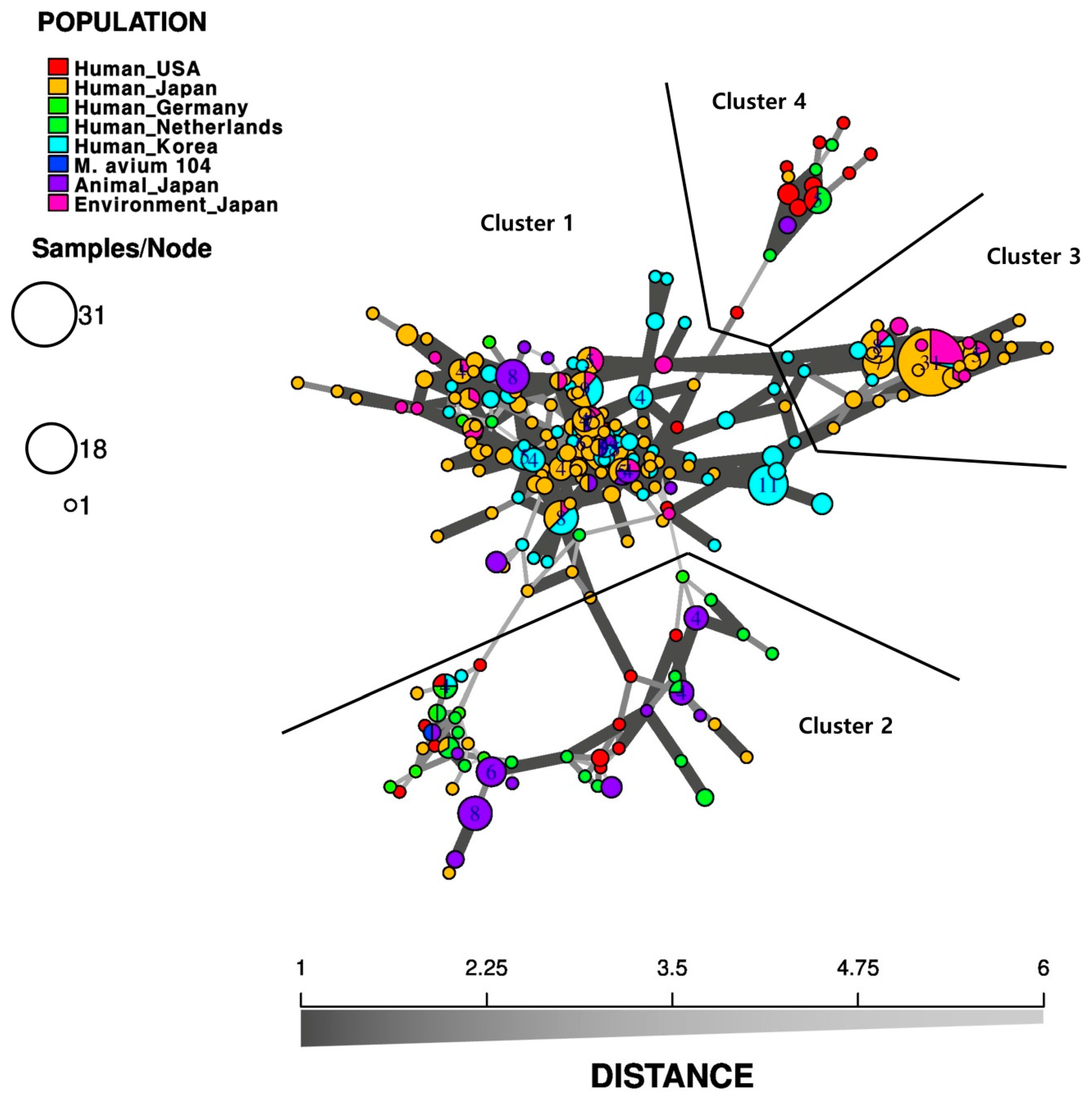{kind=link}
| Strain | Origin | Sample No. | VNTR Type | Loci No. | HGDI 1 | Reference | |
|---|---|---|---|---|---|---|---|
| Source | Country | ||||||
| M. avium | AIDS patients | France | 82 | 30 types | 8 TRs | 0.889 | [69] |
| Patients with/without pulmonary disease | Japan | 40 | 27 types | 16 MATRs | 0.945 | [76] | |
| HIV-negative patients with pulmonary MAC infection | Japan | 70 | 56 MATR, 27 TR types | 15 MATRs, 8 TRs | MATR: 0.990 TR: 0.949 | [68] | |
| Patients | Poland | 33 | 21 types | 8 TRs | 0.945 | [89] | |
| Patients with pulmonary MAC infection and residential soil samples | Japan | 88 | 78 types | 15 MATR | 0.997 | [80] | |
| Patients with pulmonary MAC infection | Japan | 310 | 93 types | 15 MATR | 0.987 | [81] | |
| Patients with pulmonary NTM infection | China | 41 | 29 types | 13 MATRs | 0.993 | [91] | |
| MAA | Bird, poultry, pig, wild animal, cat, bovine, goat | France | 31 | 8 types | 8 TRs | 0.723 | [85] |
| Diseased cattle, slaughtered pigs | Germany | 27 | 19 types | 6 MIRUs, 2 VNTRs, 6 TRs, and 1 RD | 0.966 | [92] | |
| Wild and domestic mammals, reptiles and birds | Hungary | 135 | 16 types | 4 MIRUs, 3 TRs, and 1 MATR | 0.845 | [93] | |
| MAH | Patients (HIV positive and negative), pig, bovine, kangaroo, wild animal, soil sample | France | 82 | 23 types | 8 TRs | 0.807 | [85] |
| Diseased cattle, slaughtered pigs | Germany | 16 | 15 types | 6 MIRUs, 2 VNTRs, 6 TRs, and 1 RD | 0.992 | [92] | |
| Patients | Italy | 47 | 8 types | 8 TRs | 0.862 | [94] | |
| Patients with pulmonary MAC infection (HIV positive and negative) | Japan | 64 | 55 types | 15 MATRs | 0.995 | [78] | |
| Wild and domestic mammals, reptiles and birds | Hungary | 84 | 33 types | 4 MIRUs, 3 TRs, and 1 MATR | 0.966 | [93] | |
| Patients | Argentina | 26 | 16 types | 8 TRs | 0.93 | [95] | |
| Patients | Italy | 23 | 8 types | 8 TRs | 0.870 | [79] | |
| Slaughtered cattle | Switzerland | 26 | 14 types | 15 MATRs, 5 TRs | 0.972 | [87] | |
| Slaughtered bovine with abnormal pulmonary case | Japan | 12 | 9 types | 7 TRs, 14 MATRs | 0.955 | [88] | |
| Humans, pigs and bathroom environments | Japan | 258 | 150 types | 7 TRs, 15 MATRs | 0.987 | [96] | |
| MAP | Bovine, goat, ovine, cervine, and leporine | Argentina, Czech Republic, France, Italy, Netherlands, Slovenia, Sweden, United Kingdom, USA, and Venezuela | 183 | 21 types | 8 TRs | 0.751 | [69] |
| Cattle, sheep, goat, wild boar, red deer, red fox, buffalo, mouflon, swine | Denmark, France, Germany, Hungary, Italy, Netherlands, and Slovakia | 515 | 15 types | 4 MIRU, 3 TRs | 0.598 | [97] | |
| Cattle | Argentina | 61 | 5 types | 8 TRs | 0.6984 | [95] | |
| Cattle | Korea | 27 | 4 types | 8 TRs | 0.567 | [90] | |
| MAS | Wood pigeon | France | 4 | 1 type | 8 TRs | 0 | [85] |
| Wild and domestic mammals, reptiles and birds | Hungary | 62 | 5 types | 4 MIRUs, 3 TRs, and 1 MATR | 0.172 | [93] | |
| M. intracellulare | Patients | France | 62 | 44 types | 7 MIRUs | 0.98 | [74] |
| HIV-negative patients with pulmonary disease | Japan | 74 | 50 types | 16 VNTRs | 0.988 | [73] | |
| Patients with pulmonary MAC infection and residential soil samples | Japan | 55 | 53 types | 16 VNTRs | 0.999 | [80] | |
| patients with nodular bronchiectasis | USA | 176 | 42 types | 7 MIRUs | 0.978 | [63] | |
| Patients with pulmonary MAC infection | Japan | 74 | 27 types | 16 VNTRs | 0.970 | [81] | |
| Patients with pulmonary MAC infection | Japan, Korea, Netherlands, and USA | 116 | 82 types | 16 VNTRs | 0.988 | [86] | |
| HIV-negative patients with pulmonary disease | China | 77 | 69 types | 1 MIRU, 7 VNTRs | 0.997 | [77] | |
| Patients with pulmonary NTM infection | China | 132 | 88 types | 16 VNTRs | 0.995 | [91] | |
| M. avium | MAA | MAH | MAP | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sources | Human | Human | Human | Human/Environment | Animal | Animal | Animal | Human/Animal | Human | Human | Animal | Animal | Animal | Human | Human | Environment | Animal | Human/Animal | Animal | Animal | Animal | Animal | |
| Country | Japan | France | Japan | Japan | Hungary | Germany | France | France | Italy | 4 countries 3 | Switzerland | Japan | Germany | Argentina | Japan | Japan | Japan | Finland | 10 Countries 4 | 7 Countries 5 | Korea | Argentina | |
| Sample No. | 70 | 82 | 40 | 310 | 281 | 27 | 31 | 82 | 22 | 262 | 26 | 12 | 16 | 22 | 146 | 37 | 75 | 33 | 183 | 515 | 27 | 61 | |
| Reference | [68] | [69] | [76] | [81] | [93] | [92] | [85] | [85] | [79] | [86] | [87] | [88] | [92] | [95] | [96] | [96] | [96] | [54] | [69] | [97] | [90] | [95] | |
| h2 | MIRU1 | − | − | − | − | 0.250 | − | − | − | − | − | − | − | − | − | − | − | − | − | − | 0.252 | − | − |
| MIRU2 | − | − | − | − | 0.224 | 0.43 | − | − | − | − | − | − | 0.52 | − | − | − | − | − | − | 0.564 | − | − | |
| MIRU3 | − | − | − | − | 0.710 | 0.67 | − | − | − | − | − | − | 0.7 | − | − | − | − | − | − | 0.064 | − | − | |
| MIRU4 | − | − | − | − | 0.285 | 0.07 | − | − | − | − | − | − | 0 | − | − | − | − | − | − | 0 | − | − | |
| MIRU5 | − | − | − | − | − | 0.07 | − | − | − | − | − | − | 0.42 | − | − | − | − | − | − | − | − | − | |
| MIRU6 | − | − | − | − | − | 0.58 | − | − | − | − | − | − | 0.57 | − | − | − | − | − | − | − | − | − | |
| MIRU7 | − | − | − | − | − | 0.22 | − | − | − | − | − | − | 0.5 | − | − | − | − | − | − | − | − | − | |
| TR 25 | 0.512 | 0.33 | − | − | 0.514 | 0.36 | 0 | 0.26 | 0.46 | − | 0.45 | 0.53 | 0.5 | 0.5844 | 0.517 | 0.47 | 0.593 | 0.55 | 0.07 | 0.094 | 0.446 | 0 | |
