
Identification and Characterization of Antibiotic-Resistant, Gram-Negative Bacteria Isolated from Korean Fresh Produce and Agricultural Environment
 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sampling
2.2. Microbiological Analyses
2.3. Preliminary Phenotypic Testing
2.4. Genotypic Characterization by RAPD-PCR Fingerprinting
2.5. Antibiotic Susceptibility Test
2.6. 16S rRNA Gene Sequencing and Identification
2.7. Whole Genome Sequencing
3. Results
3.1. Preliminary Physiological Tests
3.2. RAPD-PCR Fingerprinting
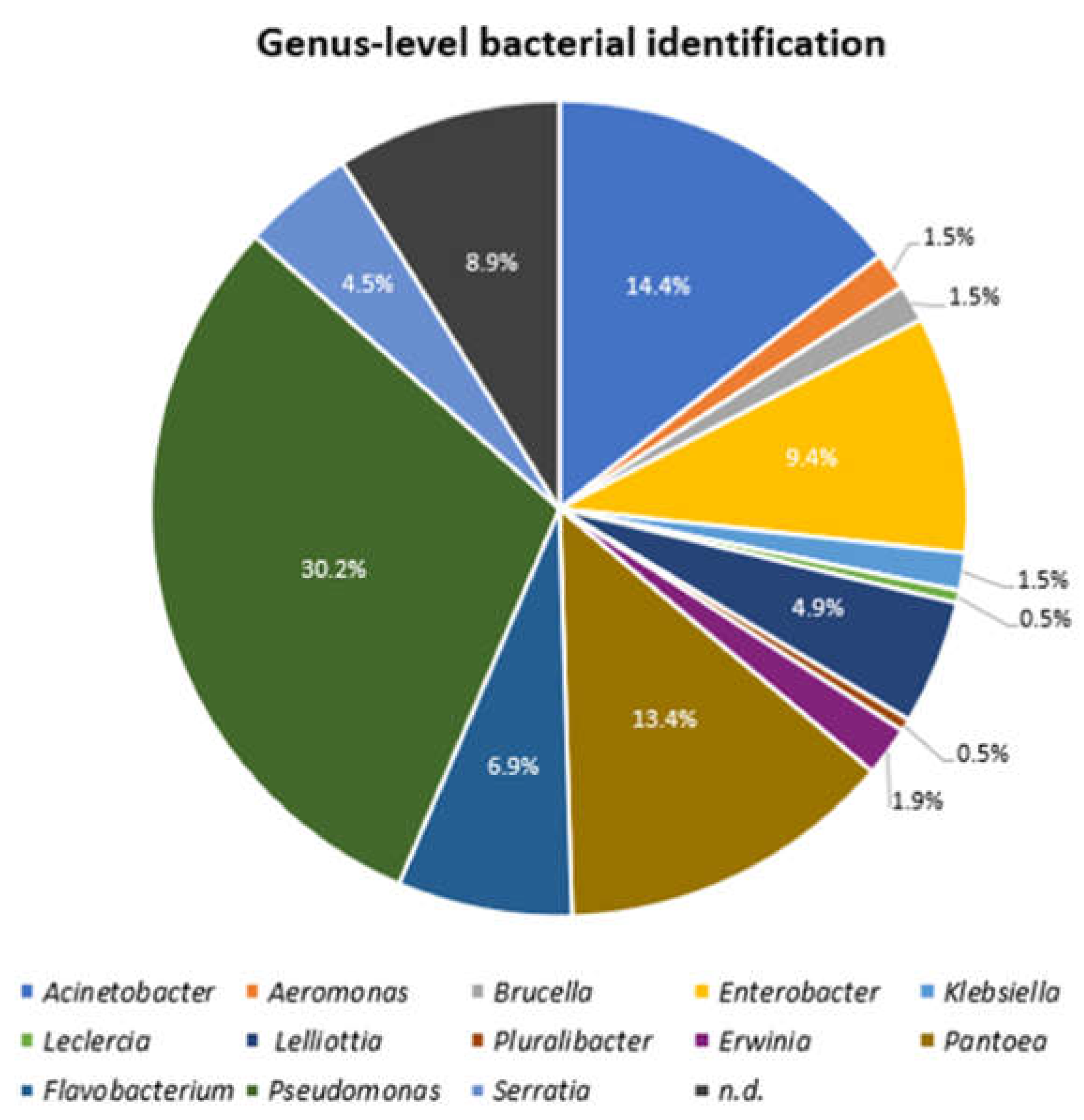
3.3. Strain Identification
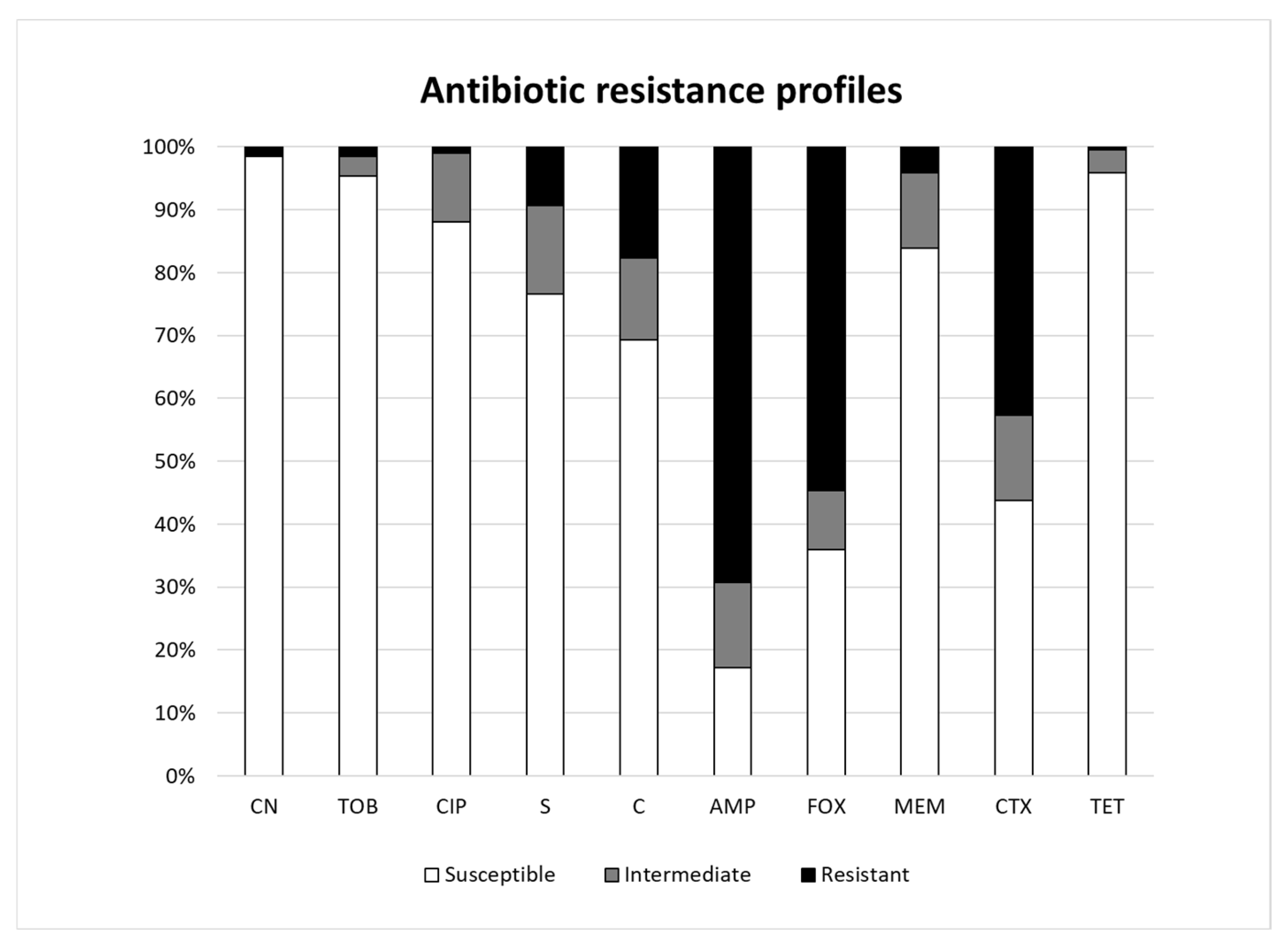
3.4. Antibiotic Resistance Profiles
3.5. Whole Genome Sequencing

{kind=link}
{kind=link}
| V98_8 | V88_4 | V104_6 | V104_10 | V89_4 | V89_7 | V106_11 | V108_6 | V87_3 | |
| No. of contigs | 113 | 48 | 58 | 66 | 30 | 34 | 37 | 58 | 35 |
| N50 | 115,019 | 382,325 | 138,943 | 195,255 | 250,587 | 215,337 | 320,651 | 163,040 | 211,224 |
| GC content (mol%) | 59.99 | 60.55 | 61.77 | 62.3 | 38.64 | 43.03 | 53.33 | 53.37 | 55.70 |
| Total length (bp) | 6,514,074 | 6,264,170 | 4,687,376 | 5,620,582 | 4,009,586 | 3,276,090 | 4,899,709 | 4,944,362 | 4,706,154 |
| Genome coverage | x 25 | x 103 | x 46 | x 32 | x 147 | x 68 | x 41 | x 63 | x 104 |
| No. of CDSs | 6043 | 5725 | 4326 | 5239 | 3771 | 3116 | 4956 | 5032 | 4443 |
| No. of tRNAs | 49 | 53 | 62 | 56 | 45 | 65 | 57 | 40 | 47 |
| No. of rRNAs | 3 | 5 | 6 | 5 | 3 | 3 | 5 | 5 | 4 |
| Acquired resistance gene(s) | n.d. | n.d. | n.d. | n.d. | blaOXA-304, blaADC-25 | n.d. | OqxB | OqxB | fosA, blaMIR-6 |
| Plasmid sequence(s) | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
| Antibiotic resistance | n.d. | C, AMP, FOX, CTX | n.d. | AMP, FOX, CTX | C, AMP, FOX, CTX | AMP, MEM, CTX | n.d. | AMP | AMP, FOX |
