
Favipiravir Suppresses Zika Virus (ZIKV) through Activity as a Mutagen
 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Virus
2.3. Antivirals
2.4. Antiviral Evaluations
2.5. Quantitative Real-Time Reverse Transcription PCR
2.6. Viral Genome Sequencing
2.7. Defective Particle Competition Assays
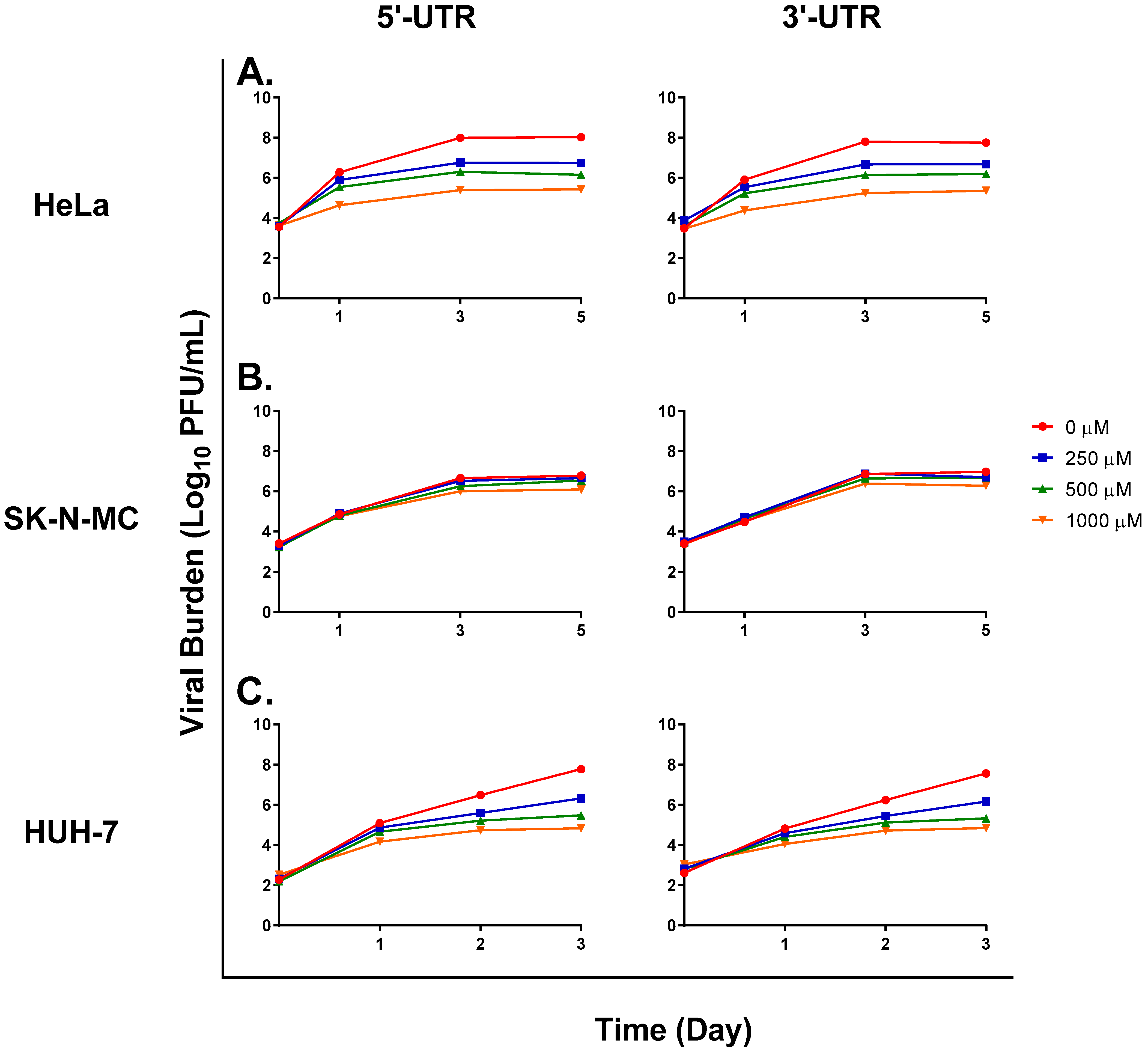
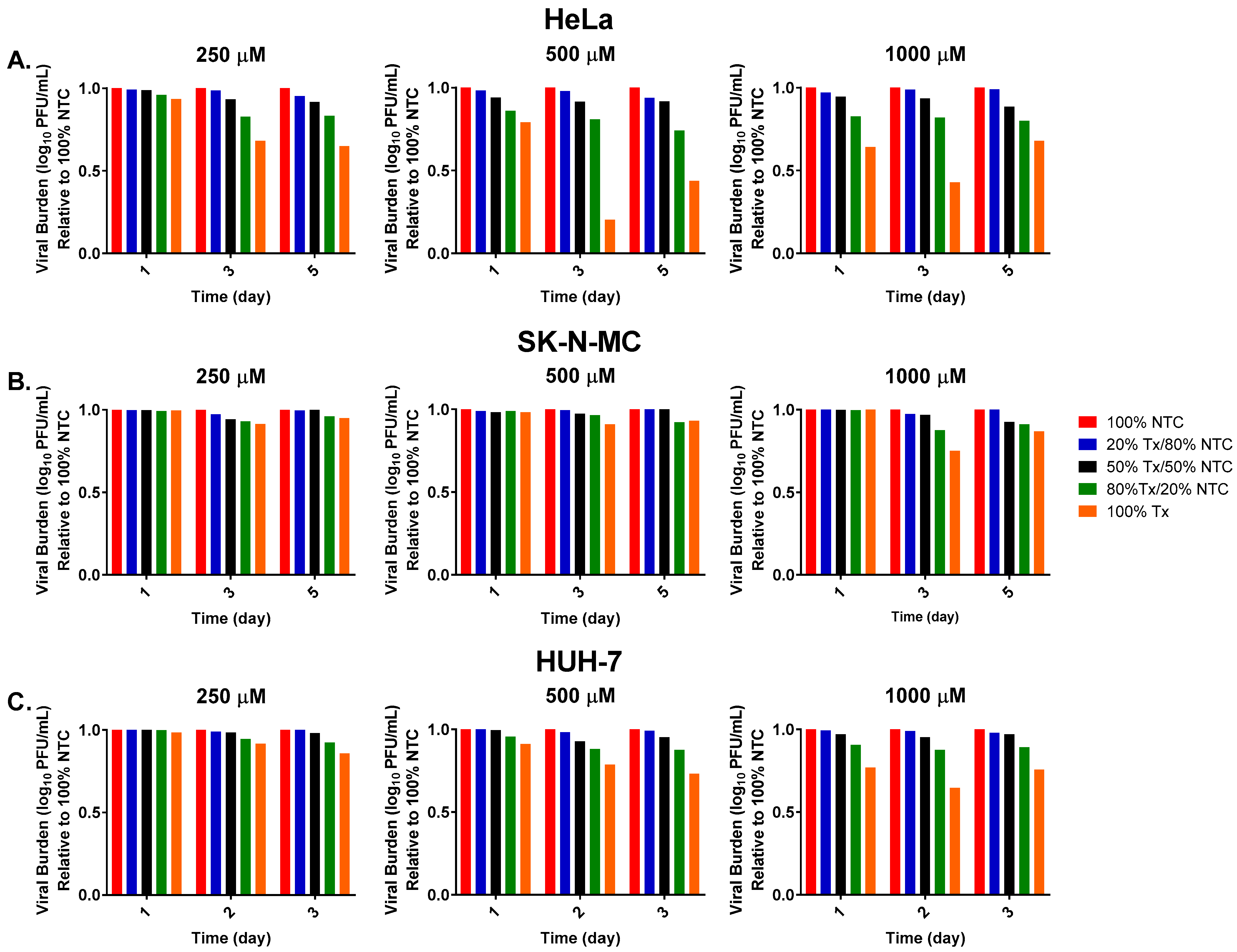
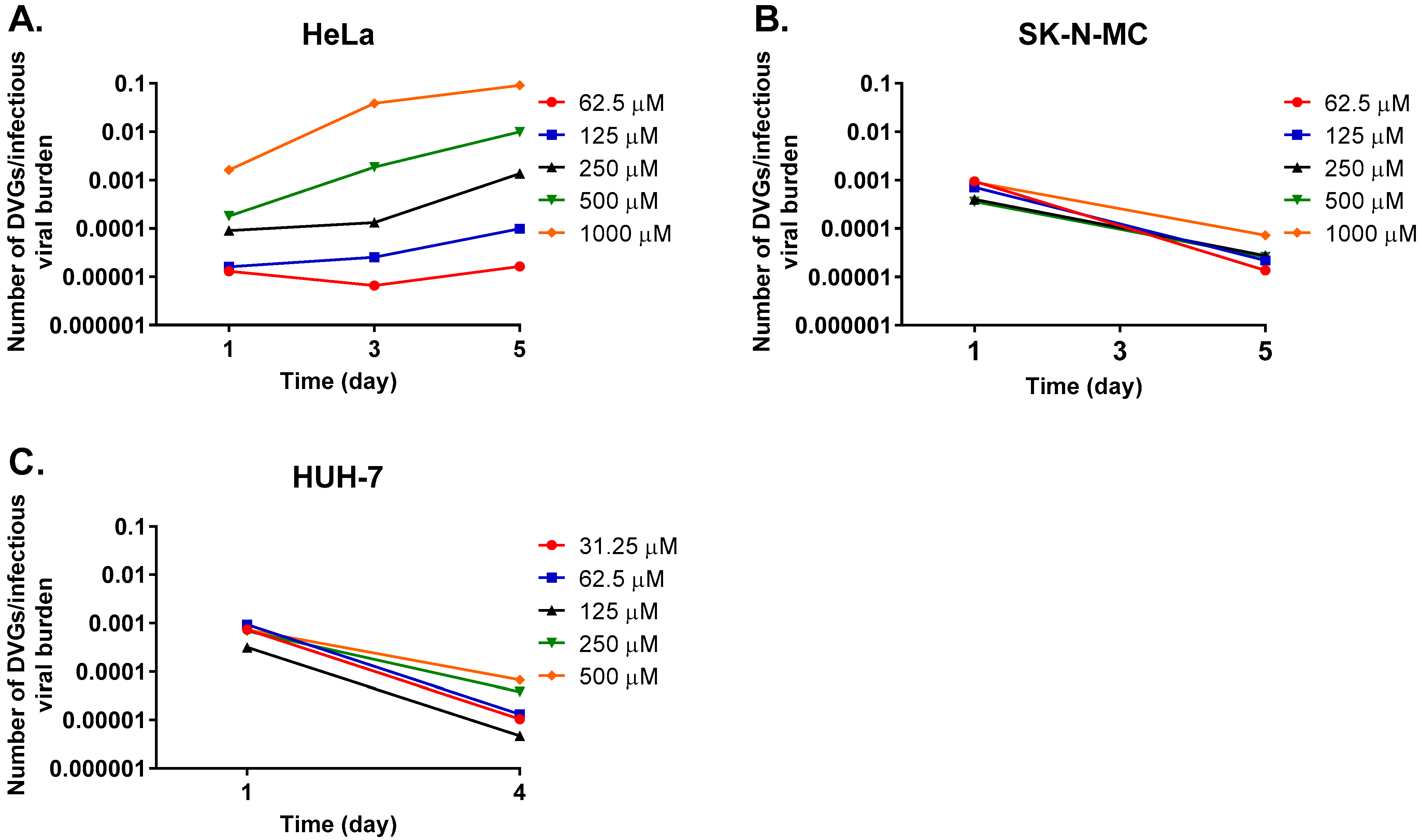
3. Results
3.1. FAV Exhibits Mutagenic Activity against ZIKV
3.2. Studies to Detect the Presence of Defective Viral Particles
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gregory, C.J.; Oduyebo, T.; Brault, A.C.; Brooks, J.T.; Chung, K.W.; Hills, S.; Kuehnert, M.J.; Mead, P.; Meaney-Delman, D.; Rabe, I.; et al. Modes of Transmission of Zika Virus. J. Infect. Dis. 2017, 216, S875–S883. [Google Scholar] [CrossRef]