| TR 32 | 0.359 | 0.3 | − | − | 0.471 | − | 0.03 | 0.17 | 0.21 | − | 0 | 0.21 | − | 0 | 0.484 | 0.497 | 0.101 | 0.19 | 0.59 | 0.032 | 0 | 0 | |
| TR 47 | 0.069 | 0.35 | − | − | − | 0.51 | 0 | 0.20 | 0.20 | − | 0.23 | 0 | 0.32 | 0.3247 | 0.116 | 0 | 0.444 | 0.43 | 0.05 | − | 0 | 0 | |
| TR 10 | 0.459 | 0.15 | − | − | − | − | 0.21 | 0.13 | 0 | − | − | 0.50 | − | 0.2554 | − | − | − | 0 | 0.18 | − | 0.036 | 0.0645 | |
| TR 259 | − | − | − | − | 0.589 | − | − | − | − | − | − | − | − | − | − | − | − | − | − | 0.063 | − | − | |
| TR 3 | − | 0 | − | − | − | 0 | 0 | 0 | 0 | − | 0 | − | 0 | 0.0909 | − | − | − | 0 | 0.005 | − | 0 | 0 | |
| TR 7 | 0 | 0.04 | − | − | − | 0 | 0 | 0 | 0 | − | 0 | 0 | 0 | 0.4848 | 0 | 0 | 0 | 0 | 0.19 | − | 0 | 0.6980 | |
| TR 8 | − | − | − | − | − | 0.49 | − | − | − | − | − | − | 0.65 | − | − | − | − | − | − | − | − | − | |
| TR 1685 | − | − | − | − | − | 0.51 | − | − | − | − | − | − | 0.7 | − | − | − | − | − | − | − | − | − | |
| RD 130 | − | − | − | − | − | 0.21 | − | − | − | − | − | − | 0.12 | − | − | − | − | − | − | − | − | − | |
| MATR−1 | 0.514 | − | 0.48 | 0.61 | − | − | − | − | − | 0.44 | 0.17 | 0 | − | − | 0.494 | 0.307 | 0.231 | − | − | − | − | − | |
| MATR−2 (= TR 292) | 0.581 | 0.27 | 0.59 | 0.63 | − | 0 | 0 | 0.19 | 0.46 | 0.71 | 0.64 | 0.53 | 0.6 | 0.5714 | 0.594 | 0.581 | 0.577 | 0.36 | 0.51 | − | 0.517 | 0.5050 | |
| MATR−3 (= TR X3) | 0.571 | 0.72 | 0.60 | 0.57 | − | 0.52 | 0.64 | 0.68 | 0.54 | 0.67 | 0.71 | 0.21 | 0.56 | 0.5844 | 0.519 | 0.485 | 0.694 | 0.66 | 0.04 | − | 0 | 0 | |
| MATR−4 | 0.096 | − | 0.60 | 0.47 | − | − | − | − | − | 0.19 | 0.23 | 0.08 | − | − | 0.08 | 0 | 0.593 | − | − | − | − | − | |
| MATR−5 | 0.096 | − | 0.12 | 0.20 | − | − | − | − | − | 0.37 | 0.52 | 0.08 | − | − | 0.079 | 0 | 0.615 | − | − | − | − | − | |
| MATR−6 | 0.420 | − | 0.12 | 0.55 | − | − | − | − | − | 0.53 | 0.41 | 0.08 | − | − | 0.492 | 0.272 | 0.47 | − | − | − | − | − | |
| MATR−7 | 0.718 | − | 0.65 | 0.68 | − | − | − | − | − | 0.79 | 0.55 | 0.23 | − | − | 0.662 | 0.498 | 0.581 | − | − | − | − | − | |
| MATR−8 | 0.376 | − | 0.49 | 0.49 | − | − | − | − | − | 0.51 | 0.46 | 0.08 | − | − | 0.463 | 0.497 | 0.657 | − | − | − | − | − | |
| MATR−9 | 0.459 | − | 0.49 | 0.65 | 0.445 | − | − | − | − | − | 0.66 | − | − | − | 0.512 | 0.272 | 0.494 | − | − | − | − | − | |
| MATR−10 | − | − | 0.56 | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | |
| MATR−11 | 0.431 | − | 0.58 | 0.53 | − | − | − | − | − | 0.63 | 0.49 | 0.08 | − | − | 0.515 | 0.482 | 0.576 | − | − | − | − | − | |
| MATR−12 | 0.000 | − | 0.02 | 0.09 | − | − | − | − | − | 0.16 | 0.47 | 0 | − | − | 0.014 | 0.053 | 0.026 | − | − | − | − | − | |
| MATR−13 | 0.525 | − | 0.46 | 0.53 | − | − | − | − | − | 0.49 | 0 | 0.50 | − | − | 0.506 | 0.456 | 0 | − | − | − | − | − | |
| MATR−14 | 0.480 | − | 0.53 | 0.52 | − | − | − | − | − | 0.50 | 0.31 | 0.08 | − | − | 0.485 | 0.52 | 0.409 | − | − | − | − | − | |
| MATR−15 | 0.070 | − | 0.16 | 0.01 | − | − | − | − | − | 0.30 | 0.17 | 0.08 | − | − | 0.118 | 0 | 0.517 | − | − | − | − | − | |
| MATR−16 | 0.400 | − | 0.53 | 0.50 | − | − | − | − | − | 0.48 | 0.35 | 0.45 | − | − | 0.508 | 0.549 | 0.655 | − | − | − | − | − | |


5. Rep−PCR Procedures and Their Implications in Epidemiological Studies
5.1. Application of Rep−PCR to Epidemiological Investigations of MAC
5.2. Implication of Rep−PCR Methods in MAC Epidemiological Investigations
5.3. Clinical Application of MAC Rep−PCR
6. Conclusions and Perspectives
| Strain | Origin | Country | Sample No. | Primers | Epidemiologic Characteristics | Reference | |
|---|---|---|---|---|---|---|---|
| MAA | Animal | cat, cattle, chukar, deer, dog, hobby, horse, pig, polecat, and peat | Sweden | 16 | s535 (IS900 specific outward primer), ERIC2 | - As a result of ERIC/IS900 PCR using the ERIC sequence and IS900 for the epidemiological analysis of MAP, MAP showed species−specific band patterns, which can be used as a method for discriminating it from other mycobacteria. - However, the MAP strains that were discriminated by RFLP cannot be distinguished and thus cannot be used as an alternative genotyping method of RFLP. | [107] |
| MAP | Human & animal | bovine, deer, goat, ovine, human | USA Europe | 60 | s535 (IS900 specific outward primer), ERIC2 | ||
| MAS | Animal | − | Norway | 1 | s535 (IS900 specific outward primer), ERIC2 | ||
| M. intracellulare | ATCC | 3 | s535 (IS900 specific outward primer), ERIC2 | ||||
| MAA | Human & environment | patients and environment | USA | 28 | DiversiLab Mycobacterium kit |
| [108] |
| M. intracellulare | Human | patients | USA | 8 | DiversiLab Mycobacterium kit |
| |
| M. avium | Human | Patients | USA Canada Netherlandss Brazil | 207 | DiversiLab Mycobacterium kit |
| [110] |
| M. avium | Human & environment | Patients, bronchoscopy preparation laboratory | USA | 22 clinical, 16 laboratory | Cangelosi et al., 2004 | - Water and biofilm samples collected from the bronchoscopy preparation laboratory yielded mycobacteria, including M. avium and M. intracellulare. - It is assumed that infection with 5/22 (23%) M. avium isolates and 42/56 (75%) M. intracellulare isolates is the result of a contaminated water supply | [111] |
| M. intracellulare | Human & environment | Patients, bronchoscopy preparation laboratory | USA | 56 clinical, 4 laboratory | Cangelosi et al., 2004 | ||
| M. avium | Human & environment | NTM patients, household sample | USA Canada | 9 clinical, 10 environmental | Cangelosi et al., 2004 |