| OrthoANI identification (% similarity of top-hit) | Pseudomonas umsongensis DSM 16611T (96.75%) | Pseudomonas glycinae MS586T (96.48%) | Pseudomonas fulva DSM 17717T (99.48%) | Pseudomonas monteilii DSM 14164T (98.0%) | Acineotbacter oleivorans JCM 16667T (96.91%) | Acinetobacter soli KCTC 22184T (98.53%) | Pantoea ananatis LMG 2665T (99.07%) | Pantoea ananatis LMG 2665T (99.16%) | Enterobacter cancerogenus ATCC 33241T (98.55%) |
| in silico DDH identification (% similarity of top-hit) | Pseudomonas umsongensis DSM 16611T (71.9%) | Pseudomonas glycinae MS586T (71.1%) | Pseudomonas fulva DSM 17717T (96.1%) | Pseudomonas monteilii DSM 14164T (83.4%) | Acineotbacter oleivorans JCM 16667T (72.9%) | Acinetobacter soli KCTC 22184T (88.2%) | Pantoea ananatis LMG 2665T (92.5%) | Pantoea ananatis LMG 2665T (93.2%) | Enterobacter cancerogenus ATCC 33241T (87.4%) |
| Accession no. | JASCAE000000000 | JASCAF000000000 | JASCAG000000000 | JASCAH000000000 | JASCAI000000000 | JASCAJ000000000 | JASCAK000000000 | JASCAL000000000 | JASCAM000000000 |
| V87_3 | V89_11 | V114_1 | V89_13 | V90_4 | V115_8 | V90_14 | |||
| No. of contigs | 35 | 58 | 21 | 46 | 79 | 52 | 49 | ||
| N50 | 211,224 | 201,295 | 2,533,332 | 173,726 | 134,120 | 194,518 | 197,099 | ||
| GC content (mol%) | 55.70 | 54.60 | 59.89 | 55.84 | 56.41 | 58.21 | 61.53 | ||
| Total length (bp) | 4,706,154 | 4,700,025 | 4,932,611 | 4,846,069 | 5,149,412 | 5,253,824 | 4,749,641 | ||
| Genome coverage | x 104 | x 115 | x 41 | x 85 | x 103 | x 40 | x 127 | ||
| No. of CDSs | 4443 | 4509 | 4720 | 4561 | 4976 | 5084 | 4422 | ||
| No. of tRNAs | 47 | 36 | 76 | 48 | 52 | 65 | 56 | ||
| No. of rRNAs | 4 | 4 | 4 | 5 | 6 | 6 | 5 | ||
| Acquired resistance gene(s) | fosA, blaMIR-6 | blaACT-12, OqxA, OqxB, fosA2 | aac(6′)-Ic, blaSST-1, OqxB, tet(41) | fosA2, OqxA, OqxB | n.d. | OqxA, OqxB, fosA, blaOKP-A-11 | ampH, blaMOX-4, cphA1 | ||
| Plasmid sequence(s) | n.d. | n.d. | n.d. | n.d. | IncFII(Yp) | n.d. | n.d. | ||
| Antibiotic resistance | AMP, FOX | AMP, FOX | AMP | n.d. | AMP | CIP, AMP, MEM, CTX | AMP | ||