- Calvez, E.; Mousson, L.; Vazeille, M.; O’Connor, O.; Cao-Lormeau, V.M.; Mathieu-Daude, F.; Pocquet, N.; Failloux, A.B.; Dupont-Rouzeyrol, M. Zika virus outbreak in the Pacific: Vector competence of regional vectors. PLoS Negl. Trop. Dis. 2018, 12, e0006637. [Google Scholar] [CrossRef] [PubMed]
- Gubler, D.J.; Vasilakis, N.; Musso, D. History and Emergence of Zika Virus. J. Infect. Dis. 2017, 216, S860–S867. [Google Scholar] [CrossRef] [PubMed]
- Duffy, M.R.; Chen, T.H.; Hancock, W.T.; Powers, A.M.; Kool, J.L.; Lanciotti, R.S.; Pretrick, M.; Marfel, M.; Holzbauer, S.; Dubray, C.; et al. Zika virus outbreak on Yap Island, Federated States of Micronesia. N. Engl. J. Med. 2009, 360, 2536–2543. [Google Scholar] [CrossRef] [PubMed]
- Song, B.H.; Yun, S.I.; Woolley, M.; Lee, Y.M. Zika virus: History, epidemiology, transmission, and clinical presentation. J. Neuroimmunol. 2017, 308, 50–64. [Google Scholar] [CrossRef]