| [61] |
| M. intracellulare | Human & environment | NTM patients, households sample | USA Canada | 6 clinical, 10 environment | Cangelosi et al., 2004 | ||
| M. avium | Human & environment | Patients with chronic rhinosinusitis, household sample | USA | 6 clinical, 33 environment | Cangelosi et al., 2004 |
| [43] |
| M. avium | Human | Patients with HIV positive | France | 8 | DiversiLab Mycobacterium kit |
| [113] |
| M. avium | Human | clinical isolates | Poland | 33 | N6(CCG)4 |
| [89] |
| MAH | Human & environment | Patients, household sample | Netherlands | 5 | DiversiLab Mycobacterium kit |
| [114] |
| M. intracellulare | Human | Patients with nodular bronchiectasis | USA | 176 | Versalovic et al. 1991 |
| [63] |
| M. avium | Human & environment | patients and environment | Brazil USA Canada, Netherlands | 127 clinical, 52 environment | Bacterial Barcodes mycobacterial kit (Athens, GA) |
| [117] |
| M. avium | Human | Patients with NB, cavitary NB, fibrocavitary disease | South Korea | 31 | DiversiLab Mycobacterium kit | - In 481 patients with MAC lung disease who received antibiotic therapy for over 12 months were associated with re−infection by other bacteria (74%), while 26% of recurrence resulted from infection by the same bacteria (26%), according to the rep−PCR results. - The NB form was also determined as a significant risk factor for the recurrence of NTM lung disease. | [115] |
| M. intracellulare | Human | Patients with NB, cavitary NB, fibrocavitary disease | South Korea | 34 | DiversiLab Mycobacterium kit | ||
| M. avium | Human | Patients with MAC lung disease | South Korea | 52 | DiversiLab Mycobacterium kit | - The therapeutic effectiveness of intermittent antibiotic therapy was evaluated in patients previously treated for MAC lung disease and receiving antibiotic treatment for recurrent noncavitary NB MAC lung disease -86% (12/14) of relapsed patients were found to be infected with a new strain of MAC -As such, intermittent antibiotic therapy was suggested intermittent antibiotic therapy as to be a reasonable treatment strategy for recurrent noncavitary NB MAC lung disease. | [116] |
| M. intracellulare | Human | Patients with MAC lung disease | South Korea | 46 | DiversiLab Mycobacterium kit | ||
| M. avium | Human | Patients with Refractory MAC lung disease | South Korea | 80 | DiversiLab Mycobacterium kit | - In 72 patients with refractory M. avium complex lung disease (MAC−LD) who received antibiotic therapy, including macrolides, for over 12 months, macrolide resistance was found in 16 patients (22%). - Of the 49 patients recorded before and after treatment, 24/49 (49%) patients were found to be infected with a new MAC strain, while 12/49 (24%) patients were infected by both the original and new strains. Only 13/49 patients (27%) showed persistent infection by the original MAC strain. - In conclusion, refractory MAC−LD is generally caused by reinfection of other strains rather than the relapse of the original strain, which is thought to be due to intermittent macrolide resistance. | [41] |
| M. intracellulare | Human | Patients with Refractory MAC lung disease | South Korea | 120 | DiversiLab Mycobacterium kit | ||
Author Contributions
Funding
Conflicts of Interest
References
- Wolinsky, E. Nontuberculous mycobacteria and associated diseases. Am. Rev. Respir. Dis. 1979, 119, 107–159. [Google Scholar] [CrossRef] [PubMed]
- Biet, F.; Boschiroli, M.L.; Thorel, M.F.; Guilloteau, L.A. Zoonotic aspects of Mycobacterium bovis and Mycobacterium avium-intracellulare complex (MAC). Vet. Res. 2005, 36, 411–436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coelho, A.C.; de Lurdes Pinto, M.; Matos, A.; Matos, M.; dos Anjos Pires, M. Mycobacterium avium complex in domestic and wild animals. In Insights from Veterinary Medicine; IntechOpen: Rijeka, Croatia, 2013. [Google Scholar]
- Miguez-Burbano, M.J.; Flores, M.; Ashkin, D.; Rodriguez, A.; Granada, A.M.; Quintero, N.; Pitchenik, A. Non-tuberculous mycobacteria disease as a cause of hospitalization in HIV-infected subjects. Int. J. Infect. Dis. 2006, 10, 47–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abubakar, I.; Myhill, D.; Aliyu, S.H.; Hunter, P.R. Detection of Mycobacterium avium subspecies paratuberculosis from patients with Crohn’s disease using nucleic acid-based techniques: A systematic review and meta-analysis. Inflamm. Bowel Dis. 2008, 14, 401–410. [Google Scholar] [CrossRef]
- Hoefsloot, W.; van Ingen, J.; Andrejak, C.; Angeby, K.; Bauriaud, R.; Bemer, P.; Beylis, N.; Boeree, M.J.; Cacho, J.; Chihota, V.; et al. The geographic diversity of nontuberculous mycobacteria isolated from pulmonary samples: An NTM-NET collaborative study. Eur. Respir. J. 2013, 42, 1604–1613. [Google Scholar] [CrossRef]
- Kendall, B.A.; Winthrop, K.L. Update on the epidemiology of pulmonary nontuberculous mycobacterial infections. Semin. Respir. Crit. Care Med. 2013, 34, 87–94. [Google Scholar] [CrossRef]
- Nightingale, S.D.; Byrd, L.T.; Southern, P.M.; Jockusch, J.D.; Cal, S.X.; Wynne, B.A. Incidence of Mycobacterium avium-intracellulare complex bacteremia in human immunodeficiency virus-positive patients. J. Infect. Dis. 1992, 165, 1082–1085. [Google Scholar] [CrossRef]
- Currier, J.S.; Gandhi, R.T. Mycobacterium Avium Complex (MAC) Infections in Persons with HIV, Post TW, ed.; UpToDate; UpToDate Inc.: Waltham, MA, USA; Available online: https://www.uptodate.com (accessed on 19 December 2019).