| OrthoANI identification (% similarity of top-hit) | Enterobacter cancerogenus ATCC 33241T (98.55%) | Enterobacter ludwigii DSM 16688T (98.88%) | Serratia marcescens ATCC 13880T (98.64%) | Lelliottia jeotgali PFL01T (91.19%), Lelliottia amnigena LMG2784T (85.24%), Lelliottia nimipressuralis CIP 104980T (84.15%) | Erwinia aphidicola JCM 21238T (99.02%) | Klebsiella quasipneumoniae subsp. quasipneumoniae 01A030T (99.19%) | Aeromonas hydrophila ATCC 7966T (96.91%) | ||
| in silico DDH identification (% similarity of top-hit) | Enterobacter cancerogenus ATCC 33241T (87.4%) | Enterobacter ludwigii DSM 16688T (91.4%) | Serratia marcescens ATCC 13880T (89.1%) | Lelliottia jeotgali PFL01T (43.6%), Lelliottia nimipressuralis CCUG 25894T (27.8%), Lelliottia amnigena LMG2784T (29.1%) | Erwinia aphidicola JCM 21238T (91.9%) | Klebsiella quasipneumoniae subsp. quasipneumoniae 01A030T (93.9%) | Aeromonas hydrophila ATCC 7966T (73.3%) | ||
| Accession no. | JASCAM000000000 | JASCAN000000000 | JASCAO000000000 | JASCAP000000000 | JASCAQ000000000 | JASCAR000000000 | JASCAS000000000 | ||
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Leff, J.W.; Fierer, N. Bacterial communities associated with the surfaces of fresh fruits and vegetables. PLoS ONE 2013, 8, e59310. [Google Scholar] [CrossRef]
- Sun, Y.; Zhao, X.; Ma, Y.; Ma, Z.; He, Z.; Zhao, W.; Wang, P.; Zhao, S.; Wang, D. Investigation on the Microbial Diversity of Fresh-Cut Lettuce during Processing and Storage Using High Throughput Sequencing and Their Relationship with Quality. Foods 2022, 11, 1683. [Google Scholar] [CrossRef] [PubMed]
- Berger, C.N.; Sodha, S.V.; Shaw, R.K.; Griffin, P.M.; Pink, D.; Hand, P.; Frankel, G. Fresh fruit and vegetables as vehicles for the transmission of human pathogens. Environ. Microbiol. 2010, 12, 2385–2397. [Google Scholar] [CrossRef] [PubMed]
- Castro-Ibáñez, I.; Gil, M.I.; Allende, A. Ready-to-eat vegetables: Current problems and potential solutions to reduce microbial risk in the production chain. Food Sci. Technol. 2017, 85, 284–292. [Google Scholar] [CrossRef]