- Sikka, V.; Chattu, V.K.; Popli, R.K.; Galwankar, S.C.; Kelkar, D.; Sawicki, S.G.; Stawicki, S.P.; Papadimos, T.J. The Emergence of Zika Virus as a Global Health Security Threat: A Review and a Consensus Statement of the INDUSEM Joint working Group (JWG). J. Glob. Infect. Dis. 2016, 8, 3–15. [Google Scholar] [CrossRef]
- Pierson, T.C.; Diamond, M.S. The emergence of Zika virus and its new clinical syndromes. Nature 2018, 560, 573–581. [Google Scholar] [CrossRef]
- Fagbami, A.H. Zika virus infections in Nigeria: Virological and seroepidemiological investigations in Oyo State. J. Hyg. 1979, 83, 213–219. [Google Scholar] [CrossRef]
- Driggers, R.W.; Ho, C.Y.; Korhonen, E.M.; Kuivanen, S.; Jaaskelainen, A.J.; Smura, T.; Rosenberg, A.; Hill, D.A.; DeBiasi, R.L.; Vezina, G.; et al. Zika Virus Infection with Prolonged Maternal Viremia and Fetal Brain Abnormalities. N. Engl. J. Med. 2016, 374, 2142–2151. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, F.M.E.; Pietrobon, A.J.; Oliveira, L.M.; Oliveira, L.; Sato, M.N. Maternal-Fetal Interplay in Zika Virus Infection and Adverse Perinatal Outcomes. Front. Immunol. 2020, 11, 175. [Google Scholar] [CrossRef]
- Teixeira, M.G.; Costa Mda, C.; de Oliveira, W.K.; Nunes, M.L.; Rodrigues, L.C. The Epidemic of Zika Virus-Related Microcephaly in Brazil: Detection, Control, Etiology, and Future Scenarios. Am. J. Public Health 2016, 106, 601–605. [Google Scholar] [CrossRef]
- Zorrilla, C.D.; Garcia Garcia, I.; Garcia Fragoso, L.; De La Vega, A. Zika Virus Infection in Pregnancy: Maternal, Fetal, and Neonatal Considerations. J. Infect. Dis. 2017, 216, S891–S896. [Google Scholar] [CrossRef] [PubMed]
- Musso, D.; Gubler, D.J. Zika Virus. Clin. Microbiol. Rev. 2016, 29, 487–524. [Google Scholar] [CrossRef] [PubMed]
- World Health Oragnization. Zika Virus. Available online: https://www.who.int/news-room/fact-sheets/detail/zika-virus (accessed on 8 February 2021).