- Turenne, C.Y.; Wallace, R., Jr.; Behr, M.A. Mycobacterium avium in the postgenomic era. Clin. Microbiol. Rev. 2007, 20, 205–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Runyon, E.H. Pathogenic mycobacteria. Bibliogr. Tuberc. 1965, 21, 235–287. [Google Scholar]
- Tortoli, E.; Rindi, L.; Garcia, M.J.; Chiaradonna, P.; Dei, R.; Garzelli, C.; Kroppenstedt, R.M.; Lari, N.; Mattei, R.; Mariottini, A.; et al. Proposal to elevate the genetic variant MAC-A, included in the Mycobacterium avium complex, to species rank as Mycobacterium chimaera sp. nov. Int. J. Syst. Evol. Microbiol. 2004, 54, 1277–1285. [Google Scholar] [CrossRef]
- Murcia, M.I.; Tortoli, E.; Menendez, M.C.; Palenque, E.; Garcia, M.J. Mycobacterium colombiense sp. nov., a novel member of the Mycobacterium avium complex and description of MAC-X as a new ITS genetic variant. Int. J. Syst. Evol. Microbiol. 2006, 56, 2049–2054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bang, D.; Herlin, T.; Stegger, M.; Andersen, A.B.; Torkko, P.; Tortoli, E.; Thomsen, V.O. Mycobacterium arosiense sp. nov., a slowly growing, scotochromogenic species causing osteomyelitis in an immunocompromised child. Int. J. Syst. Evol. Microbiol. 2008, 58, 2398–2402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Ingen, J.; Boeree, M.J.; Kosters, K.; Wieland, A.; Tortoli, E.; Dekhuijzen, P.N.; van Soolingen, D. Proposal to elevate Mycobacterium avium complex ITS sequevar MAC-Q to Mycobacterium vulneris sp. nov. Int. J. Syst. Evol. Microbiol. 2009, 59, 2277–2282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben Salah, I.; Cayrou, C.; Raoult, D.; Drancourt, M. Mycobacterium marseillense sp. nov., Mycobacterium timonense sp. nov. and Mycobacterium bouchedurhonense sp. nov., members of the Mycobacterium avium complex. Int. J. Syst. Evol. Microbiol. 2009, 59, 2803–2808. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.Y.; Kim, B.J.; Kim, H.; Won, Y.S.; Jeon, C.O.; Jeong, J.; Lee, S.H.; Lim, J.H.; Lee, S.H.; Kim, C.K.; et al. Mycobacterium paraintracellulare sp. nov., for the genotype INT-1 of Mycobacterium intracellulare. Int. J. Syst. Evol. Microbiol. 2016, 66, 3132–3141. [Google Scholar] [CrossRef] [Green Version]
- Kim, B.J.; Math, R.K.; Jeon, C.O.; Yu, H.K.; Park, Y.G.; Kook, Y.H.; Kim, B.J. Mycobacterium yongonense sp. nov., a slow-growing non-chromogenic species closely related to Mycobacterium intracellulare. Int. J. Syst. Evol. Microbiol. 2013, 63, 192–199. [Google Scholar] [CrossRef]
- Castejon, M.; Menendez, M.C.; Comas, I.; Vicente, A.; Garcia, M.J. Whole-genome sequence analysis of the Mycobacterium avium complex and proposal of the transfer of Mycobacterium yongonense to Mycobacterium intracellulare subsp. yongonense subsp. nov. Int. J. Syst. Evol. Microbiol. 2018, 68, 1998–2005. [Google Scholar] [CrossRef]
- Thorel, M.F.; Krichevsky, M.; Levy-Frebault, V.V. Numerical taxonomy of mycobactin-dependent mycobacteria, emended description of Mycobacterium avium, and description of Mycobacterium avium subsp. avium subsp. nov., Mycobacterium avium subsp. paratuberculosis subsp. nov., and Mycobacterium avium subsp. silvaticum subsp. nov. Int. J. Syst. Bacteriol. 1990, 40, 254–260. [Google Scholar] [CrossRef] [Green Version]
- Mijs, W.; de Haas, P.; Rossau, R.; Van der Laan, T.; Rigouts, L.; Portaels, F.; van Soolingen, D. Molecular evidence to support a proposal to reserve the designation Mycobacterium avium subsp. avium for bird-type isolates and ‘M. avium subsp. hominissuis’ for the human/porcine type of M. avium. Int. J. Syst. Evol. Microbiol. 2002, 52, 1505–1518. [Google Scholar] [CrossRef]
- Ben Salah, I.; Adekambi, T.; Raoult, D.; Drancourt, M. rpoB sequence-based identification of Mycobacterium avium complex species. Microbiology 2008, 154, 3715–3723. [Google Scholar] [CrossRef] [Green Version]
- Turenne, C.Y.; Semret, M.; Cousins, D.V.; Collins, D.M.; Behr, M.A. Sequencing of hsp65 distinguishes among subsets of the Mycobacterium avium complex. J. Clin. Microbiol. 2006, 44, 433–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez, J.; Garcia, I.G.; Aranaz, A.; Bezos, J.; Romero, B.; de Juan, L.; Mateos, A.; Gomez-Mampaso, E.; Dominguez, L. Genetic diversity of Mycobacterium avium isolates recovered from clinical samples and from the environment: Molecular characterization for diagnostic purposes. J. Clin. Microbiol. 2008, 46, 1246–1251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Runyon, E.H. Micobacterium intracellulare. Am. Rev. Respir. Dis. 1967, 95, 861–865. [Google Scholar] [CrossRef] [PubMed]
- Primm, T.P.; Lucero, C.A.; Falkinham, J.O., 3rd. Health impacts of environmental mycobacteria. Clin. Microbiol. Rev. 2004, 17, 98–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du Moulin, G.C.; Stottmeier, K.D.; Pelletier, P.A.; Tsang, A.Y.; Hedley-Whyte, J. Concentration of Mycobacterium avium by hospital hot water systems. JAMA 1988, 260, 1599–1601. [Google Scholar] [CrossRef] [PubMed]
- Falkinham, J.O., 3rd; Norton, C.D.; LeChevallier, M.W. Factors influencing numbers of Mycobacterium avium, Mycobacterium intracellulare, and other Mycobacteria in drinking water distribution systems. Appl. Environ. Microbiol. 2001, 67, 1225–1231. [Google Scholar] [CrossRef] [Green Version]
- Von Reyn, C.F.; Maslow, J.N.; Barber, T.W.; Falkinham, J.O., 3rd; Arbeit, R.D. Persistent colonisation of potable water as a source of Mycobacterium avium infection in AIDS. Lancet 1994, 343, 1137–1141. [Google Scholar] [CrossRef]
- Cayrou, C.; Turenne, C.; Behr, M.A.; Drancourt, M. Genotyping of Mycobacterium avium complex organisms using multispacer sequence typing. Microbiology 2010, 156, 687–694. [Google Scholar] [CrossRef] [Green Version]
- Nishiuchi, Y.; Maekura, R.; Kitada, S.; Tamaru, A.; Taguri, T.; Kira, Y.; Hiraga, T.; Hirotani, A.; Yoshimura, K.; Miki, M.; et al. The recovery of Mycobacterium avium-intracellulare complex (MAC) from the residential bathrooms of patients with pulmonary MAC. Clin. Infect. Dis. 2007, 45, 347–351. [Google Scholar] [CrossRef] [Green Version]
- Sharma-Kuinkel, B.K.; Rude, T.H.; Fowler, V.G. Pulse field gel electrophoresis. In The Genetic Manipulation of Staphylococci; Springer: Berlin/Heidelberg, Germany, 2014; pp. 117–130. [Google Scholar]
- Tenover, F.C.; Arbeit, R.D.; Goering, R.V.; Mickelsen, P.A.; Murray, B.E.; Persing, D.H.; Swaminathan, B. Interpreting chromosomal DNA restriction patterns produced by pulsed-field gel electrophoresis: Criteria for bacterial strain typing. J. Clin. Microbiol. 1995, 33, 2233–2239. [Google Scholar] [CrossRef] [Green Version]