- Yu, Y.C.; Yum, S.J.; Jeon, D.Y.; Jeong, H.G. Analysis of the Microbiota on Lettuce (Lactuca sativa L.) Cultivated in South Korea to Identify Foodborne Pathogens. J. Microbiol. Biotechnol. 2018, 28, 1318–1331. [Google Scholar] [CrossRef]
- Abdelfattah, A.; Freilich, S.; Bartuv, R.; Zhimo, V.Y.; Kumar, A.; Biasi, A.; Salim, S.; Feygenberg, O.; Burchard, E.; Dardick, C.; et al. Global analysis of the apple fruit microbiome: Are all apples the same? Environ. Microbiol. 2021, 23, 6038–6055. [Google Scholar] [CrossRef]
- Food and Agriculture Organization of the United Nations. Effects of Fruit and Vegetable Intakes on Direct and Indirect Health Outcomes: Background Paper for the FAO/WHO International Workshop on Fruits and Vegetables 2020; FAO: Rome, Italy, 2021. [Google Scholar]
- Aiyedun, S.O.; Onarinde, B.A.; Swainson, M.; Dixon, R.A. Foodborne outbreaks of microbial infection from fresh produce in Europe and North America: A systematic review of data from this millennium. Int. J. Food Sci. Technol. 2021, 56, 2215–2223. [Google Scholar] [CrossRef]
- FAO/WHO. Summary Report of the Joint FAO/WHO Expert Meeting on Microbiological Risk Assessment on the Prevention and Control of Microbiological Hazards in Fresh Fruits and Vegetables (Part 1: Administrative Procedures, Meeting Scope/Objectives, Data Collection; Part 2 General Principle and Fresh Fruits and Vegetables); FAO: Rome, Italy, 2021. [Google Scholar]
- Fiedler, G.; Kabisch, J.; Bohnlein, C.; Huch, M.; Becker, B.; Cho, G.S.; Franz, C. Presence of Human Pathogens in Produce from Retail Markets in Northern Germany. Foodborne Pathog. Dis. 2017, 14, 502–509. [Google Scholar] [CrossRef]
- Carstens, C.K.; Salazar, J.K.; Darkoh, C. Multistate Outbreaks of Foodborne Illness in the United States Associated with Fresh Produce From 2010 to 2017. Front. Microbiol. 2019, 10, 2667. [Google Scholar] [CrossRef]
- Authority, E.F.S. Shiga toxin-producing E. coli (STEC) O104:H4 2011 outbreaks in Europe: Taking Stock. EFSA J. 2011, 9, 2390. [Google Scholar] [CrossRef]