- Furuta, Y.; Komeno, T.; Nakamura, T. Favipiravir (T-705), a broad spectrum inhibitor of viral RNA polymerase. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2017, 93, 449–463. [Google Scholar] [CrossRef]
- Furuta, Y.; Takahashi, K.; Shiraki, K.; Sakamoto, K.; Smee, D.F.; Barnard, D.L.; Gowen, B.B.; Julander, J.G.; Morrey, J.D. T-705 (favipiravir) and related compounds: Novel broad-spectrum inhibitors of RNA viral infections. Antivir. Res. 2009, 82, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Franco, E.J.; Pires de Mello, C.P.; Brown, A.N. Antiviral Evaluation of UV-4B and Interferon-Alpha Combination Regimens against Dengue Virus. Viruses 2021, 13, 771. [Google Scholar] [CrossRef]
- Franco, E.J.; Rodriquez, J.L.; Pomeroy, J.J.; Hanrahan, K.C.; Brown, A.N. The effectiveness of antiviral agents with broad-spectrum activity against chikungunya virus varies between host cell lines. Antivir. Chem. Chemother. 2018, 26, 2040206618807580. [Google Scholar] [CrossRef] [PubMed]
- Franco, E.J.; Tao, X.; Hanrahan, K.C.; Zhou, J.; Bulitta, J.B.; Brown, A.N. Combination Regimens of Favipiravir Plus Interferon Alpha Inhibit Chikungunya Virus Replication in Clinically Relevant Human Cell Lines. Microorganisms 2021, 9, 307. [Google Scholar] [CrossRef]
- Cai, L.; Sun, Y.; Song, Y.; Xu, L.; Bei, Z.; Zhang, D.; Dou, Y.; Wang, H. Viral polymerase inhibitors T-705 and T-1105 are potential inhibitors of Zika virus replication. Arch. Virol. 2017, 162, 2847–2853. [Google Scholar] [CrossRef]
- Franco, E.J.; Hanrahan, K.C.; Brown, A.N. Favipiravir Inhibits Zika Virus (ZIKV) Replication in HeLa Cells by Altering Viral Infectivity. Microorganisms 2023, 11, 1097. [Google Scholar] [CrossRef]
- Goldhill, D.H.; Te Velthuis, A.J.W.; Fletcher, R.A.; Langat, P.; Zambon, M.; Lackenby, A.; Barclay, W.S. The mechanism of resistance to favipiravir in influenza. Proc. Natl. Acad. Sci. USA 2018, 115, 11613–11618. [Google Scholar] [CrossRef]
- Venkataraman, S.; Prasad, B.; Selvarajan, R. RNA Dependent RNA Polymerases: Insights from Structure, Function and Evolution. Viruses 2018, 10, 76. [Google Scholar] [CrossRef] [PubMed]
- Shu, B.; Gong, P. Structural basis of viral RNA-dependent RNA polymerase catalysis and translocation. Proc. Natl. Acad. Sci. USA 2016, 113, E4005–E4014. [Google Scholar] [CrossRef]
- Baranovich, T.; Wong, S.S.; Armstrong, J.; Marjuki, H.; Webby, R.J.; Webster, R.G.; Govorkova, E.A. T-705 (favipiravir) induces lethal mutagenesis in influenza A H1N1 viruses in vitro. J. Virol. 2013, 87, 3741–3751. [Google Scholar] [CrossRef]
- de Avila, A.I.; Gallego, I.; Soria, M.E.; Gregori, J.; Quer, J.; Esteban, J.I.; Rice, C.M.; Domingo, E.; Perales, C. Lethal Mutagenesis of Hepatitis C Virus Induced by Favipiravir. PLoS ONE 2016, 11, e0164691. [Google Scholar] [CrossRef] [PubMed]
- Escribano-Romero, E.; Jimenez de Oya, N.; Domingo, E.; Saiz, J.C. Extinction of West Nile Virus by Favipiravir through Lethal Mutagenesis. Antimicrob. Agents Chemother. 2017, 61, e01400-17. [Google Scholar] [CrossRef]
- Borrego, B.; de Avila, A.I.; Domingo, E.; Brun, A. Lethal Mutagenesis of Rift Valley Fever Virus Induced by Favipiravir. Antimicrob. Agents Chemother. 2019, 63, e00669-19. [Google Scholar] [CrossRef] [PubMed]