- Arbeit, R.D.; Slutsky, A.; Barber, T.W.; Maslow, J.N.; Niemczyk, S.; Falkinham, J.O., 3rd; O’Connor, G.T.; von Reyn, C.F. Genetic diversity among strains of Mycobacterium avium causing monoclonal and polyclonal bacteremia in patients with AIDS. J. Infect. Dis. 1993, 167, 1384–1390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazurek, G.H.; Hartman, S.; Zhang, Y.; Brown, B.A.; Hector, J.S.; Murphy, D.; Wallace, R.J., Jr. Large DNA restriction fragment polymorphism in the Mycobacterium avium-M. intracellulare complex: A potential epidemiologic tool. J. Clin. Microbiol. 1993, 31, 390–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tenover, F.C.; Arbeit, R.D.; Goering, R.V. How to select and interpret molecular strain typing methods for epidemiological studies of bacterial infections: A review for healthcare epidemiologists. Molecular Typing Working Group of the Society for Healthcare Epidemiology of America. Infect. Control Hosp. Epidemiol. 1997, 18, 426–439. [Google Scholar] [CrossRef] [PubMed]
- Slutsky, A.M.; Arbeit, R.D.; Barber, T.W.; Rich, J.; von Reyn, C.F.; Pieciak, W.; Barlow, M.A.; Maslow, J.N. Polyclonal infections due to Mycobacterium avium complex in patients with AIDS detected by pulsed-field gel electrophoresis of sequential clinical isolates. J. Clin. Microbiol. 1994, 32, 1773–1778. [Google Scholar] [CrossRef] [Green Version]
- Wallace, R.J., Jr.; Zhang, Y.; Brown, B.A.; Dawson, D.; Murphy, D.T.; Wilson, R.; Griffith, D.E. Polyclonal Mycobacterium avium complex infections in patients with nodular bronchiectasis. Am. J. Respir. Crit. Care Med. 1998, 158, 1235–1244. [Google Scholar] [CrossRef]
- Picardeau, M.; Varnerot, A.; Lecompte, T.; Brel, F.; May, T.; Vincent, V. Use of different molecular typing techniques for bacteriological follow-up in a clinical trial with AIDS patients with Mycobacterium avium bacteremia. J. Clin. Microbiol. 1997, 35, 2503–2510. [Google Scholar] [CrossRef] [Green Version]
- Wallace, R.J., Jr.; Zhang, Y.; Brown-Elliott, B.A.; Yakrus, M.A.; Wilson, R.W.; Mann, L.; Couch, L.; Girard, W.M.; Griffith, D.E. Repeat positive cultures in Mycobacterium intracellulare lung disease after macrolide therapy represent new infections in patients with nodular bronchiectasis. J. Infect. Dis. 2002, 186, 266–273. [Google Scholar] [CrossRef] [Green Version]
- Jhun, B.W.; Kim, S.Y.; Moon, S.M.; Jeon, K.; Kwon, O.J.; Huh, H.J.; Ki, C.S.; Lee, N.Y.; Shin, S.J.; Daley, C.L.; et al. Development of Macrolide Resistance and Reinfection in Refractory Mycobacterium avium Complex Lung Disease. Am. J. Respir. Crit. Care Med. 2018, 198, 1322–1330. [Google Scholar] [CrossRef]
- Nishiuchi, Y.; Tamura, A.; Kitada, S.; Taguri, T.; Matsumoto, S.; Tateishi, Y.; Yoshimura, M.; Ozeki, Y.; Matsumura, N.; Ogura, H.; et al. Mycobacterium avium complex organisms predominantly colonize in the bathtub inlets of patients’ bathrooms. Jpn. J. Infect. Dis. 2009, 62, 182–186. [Google Scholar]
- Tichenor, W.S.; Thurlow, J.; McNulty, S.; Brown-Elliott, B.A.; Wallace, R.J.; Falkinham, J.O., 3rd. Nontuberculous Mycobacteria in household plumbing as possible cause of chronic rhinosinusitis. Emerg. Infect. Dis. 2012, 18, 1612–1617. [Google Scholar] [CrossRef]
- Moraga-McHaley, S.A.; Landen, M.; Krapfl, H.; Sewell, C.M. Hypersensitivity pneumonitis with Mycobacterium avium complex among spa workers. Int. J. Occup. Environ. Health 2013, 19, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Lyman, M.M.; Grigg, C.; Kinsey, C.B.; Keckler, M.S.; Moulton-Meissner, H.; Cooper, E.; Soe, M.M.; Noble-Wang, J.; Longenberger, A.; Walker, S.R. Invasive nontuberculous mycobacterial infections among cardiothoracic surgical patients exposed to heater–cooler devices. Emerg. Infect. Dis. 2017, 23, 796. [Google Scholar] [CrossRef] [PubMed]
- Aronson, T.; Holtzman, A.; Glover, N.; Boian, M.; Froman, S.; Berlin, O.G.; Hill, H.; Stelma, G., Jr. Comparison of large restriction fragments of Mycobacterium avium isolates recovered from AIDS and non-AIDS patients with those of isolates from potable water. J. Clin. Microbiol. 1999, 37, 1008–1012. [Google Scholar] [CrossRef] [Green Version]
- Kyriakopoulos, A.M.; Matsiota-Bernard, P.; Marinis, E.; Legakis, N.J.; Tassios, P.T. Comparison of Mycobacterium avium isolates from Greek AIDS and human immunodeficiency virus-negative patients by pulsed-field gel electrophoresis. Clin. Microbiol. Infect. 2000, 6, 490–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dvorska, L.; Bull, T.J.; Bartos, M.; Matlova, L.; Svastova, P.; Weston, R.T.; Kintr, J.; Parmova, I.; Van Soolingen, D.; Pavlik, I. A standardised restriction fragment length polymorphism (RFLP) method for typing Mycobacterium avium isolates links IS901 with virulence for birds. J. Microbiol. Methods 2003, 55, 11–27. [Google Scholar] [CrossRef]
- Dvorska, L.; Matlova, L.; Ayele, W.Y.; Fischer, O.A.; Amemori, T.; Weston, R.T.; Alvarez, J.; Beran, V.; Moravkova, M.; Pavlik, I. Avian tuberculosis in naturally infected captive water birds of the Ardeideae and Threskiornithidae families studied by serotyping, IS901 RFLP typing, and virulence for poultry. Vet. Microbiol. 2007, 119, 366–374. [Google Scholar] [CrossRef] [PubMed]
- Fischer, O.A.; Matlova, L.; Dvorska, L.; Svastova, P.; Bartl, J.; Weston, R.T.; Pavlik, I. Blowflies Calliphora vicina and Lucilia sericata as passive vectors of Mycobacterium avium subsp. avium, M. a. paratuberculosis and M. a. hominissuis. Med. Vet. Entomol. 2004, 18, 116–122. [Google Scholar] [CrossRef]
- Feizabadi, M.M.; Robertson, I.D.; Cousins, D.V.; Dawson, D.; Chew, W.; Gilbert, G.L.; Hampson, D.J. Genetic characterization of Mycobacterium avium isolates recovered from humans and animals in Australia. Epidemiol. Infect. 1996, 116, 41–49. [Google Scholar] [CrossRef] [Green Version]
- Johansen, T.B.; Olsen, I.; Jensen, M.R.; Dahle, U.R.; Holstad, G.; Djonne, B. New probes used for IS1245 and IS1311 restriction fragment length polymorphism of Mycobacterium avium subsp. avium and Mycobacterium avium subsp. hominissuis isolates of human and animal origin in Norway. BMC Microbiol. 2007, 7, 14. [Google Scholar] [CrossRef] [Green Version]
- Sevilla, I.; Garrido, J.M.; Geijo, M.; Juste, R.A. Pulsed-field gel electrophoresis profile homogeneity of Mycobacterium avium subsp. paratuberculosis isolates from cattle and heterogeneity of those from sheep and goats. BMC Microbiol. 2007, 7, 18. [Google Scholar] [CrossRef] [Green Version]