- Mellmann, A.; Harmsen, D.; Cummings, C.A.; Zentz, E.B.; Leopold, S.R.; Rico, A.; Prior, K.; Szczepanowski, R.; Ji, Y.; Zhang, W.; et al. Prospective genomic characterization of the German enterohemorrhagic Escherichia coli O104:H4 outbreak by rapid next generation sequencing technology. PLoS ONE 2011, 6, e22751. [Google Scholar] [CrossRef]
- Österblad, M.; Pensala, O.; Peterzéns, M.; Heleniusc, H.; Huovinen, P. Antimicrobial susceptibility of Enterobacteriaceae isolated from vegetables. J. Antimicrob. Chemother. 1999, 43, 503–509. [Google Scholar] [CrossRef] [PubMed]
- Al-Kharousi, Z.S.; Guizani, N.; Al-Sadi, A.M.; Al-Bulushi, I.M.; Shaharoona, B. Hiding in Fresh Fruits and Vegetables: Opportunistic Pathogens May Cross Geographical Barriers. Int. J. Microbiol. 2016, 2016, 4292417. [Google Scholar] [CrossRef]
- Rahman, M.; Alam, M.-U.; Luies, S.K.; Kamal, A.; Ferdous, S.; Lin, A.; Sharior, F.; Khan, R.; Rahman, Z.; Parvez, S.M.; et al. Contamination of Fresh Produce with Antibiotic-Resistant Bacteria and Associated Risks to Human Health: A Scoping Review. Int. J. Environ. Res. Public Health 2022, 19, 360. [Google Scholar] [CrossRef]
- Bezanson, G.S.; MacInnis, R.; Potter, G.; Hughes, T. Presence and potential for horizontal transfer of antibiotic resistance in oxidase-positive bacteria populating raw salad vegetables. Int. J. Food Microbiol. 2008, 127, 37–42. [Google Scholar] [CrossRef]
- Pleva, P.; Janalíková, M.; Pavlíčková, S.; Lecomte, M.; Godillon, T.; Holko, I. Characterization of Escherichia coli strains isolated from raw vegetables. Potravin. Slovak J. Food Sci. 2018, 12, 304–312. [Google Scholar] [CrossRef]
- Liu, S.; Kilonzo-Nthenge, A. Prevalence of Multidrug-Resistant Bacteria from U.S.-Grown and Imported Fresh Produce Retailed in Chain Supermarkets and Ethnic Stores of Davidson County, Tennessee. J. Food Prot. 2017, 80, 506–514. [Google Scholar] [CrossRef] [PubMed]
- Abatcha, M.G.; Effarizah, M.E.; Rusul, G. Prevalence, antimicrobial resistance, resistance genes and class 1 integrons of Salmonella serovars in leafy vegetables, chicken carcasses and related processing environments in Malaysian fresh food markets. Food Control 2018, 91, 170–180. [Google Scholar] [CrossRef]