- de Avila, A.I.; Moreno, E.; Perales, C.; Domingo, E. Favipiravir can evoke lethal mutagenesis and extinction of foot-and-mouth disease virus. Virus Res. 2017, 233, 105–112. [Google Scholar] [CrossRef]
- Sangawa, H.; Komeno, T.; Nishikawa, H.; Yoshida, A.; Takahashi, K.; Nomura, N.; Furuta, Y. Mechanism of action of T-705 ribosyl triphosphate against influenza virus RNA polymerase. Antimicrob. Agents Chemother. 2013, 57, 5202–5208. [Google Scholar] [CrossRef]
- Jin, Z.; Smith, L.K.; Rajwanshi, V.K.; Kim, B.; Deval, J. The ambiguous base-pairing and high substrate efficiency of T-705 (Favipiravir) Ribofuranosyl 5’-triphosphate towards influenza A virus polymerase. PLoS ONE 2013, 8, e68347. [Google Scholar] [CrossRef]
- Vignuzzi, M.; Lopez, C.B. Defective viral genomes are key drivers of the virus-host interaction. Nat. Microbiol. 2019, 4, 1075–1087. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Lyu, T.; Zhou, R.; He, X.; Ye, K.; Xie, Q.; Zhu, L.; Chen, T.; Shen, C.; Wu, Q.; et al. The Antiviral and Antitumor Effects of Defective Interfering Particles/Genomes and Their Mechanisms. Front. Microbiol. 2019, 10, 1852. [Google Scholar] [CrossRef]
- Stauffer Thompson, K.A.; Rempala, G.A.; Yin, J. Multiple-hit inhibition of infection by defective interfering particles. J. Gen. Virol. 2009, 90, 888–899. [Google Scholar] [CrossRef]
- Pires de Mello, C.P.; Tao, X.; Kim, T.H.; Bulitta, J.B.; Rodriquez, J.L.; Pomeroy, J.J.; Brown, A.N. Zika Virus Replication Is Substantially Inhibited by Novel Favipiravir and Interferon Alpha Combination Regimens. Antimicrob. Agents Chemother. 2018, 62, e01983-17. [Google Scholar] [CrossRef]
- Chan, J.F.; Yip, C.C.; Tee, K.M.; Zhu, Z.; Tsang, J.O.; Chik, K.K.; Tsang, T.G.; Chan, C.C.; Poon, V.K.; Sridhar, S.; et al. Improved detection of Zika virus RNA in human and animal specimens by a novel, highly sensitive and specific real-time RT-PCR assay targeting the 5’-untranslated region of Zika virus. Trop. Med. Int. Health 2017, 22, 594–603. [Google Scholar] [CrossRef] [PubMed]
- Quick, J.; Grubaugh, N.D.; Pullan, S.T.; Claro, I.M.; Smith, A.D.; Gangavarapu, K.; Oliveira, G.; Robles-Sikisaka, R.; Rogers, T.F.; Beutler, N.A.; et al. Multiplex PCR method for MinION and Illumina sequencing of Zika and other virus genomes directly from clinical samples. Nat. Protoc. 2017, 12, 1261–1276. [Google Scholar] [CrossRef]
- Chen, C.; Khaleel, S.S.; Huang, H.; Wu, C.H. Software for pre-processing Illumina next-generation sequencing short read sequences. Source Code Biol. Med. 2014, 9, 8. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Grubaugh, N.D.; Gangavarapu, K.; Quick, J.; Matteson, N.L.; De Jesus, J.G.; Main, B.J.; Tan, A.L.; Paul, L.M.; Brackney, D.E.; Grewal, S.; et al. An amplicon-based sequencing framework for accurately measuring intrahost virus diversity using PrimalSeq and iVar. Genome Biol. 2019, 20, 8. [Google Scholar] [CrossRef] [PubMed]
- Team R Core. R: A Language and Environment for Statistical Computing. Available online: https://www.r-project.org/ (accessed on 1 May 2023).