- Tirkkonen, T.; Pakarinen, J.; Rintala, E.; Ali-Vehmas, T.; Marttila, H.; Peltoniemi, O.A.; Makinen, J. Comparison of variable-number tandem-repeat markers typing and IS1245 restriction fragment length polymorphism fingerprinting of Mycobacterium avium subsp. hominissuis from human and porcine origins. Acta Vet. Scand. 2010, 52, 21. [Google Scholar] [CrossRef] [PubMed]
- Mobius, P.; Lentzsch, P.; Moser, I.; Naumann, L.; Martin, G.; Kohler, H. Comparative macrorestriction and RFLP analysis of Mycobacterium avium subsp. avium and Mycobacterium avium subsp. hominissuis isolates from man, pig, and cattle. Vet. Microbiol. 2006, 117, 284–291. [Google Scholar] [CrossRef] [PubMed]
- Agdestein, A.; Johansen, T.B.; Polacek, V.; Lium, B.; Holstad, G.; Vidanovic, D.; Aleksic-Kovacevic, S.; Jorgensen, A.; Zultauskas, J.; Nilsen, S.F.; et al. Investigation of an outbreak of mycobacteriosis in pigs. BMC Vet. Res. 2011, 7, 63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez, J.; Castellanos, E.; Romero, B.; Aranaz, A.; Bezos, J.; Rodriguez, S.; Mateos, A.; Dominguez, L.; de Juan, L. Epidemiological investigation of a Mycobacterium avium subsp. hominissuis outbreak in swine. Epidemiol. Infect. 2011, 139, 143–148. [Google Scholar] [CrossRef]
- Matlova, L.; Dvorska, L.; Palecek, K.; Maurenc, L.; Bartos, M.; Pavlik, I. Impact of sawdust and wood shavings in bedding on pig tuberculous lesions in lymph nodes, and IS1245 RFLP analysis of Mycobacterium avium subsp. hominissuis of serotypes 6 and 8 isolated from pigs and environment. Vet. Microbiol. 2004, 102, 227–236. [Google Scholar] [CrossRef]
- Agdestein, A.; Johansen, T.B.; Kolbjornsen, O.; Jorgensen, A.; Djonne, B.; Olsen, I. A comparative study of Mycobacterium avium subsp. avium and Mycobacterium avium subsp. hominissuis in experimentally infected pigs. BMC Vet. Res. 2012, 8, 11. [Google Scholar] [CrossRef] [Green Version]
- Agdestein, A.; Olsen, I.; Jørgensen, A.; Djønne, B.; Johansen, T.B. Novel insights into transmission routes of Mycobacterium avium in pigs and possible implications for human health. Vet. Res. 2014, 45, 46. [Google Scholar] [CrossRef] [Green Version]
- Falkinham, J.O., 3rd. Nontuberculous mycobacteria from household plumbing of patients with nontuberculous mycobacteria disease. Emerg. Infect. Dis. 2011, 17, 419–424. [Google Scholar] [CrossRef]
- Pestel-Caron, M.; Graff, G.; Berthelot, G.; Pons, J.L.; Lemeland, J.F. Molecular analysis of Mycobacterium avium isolates by using pulsed-field gel electrophoresis and PCR. J. Clin. Microbiol. 1999, 37, 2450–2455. [Google Scholar] [CrossRef] [Green Version]
- Iakhiaeva, E.; McNulty, S.; Brown Elliott, B.A.; Falkinham, J.O., 3rd; Williams, M.D.; Vasireddy, R.; Wilson, R.W.; Turenne, C.; Wallace, R.J., Jr. Mycobacterial interspersed repetitive-unit-variable-number tandem-repeat (MIRU-VNTR) genotyping of mycobacterium intracellulare for strain comparison with establishment of a PCR-based database. J. Clin. Microbiol. 2013, 51, 409–416. [Google Scholar] [CrossRef] [Green Version]
- Jeon, S.; Lim, N.; Kwon, S.; Shim, T.; Park, M.; Kim, B.J.; Kim, S. Molecular Typing of Mycobacterium intracellulare Using Pulsed-Field Gel Electrophoresis, Variable-Number Tandem-Repeat Analysis, Mycobacteria Interspersed Repetitive-Unit-Variable-Number Tandem Repeat Typing, and Multilocus Sequence Typing: Molecular Characterization and Comparison of Each Typing Methods. Osong Public Health Res. Perspect. 2014, 5, 119–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kannan, N.; Lai, Y.P.; Haug, M.; Lilleness, M.K.; Bakke, S.S.; Marstad, A.; Hov, H.; Naustdal, T.; Afset, J.E.; Ioerger, T.R.; et al. Genetic Variation/Evolution and Differential Host Responses Resulting from In-Patient Adaptation of Mycobacterium avium. Infect. Immun. 2019, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazars, E.; Lesjean, S.; Banuls, A.L.; Gilbert, M.; Vincent, V.; Gicquel, B.; Tibayrenc, M.; Locht, C.; Supply, P. High-resolution minisatellite-based typing as a portable approach to global analysis of Mycobacterium tuberculosis molecular epidemiology. Proc. Natl. Acad. Sci. USA 2001, 98, 1901–1906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frothingham, R.; Meeker-O’Connell, W.A. Genetic diversity in the Mycobacterium tuberculosis complex based on variable numbers of tandem DNA repeats. Microbiology 1998, 144 Pt 5, 1189–1196. [Google Scholar] [CrossRef] [Green Version]
- Inagaki, T.; Nishimori, K.; Yagi, T.; Ichikawa, K.; Moriyama, M.; Nakagawa, T.; Shibayama, T.; Uchiya, K.; Nikai, T.; Ogawa, K. Comparison of a variable-number tandem-repeat (VNTR) method for typing Mycobacterium avium with mycobacterial interspersed repetitive-unit-VNTR and IS1245 restriction fragment length polymorphism typing. J. Clin. Microbiol. 2009, 47, 2156–2164. [Google Scholar] [CrossRef] [Green Version]
- Thibault, V.C.; Grayon, M.; Boschiroli, M.L.; Hubbans, C.; Overduin, P.; Stevenson, K.; Gutierrez, M.C.; Supply, P.; Biet, F. New variable-number tandem-repeat markers for typing Mycobacterium avium subsp. paratuberculosis and M. avium strains: Comparison with IS900 and IS1245 restriction fragment length polymorphism typing. J. Clin. Microbiol. 2007, 45, 2404–2410. [Google Scholar] [CrossRef] [Green Version]
- Bull, T.J.; Sidi-Boumedine, K.; McMinn, E.J.; Stevenson, K.; Pickup, R.; Hermon-Taylor, J. Mycobacterial interspersed repetitive units (MIRU) differentiate Mycobacterium avium subspecies paratuberculosis from other species of the Mycobacterium avium complex. Mol. Cell. Probes 2003, 17, 157–164. [Google Scholar] [CrossRef]
- Overduin, P.; Schouls, L.; Roholl, P.; van der Zanden, A.; Mahmmod, N.; Herrewegh, A.; van Soolingen, D. Use of multilocus variable-number tandem-repeat analysis for typing Mycobacterium avium subsp. paratuberculosis. J. Clin. Microbiol. 2004, 42, 5022–5028. [Google Scholar] [CrossRef] [Green Version]
- Romano, M.I.; Amadio, A.; Bigi, F.; Klepp, L.; Etchechoury, I.; Llana, M.N.; Morsella, C.; Paolicchi, F.; Pavlik, I.; Bartos, M.; et al. Further analysis of VNTR and MIRU in the genome of Mycobacterium avium complex, and application to molecular epidemiology of isolates from South America. Vet. Microbiol. 2005, 110, 221–237. [Google Scholar] [CrossRef]
- Ichikawa, K.; Yagi, T.; Inagaki, T.; Moriyama, M.; Nakagawa, T.; Uchiya, K.; Nikai, T.; Ogawa, K. Molecular typing of Mycobacterium intracellulare using multilocus variable-number of tandem-repeat analysis: Identification of loci and analysis of clinical isolates. Microbiology 2010, 156, 496–504. [Google Scholar] [CrossRef] [Green Version]