- Freitag, C.; Michael, G.B.; Li, J.; Kadlec, K.; Wang, Y.; Hassel, M.; Schwarz, S. Occurrence and characterisation of ESBL-encoding plasmids among Escherichia coli isolates from fresh vegetables. Vet. Microbiol. 2018, 219, 63–69. [Google Scholar] [CrossRef]
- Li, Y.; Cao, W.; Liang, S.; Yamasaki, S.; Chen, X.; Shi, L.; Ye, L. Metagenomic characterization of bacterial community and antibiotic resistance genes in representative ready-to-eat food in southern China. Sci. Rep. 2020, 10, 15175. [Google Scholar] [CrossRef]
- Song, J.; Oh, S.S.; Kim, J.; Shin, J. Extended-spectrum β-lactamase-producing Escherichia coli isolated from raw vegetables in South Korea. Sci. Rep. 2020, 10, 19721. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Chon, J.W.; Kim, Y.J.; Kim, D.H.; Kim, M.S.; Seo, K.H. Prevalence and characterization of extended-spectrum-β-lactamase-producing Escherichia coli and Klebsiella pneumoniae in ready-to-eat vegetables. Int. J. Food Microbiol. 2015, 207, 83–86. [Google Scholar] [CrossRef]
- Buck, J.D. Nonstaining (KOH) method for determination of gram reactions of marine bacteria. Appl. Environ. Microbiol. 1982, 44, 992–993. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.W.; Kotiw, M.; Daggard, G. A RAPD-PCR genotyping assay which correlates with serotypes of group B streptococci. Lett. Appl. Microbiol. 2002, 35, 247–250. [Google Scholar] [CrossRef]
- Araújo, S.; Silva, I.A.; Tacão, M.; Patinha, C.; Alves, A.; Henriques, I. Characterization of antibiotic resistant and pathogenic Escherichia coli in irrigation water and vegetables in household farms. Int. J. Food Microbiol. 2017, 257, 192–200. [Google Scholar] [CrossRef]
- CLSI. Performance Standards for Antimicrobial Susbeptibility Testing. 30th ed. CLSI supplement M100. Clinical and Laboratory Standards Institute. Available online: https://clsi.org/media/3481/m100ed30_sample.pdf (accessed on 15 October 2020).