- Wickham, H. ggplot2. WIREs Comput. Stat. 2011, 3, 180–185. [Google Scholar] [CrossRef]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Beauclair, G.; Mura, M.; Combredet, C.; Tangy, F.; Jouvenet, N.; Komarova, A.V. DI-tector: Defective interfering viral genomes’ detector for next-generation sequencing data. RNA 2018, 24, 1285–1296. [Google Scholar] [CrossRef] [PubMed]
- Grande-Perez, A.; Lazaro, E.; Lowenstein, P.; Domingo, E.; Manrubia, S.C. Suppression of viral infectivity through lethal defection. Proc. Natl. Acad. Sci. USA 2005, 102, 4448–4452. [Google Scholar] [CrossRef]
- Martin, V.; Abia, D.; Domingo, E.; Grande-Perez, A. An interfering activity against lymphocytic choriomeningitis virus replication associated with enhanced mutagenesis. J. Gen. Virol. 2010, 91, 990–1003. [Google Scholar] [CrossRef]
- Grande-Perez, A.; Gomez-Mariano, G.; Lowenstein, P.R.; Domingo, E. Mutagenesis-induced, large fitness variations with an invariant arenavirus consensus genomic nucleotide sequence. J. Virol. 2005, 79, 10451–10459. [Google Scholar] [CrossRef] [PubMed]
- Moreno, H.; Tejero, H.; de la Torre, J.C.; Domingo, E.; Martin, V. Mutagenesis-mediated virus extinction: Virus-dependent effect of viral load on sensitivity to lethal defection. PLoS ONE 2012, 7, e32550. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Martinez, L.; Brichette-Mieg, I.; Pineno-Ramos, A.; Dominguez-Huerta, G.; Grande-Perez, A. Lethal mutagenesis of an RNA plant virus via lethal defection. Sci. Rep. 2018, 8, 1444. [Google Scholar] [CrossRef] [PubMed]
- Bixler, S.L.; Bocan, T.M.; Wells, J.; Wetzel, K.S.; Van Tongeren, S.A.; Garza, N.L.; Donnelly, G.; Cazares, L.H.; Soloveva, V.; Welch, L.; et al. Intracellular conversion and in vivo dose response of favipiravir (T-705) in rodents infected with Ebola virus. Antivir. Res. 2018, 151, 50–54. [Google Scholar] [CrossRef]
- Garcia-Lerma, J.G.; Aung, W.; Cong, M.E.; Zheng, Q.; Youngpairoj, A.S.; Mitchell, J.; Holder, A.; Martin, A.; Kuklenyik, S.; Luo, W.; et al. Natural substrate concentrations can modulate the prophylactic efficacy of nucleotide HIV reverse transcriptase inhibitors. J. Virol. 2011, 85, 6610–6617. [Google Scholar] [CrossRef]
- Prisant, N.; Bujan, L.; Benichou, H.; Hayot, P.H.; Pavili, L.; Lurel, S.; Herrmann, C.; Janky, E.; Joguet, G. Zika virus in the female genital tract. Lancet Infect. Dis. 2016, 16, 1000–1001. [Google Scholar] [CrossRef]
- Almeida, R.D.N.; Braz-de-Melo, H.A.; Santos, I.O.; Correa, R.; Kobinger, G.P.; Magalhaes, K.G. The Cellular Impact of the ZIKA Virus on Male Reproductive Tract Immunology and Physiology. Cells 2020, 9, 1006. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| HeLa | SK-N-MC | HUH-7 | ||||||
|---|---|---|---|---|---|---|---|---|
| Mutations (n) | Mutations (n) | Mutations (n) | ||||||
| FAV Exposure (μM) | D1 | D3 | D5 | D1 | D5 | FAV Exposure (μM) | D1 | D4 |