- Dauchy, F.A.; Degrange, S.; Charron, A.; Dupon, M.; Xin, Y.; Bebear, C.; Maugein, J. Variable-number tandem-repeat markers for typing Mycobacterium intracellulare strains isolated in humans. BMC Microbiol. 2010, 10, 93. [Google Scholar] [CrossRef] [Green Version]
- Selander, R.K.; Caugant, D.A.; Ochman, H.; Musser, J.M.; Gilmour, M.N.; Whittam, T.S. Methods of multilocus enzyme electrophoresis for bacterial population genetics and systematics. Appl. Environ. Microbiol. 1986, 51, 873–884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kikuchi, T.; Watanabe, A.; Gomi, K.; Sakakibara, T.; Nishimori, K.; Daito, H.; Fujimura, S.; Tazawa, R.; Inoue, A.; Ebina, M.; et al. Association between mycobacterial genotypes and disease progression in Mycobacterium avium pulmonary infection. Thorax 2009, 64, 901–907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, K.; Zhang, Y.; Peng, Y. Variable-number tandem repeat markers for Mycobacterium intracellulare genotyping: Comparison to the 16S rRNA gene sequencing. J. Infect. Dev. Ctries. 2017, 11, 158–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uchiya, K.; Takahashi, H.; Nakagawa, T.; Yagi, T.; Moriyama, M.; Inagaki, T.; Ichikawa, K.; Nikai, T.; Ogawa, K. Characterization of a novel plasmid, pMAH135, from Mycobacterium avium subsp. hominissuis. PLoS ONE 2015, 10, e0117797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rindi, L.; Lari, N.; Garzelli, C. Virulence of Mycobacterium avium Subsp. hominissuis Human Isolates in an in vitro Macrophage Infection Model. Int. J. Mycobacteriol. 2018, 7, 48–52. [Google Scholar] [CrossRef]
- Fujita, K.; Ito, Y.; Hirai, T.; Maekawa, K.; Imai, S.; Tatsumi, S.; Niimi, A.; Iinuma, Y.; Ichiyama, S.; Mishima, M. Genetic relatedness of Mycobacterium avium-intracellulare complex isolates from patients with pulmonary MAC disease and their residential soils. Clin. Microbiol. Infect. 2013, 19, 537–541. [Google Scholar] [CrossRef] [Green Version]
- Fujita, K.; Ito, Y.; Hirai, T.; Kubo, T.; Maekawa, K.; Togashi, K.; Ichiyama, S.; Mishima, M. Association between polyclonal and mixed mycobacterial Mycobacterium avium complex infection and environmental exposure. Ann. Am. Thorac. Soc. 2014, 11, 45–53. [Google Scholar] [CrossRef] [Green Version]
- Kamvar, Z.N.; Tabima, J.F.; Grunwald, N.J. Poppr: An R package for genetic analysis of populations with clonal, partially clonal, and/or sexual reproduction. PeerJ 2014, 2, e281. [Google Scholar] [CrossRef] [Green Version]
- Turenne, C.Y.; Collins, D.M.; Alexander, D.C.; Behr, M.A. Mycobacterium avium subsp. paratuberculosis and M. avium subsp. avium are independently evolved pathogenic clones of a much broader group of M. avium organisms. J. Bacteriol. 2008, 190, 2479–2487. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Moolji, J.; Dufort, A.; Staffa, A.; Domenech, P.; Reed, M.B.; Behr, M.A. Iron Acquisition in Mycobacterium avium subsp. paratuberculosis. J. Bacteriol. 2015, 198, 857–866. [Google Scholar] [CrossRef] [Green Version]
- Radomski, N.; Thibault, V.C.; Karoui, C.; de Cruz, K.; Cochard, T.; Gutierrez, C.; Supply, P.; Biet, F.; Boschiroli, M.L. Determination of genotypic diversity of Mycobacterium avium subspecies from human and animal origins by mycobacterial interspersed repetitive-unit-variable-number tandem-repeat and IS1311 restriction fragment length polymorphism typing methods. J. Clin. Microbiol. 2010, 48, 1026–1034. [Google Scholar] [CrossRef] [Green Version]
- Ichikawa, K.; van Ingen, J.; Koh, W.J.; Wagner, D.; Salfinger, M.; Inagaki, T.; Uchiya, K.I.; Nakagawa, T.; Ogawa, K.; Yamada, K.; et al. Genetic diversity of clinical Mycobacterium avium subsp. hominissuis and Mycobacterium intracellulare isolates causing pulmonary diseases recovered from different geographical regions. Infect. Genet. Evol. 2015, 36, 250–255. [Google Scholar] [CrossRef]
- Scherrer, S.; Landolt, P.; Carroli, N.; Stephan, R. Molecular Characterization of Mycobacterium avium subsp. hominissuis of Two Groups of Lymph Nodes, Being Intradermal Tuberculin or Interferon-Gamma Test Positive and Negative, Isolated from Swiss Cattle at Slaughter. Front. Vet. Sci. 2018, 5, 32. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, S.; Araki, T.; Asai, T.; Tsuyuguchi, K.; Arikawa, K.; Iwamoto, T.; Nakajima, C.; Suzuki, Y.; Ohya, K.; Yanai, T.; et al. Phylogenetic uniqueness of Mycobacterium avium subspecies hominissuis isolated from an abnormal pulmonary bovine case. Infect. Genet. Evol. 2018, 62, 122–129. [Google Scholar] [CrossRef]
- Wojtasik, A.; Kubiak, A.B.; Krzyzanowska, A.; Majchrzak, M.; Augustynowicz-Kopec, E.; Parniewski, P. Comparison of the (CCG)4-based PCR and MIRU-VNTR for molecular typing of Mycobacterium avium strains. Mol. Biol. Rep. 2012, 39, 7681–7686. [Google Scholar] [CrossRef]
- Park, H.T.; Park, H.E.; Park, W.B.; Kim, S.; Hur, T.Y.; Jung, Y.H.; Yoo, H.S. Genetic diversity of bovine Mycobacterium avium subsp. paratuberculosis discriminated by IS1311 PCR-REA, MIRU-VNTR, and MLSSR genotyping. J. Vet. Sci. 2018, 19, 627–634. [Google Scholar] [CrossRef]
- Zheng, H.W.; Pang, Y.; He, G.X.; Song, Y.Y.; Zhao, Y.L. Comparing the Genotype and Drug Susceptibilities between Mycobacterium avium and Mycobacterium intracellulare in China. Biomed. Environ. Sci. 2017, 30, 517–525. [Google Scholar] [CrossRef]
- El-Sayed, A.; Natur, S.; Abdou, N.-E.M.; Salem, M.; Hassan, A.; Zschöck, M. Genotyping of Mycobacterium avium field isolates based on repetitive elements. Int. J. Vet. Sci. Med. 2013, 1, 36–42. [Google Scholar] [CrossRef] [Green Version]
- Ronai, Z.; Csivincsik, A.; Dan, A.; Gyuranecz, M. Molecular analysis and MIRU-VNTR typing of Mycobacterium avium subsp. avium, ‘hominissuis’ and silvaticum strains of veterinary origin. Infect. Genet. Evol. 2016, 40, 192–199. [Google Scholar] [CrossRef]
- Rindi, L.; Buzzigoli, A.; Medici, C.; Garzelli, C. High phylogenetic proximity of isolates of Mycobacterium avium subsp. hominissuis over a two decades-period. Infect. Genet. Evol. 2013, 16, 99–102. [Google Scholar] [CrossRef] [PubMed]
- Imperiale, B.R.; Moyano, R.D.; Di Giulio, A.B.; Romero, M.A.; Alvarado Pinedo, M.F.; Santangelo, M.P.; Traveria, G.E.; Morcillo, N.S.; Romano, M.I. Genetic diversity of Mycobacterium avium complex strains isolated in Argentina by MIRU-VNTR. Epidemiol. Infect. 2017, 145, 1382–1391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwamoto, T.; Nakajima, C.; Nishiuchi, Y.; Kato, T.; Yoshida, S.; Nakanishi, N.; Tamaru, A.; Tamura, Y.; Suzuki, Y.; Nasu, M. Genetic diversity of Mycobacterium avium subsp. hominissuis strains isolated from humans, pigs, and human living environment. Infect. Genet. Evol. 2012, 12, 846–852. [Google Scholar] [CrossRef] [PubMed]
- Ronai, Z.; Csivincsik, A.; Gyuranecz, M.; Kreizinger, Z.; Dan, A.; Janosi, S. Molecular analysis and MIRU-VNTR typing of Mycobacterium avium subsp. paratuberculosis strains from various sources. J. Appl. Microbiol. 2015, 118, 275–283. [Google Scholar] [CrossRef]
- Keim, P.; Price, L.B.; Klevytska, A.M.; Smith, K.L.; Schupp, J.M.; Okinaka, R.; Jackson, P.J.; Hugh-Jones, M.E. Multiple-locus variable-number tandem repeat analysis reveals genetic relationships within Bacillus anthracis. J. Bacteriol. 2000, 182, 2928–2936. [Google Scholar] [CrossRef] [Green Version]
- Nei, M. Estimation of average heterozygosity and genetic distance from a small number of individuals. Genetics 1978, 89, 583–590. [Google Scholar]
- Versalovic, J.; de Bruijn, F.J.; Lupski, J.R. Repetitive sequence-based PCR (rep-PCR) DNA fingerprinting of bacterial genomes. In Bacterial Genomes; Springer: Berlin/Heidelberg, Germany, 1998; pp. 437–454. [Google Scholar]
- Koeuth, T.; Versalovic, J.; Lupski, J.R. Differential subsequence conservation of interspersed repetitive Streptococcus pneumoniae BOX elements in diverse bacteria. Genome Res. 1995, 5, 408–418. [Google Scholar] [CrossRef] [Green Version]
- Hulton, C.S.; Higgins, C.F.; Sharp, P.M. ERIC sequences: A novel family of repetitive elements in the genomes of Escherichia coli, Salmonella typhimurium and other enterobacteria. Mol. Microbiol. 1991, 5, 825–834. [Google Scholar] [CrossRef]
- Stern, M.J.; Ames, G.F.; Smith, N.H.; Robinson, E.C.; Higgins, C.F. Repetitive extragenic palindromic sequences: A major component of the bacterial genome. Cell 1984, 37, 1015–1026. [Google Scholar] [CrossRef]
- Versalovic, J.; Koeuth, T.; Lupski, J.R. Distribution of repetitive DNA sequences in eubacteria and application to fingerprinting of bacterial genomes. Nucleic Acids Res. 1991, 19, 6823–6831. [Google Scholar] [CrossRef]
- Doll, L.; Moshitch, S.; Frankel, G. Poly(GTG)5-associated profiles of Salmonella and Shigella genomic DNA. Res. Microbiol. 1993, 144, 17–24. [Google Scholar] [CrossRef]
- Doran, T.J.; Hodgson, A.L.; Davies, J.K.; Radford, A.J. Characterisation of a highly repeated DNA sequence from Mycobacterium bovis. FEMS Microbiol. Lett. 1993, 111, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Englund, S. IS900/ERIC-PCR as a tool to distinguish Mycobacterium avium subsp. paratuberculosis from closely related mycobacteria. Vet. Microbiol. 2003, 96, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Cangelosi, G.A.; Freeman, R.J.; Lewis, K.N.; Livingston-Rosanoff, D.; Shah, K.S.; Milan, S.J.; Goldberg, S.V. Evaluation of a high-throughput repetitive-sequence-based PCR system for DNA fingerprinting of Mycobacterium tuberculosis and Mycobacterium avium complex strains. J. Clin. Microbiol. 2004, 42, 2685–2693. [Google Scholar] [CrossRef] [Green Version]
- Otsuka, Y.; Parniewski, P.; Zwolska, Z.; Kai, M.; Fujino, T.; Kirikae, F.; Toyota, E.; Kudo, K.; Kuratsuji, T.; Kirikae, T. Characterization of a trinucleotide repeat sequence (CGG)5 and potential use in restriction fragment length polymorphism typing of Mycobacterium tuberculosis. J. Clin. Microbiol. 2004, 42, 3538–3548. [Google Scholar] [CrossRef] [Green Version]
- Horan, K.L.; Freeman, R.; Weigel, K.; Semret, M.; Pfaller, S.; Covert, T.C.; van Soolingen, D.; Leao, S.C.; Behr, M.A.; Cangelosi, G.A. Isolation of the genome sequence strain Mycobacterium avium 104 from multiple patients over a 17-year period. J. Clin. Microbiol. 2006, 44, 783–789. [Google Scholar] [CrossRef] [Green Version]
- Falkinham, J.O., 3rd. Hospital water filters as a source of Mycobacterium avium complex. J. Med. Microbiol. 2010, 59, 1198–1202. [Google Scholar] [CrossRef]
- Schulze-Robbecke, R.; Buchholtz, K. Heat susceptibility of aquatic mycobacteria. Appl. Environ. Microbiol. 1992, 58, 1869–1873. [Google Scholar] [CrossRef] [Green Version]
- Bussone, G.; Brossier, F.; Roudiere, L.; Bille, E.; Sekkal, N.; Charlier, C.; Gilquin, J.; Lanternier, F.; Lecuit, M.; Lortholary, O.; et al. Recurrent Mycobacterium avium infection after seven years of latency in a HIV-infected patient receiving efficient antiretroviral therapy. J. Infect. 2012, 64, 613–617. [Google Scholar] [CrossRef]
- Van der Zanden, R.J.; Magis-Escurra, C.; de Lange, W.C.; Hoefsloot, W.; Boeree, M.J.; van Ingen, J.; van Soolingen, D. Hypersensitivity pneumonitis caused by Mycobacterium avium subsp. hominissuis in a hot tub, as proven by IS1245 RFLP and rep-PCR typing. Int. J. Mycobacteriol. 2012, 1, 152–154. [Google Scholar] [CrossRef] [Green Version]
- Koh, W.J.; Moon, S.M.; Kim, S.Y.; Woo, M.A.; Kim, S.; Jhun, B.W.; Park, H.Y.; Jeon, K.; Huh, H.J.; Ki, C.S.; et al. Outcomes of Mycobacterium avium complex lung disease based on clinical phenotype. Eur. Respir. J. 2017, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jhun, B.W.; Moon, S.M.; Kim, S.Y.; Park, H.Y.; Jeon, K.; Kwon, O.J.; Huh, H.J.; Ki, C.S.; Lee, N.Y.; Chung, M.J.; et al. Intermittent Antibiotic Therapy for Recurrent Nodular Bronchiectatic Mycobacterium avium Complex Lung Disease. Antimicrob. Agents Chemother. 2018, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dirac, M.A.; Weigel, K.M.; Yakrus, M.A.; Becker, A.L.; Chen, H.L.; Fridley, G.; Sikora, A.; Speake, C.; Hilborn, E.D.; Pfaller, S.; et al. Shared Mycobacterium avium genotypes observed among unlinked clinical and environmental isolates. Appl. Environ. Microbiol. 2013, 79, 5601–5607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shin, J.-I.; Shin, S.J.; Shin, M.-K. Differential Genotyping of Mycobacterium avium Complex and Its Implications in Clinical and Environmental Epidemiology. Microorganisms 2020, 8, 98. https://doi.org/10.3390/microorganisms8010098
Shin J-I, Shin SJ, Shin M-K. Differential Genotyping of Mycobacterium avium Complex and Its Implications in Clinical and Environmental Epidemiology. Microorganisms. 2020; 8(1):98. https://doi.org/10.3390/microorganisms8010098
Chicago/Turabian StyleShin, Jeong-Ih, Sung Jae Shin, and Min-Kyoung Shin. 2020. "Differential Genotyping of Mycobacterium avium Complex and Its Implications in Clinical and Environmental Epidemiology" Microorganisms 8, no. 1: 98. https://doi.org/10.3390/microorganisms8010098