| 0 | 21 | 22 | 25 | 40 | 78 | 0 | 30 | 92 |
| 62.5 | 22 | 38 | 36 | 48 | 194 | 31.25 | 74 | 82 |
| 125 | 21 | 34 | 36 | 48 | 131 | 62.5 | 59 | 48 |
| 250 | 21 | 29 | 35 | 73 | 175 | 125 | 113 | 111 |
| 500 | 24 | 37 | 27 | 76 | 182 | 250 | 130 | 125 |
| 1000 | 27 | 22 | 31 | 66 | 176 | 500 | 125 | 130 |
| Mean Drug (95% CI) | 23 (19.8–26.2) | 32 (23.8–40.2) | 33 (28.1–37.9) | 62.2 (45.5–78.9) | 171.6 (141.9–201.3) | Mean Drug (95% CI) | 100.2 (60.7–139.7) | 99.2 (56.8–141.6) |
| HeLa | SK-N-MC | HUH-7 | ||||||
|---|---|---|---|---|---|---|---|---|
| Ts/Tv Ratio | Ts/Tv Ratio | Ts/Tv Ratio | ||||||
| FAV Exposure (μM) | D1 | D3 | D5 | D1 | D5 | FAV Exposure (μM) | D1 | D4 |
| 0 | 4.25 | 3.4 | 3 | 1.7 | 2.7 | 0 | 1.4 | 1.2 |
| 62.5 | 7 | 2 | 3 | 2.17 | 2.64 | 31.25 | 2.2 | 2.5 |
| 125 | 3 | 3.8 | 3.6 | 1.43 | 2.9 | 62.5 | 1.7 | 4.1 |
| 250 | 3.2 | 4.6 | 5.5 | 2 | 2.7 | 125 | 3.2 | 3.5 |
| 500 | 3.4 | 3.6 | 3.75 | 1.7 | 2.2 | 250 | 2.1 | 4.3 |
| 1000 | 3.5 | 3.2 | 2.83 | 2.1 | 3.3 | 500 | 1.5 | 5.4 |
| Mean Drug (95% CI) | 4 (1.9–6.1) | 3.4 (2.3–4.6) | 3.7 (2.4–5) | 1.9 (1.5–2.3) | 2.8 (2.3–3.2) | Mean Drug (95% CI) | 2.1 (1.3–2.9) | 4 (2.6–5.3) |
| Number of Mutations | ||||
|---|---|---|---|---|
| FAV Exposure (μM) | C→U | U→C | A→G | G→A |
| 0 | 8 | 2 | 3 | 4 |
| 62.5 | 7 | 2 | 3 | 5 |
| 125 | 7 | 2 | 3 | 6 |
| 250 | 8 | 2 | 4 | 8 |
| 500 | 7 | 1 | 3 | 4 |
| 1000 | 8 | 1 | 3 | 5 |
| Mean Drug (95% CI) | 7.4 (6.7–8.1) | 1.6 (0.9–2.3) | 3.2 (2.6–3.8) | 5.6 (3.7–7.5) |
| Number of Mutations | ||||
|---|---|---|---|---|
| FAV Exposure (μM) | C→U | U→C | A→G | G→A |
| 0 | 9 | 3 | 3 | 4 |
| 62.5 | 11 | 2 | 4 | 5 |
| 125 | 9 | 1 | 3 | 8 |
| 250 | 9 | 3 | 2 | 5 |
| 500 | 6 | 2 | 7 | 6 |
| 1000 | 8 | 1 | 3 | 8 |
| Mean Drug (95% CI) | 8.6 (6.3–10.9) | 1.8 (0.8–2.8) | 3.8 (1.4–6.2) | 6.4 (4.5–8.3) |
| Number of Mutations | ||||
|---|---|---|---|---|
| FAV Exposure (μM) | C→U | U→C | A→G | G→A |
| 0 | 8 | 2 | 3 | 5 |
| 31.25 | 8 | 1 | 3 | 6 |
| 62.5 | 11 | 7 | 4 | 12 |
| 125 | 7 | 4 | 1 | 8 |
| 250 | 12 | 3 | 4 | 10 |
| 500 | 12 | 6 | 5 | 14 |
| Mean Drug (95% CI) | 10 (7.1–12.9) | 4.2 (1.2–7.2) | 3.4 (1.5–5.3) | 10 (6.1–13.9) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Franco, E.J.; Cella, E.; Tao, X.; Hanrahan, K.C.; Azarian, T.; Brown, A.N. Favipiravir Suppresses Zika Virus (ZIKV) through Activity as a Mutagen. Microorganisms 2023, 11, 1342. https://doi.org/10.3390/microorganisms11051342
Franco EJ, Cella E, Tao X, Hanrahan KC, Azarian T, Brown AN. Favipiravir Suppresses Zika Virus (ZIKV) through Activity as a Mutagen. Microorganisms. 2023; 11(5):1342. https://doi.org/10.3390/microorganisms11051342
Chicago/Turabian StyleFranco, Evelyn J., Eleonora Cella, Xun Tao, Kaley C. Hanrahan, Taj Azarian, and Ashley N. Brown. 2023. "Favipiravir Suppresses Zika Virus (ZIKV) through Activity as a Mutagen" Microorganisms 11, no. 5: 1342. https://doi.org/10.3390/microorganisms11051342