- Borges, A.S.G.; Basu, M.; Brinks, E.; Bang, C.; Cho, G.-S.; Baines, J.F.; Franke, A.; Franz, C.M.A.P. Fast Identification Method for Screening Bacteria from Faecal Samples Using Oxford Nanopore Technologies MinION Sequencing. Curr. Microbiol. 2023, 80, 101. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Perez, H.; Ciuffreda, L.; Flores, C. NanoCLUST: A species-level analysis of 16S rRNA nanopore sequencing data. Bioinformatics 2021, 37, 1600–1601. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Bharat, A.; Petkau, A.; Avery, B.P.; Chen, J.C.; Folster, J.P.; Carson, C.A.; Kearney, A.; Nadon, C.; Mabon, P.; Thiessen, J.; et al. Correlation between Phenotypic and In Silico Detection of Antimicrobial Resistance in Salmonella enterica in Canada Using Staramr. Microorganisms 2022, 10, 292. [Google Scholar] [CrossRef] [PubMed]
- Carattoli, A.; Zankari, E.; Garcia-Fernandez, A.; Voldby Larsen, M.; Lund, O.; Villa, L.; Moller Aarestrup, F.; Hasman, H. In silico detection and typing of plasmids using PlasmidFinder and plasmid multilocus sequence typing. Antimicrob. Agents Chemother. 2014, 58, 3895–3903. [Google Scholar] [CrossRef] [PubMed]
- Zankari, E.; Hasman, H.; Cosentino, S.; Vestergaard, M.; Rasmussen, S.; Lund, O.; Aarestrup, F.M.; Larsen, M.V. Identification of acquired antimicrobial resistance genes. J. Antimicrob. Chemother. 2012, 67, 2640–2644. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.; Ouk Kim, Y.; Park, S.C.; Chun, J. OrthoANI: An improved algorithm and software for calculating average nucleotide identity. Int. J. Syst. Evol. Microbiol. 2016, 66, 1100–1103. [Google Scholar] [CrossRef] [PubMed]
- Meier-Kolthoff, J.P.; Auch, A.F.; Klenk, H.P.; Goker, M. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinform. 2013, 14, 60. [Google Scholar] [CrossRef] [PubMed]
- Kim, O.S.; Cho, Y.J.; Lee, K.; Yoon, S.H.; Kim, M.; Na, H.; Park, S.C.; Jeon, Y.S.; Lee, J.H.; Yi, H.; et al. Introducing EzTaxon-e: A prokaryotic 16S rRNA gene sequence database with phylotypes that represent uncultured species. Int. J. Syst. Evol. Microbiol. 2012, 62, 716–721. [Google Scholar] [CrossRef] [PubMed]
- Blau, K.; Bettermann, A.; Jechalke, S.; Fornefeld, E.; Vanrobaeys, Y.; Stalder, T.; Top, E.M.; Smalla, K. The Transferable Resistome of Produce. mBio 2018, 9, e01300-18. [Google Scholar] [CrossRef]
- Chun, J.; Oren, A.; Ventosa, A.; Christensen, H.; Arahal, D.R.; da Costa, M.S.; Rooney, A.P.; Yi, H.; Xu, X.-W.; De Meyer, S.; et al. Proposed minimal standards for the use of genome data for the taxonomy of prokaryotes. Int. J. Syst. Evol. Microbiol. 2018, 68, 461–466. [Google Scholar] [CrossRef]
- Ghaith, D.M.; Zafer, M.M.; Ismail, D.K.; Al-Agamy, M.H.; Bohol, M.F.F.; Al-Qahtani, A.; Al-Ahdal, M.N.; Elnagdy, S.M.; Mostafa, I.Y. First reported nosocomial outbreak of Serratia marcescens harboring bla (IMP-4) and bla (VIM-2) in a neonatal intensive care unit in Cairo, Egypt. Infect. Drug Resist. 2018, 11, 2211–2217. [Google Scholar] [CrossRef]
- Kim, D.; Hong, S.; Kim, Y.T.; Ryu, S.; Kim, H.B.; Lee, J.H. Metagenomic Approach to Identifying Foodborne Pathogens on Chinese Cabbage. J. Microbiol. Biotechnol. 2018, 28, 227–235. [Google Scholar] [CrossRef]
- Vasala, A.; Hytönen, V.P.; Laitinen, O.H. Modern Tools for Rapid Diagnostics of Antimicrobial Resistance. Front. Cell. Infect. Microbiol. 2020, 10, 308. [Google Scholar] [CrossRef] [PubMed]
- Allydice-Francis, K.; Brown, P.D. Diversity of Antimicrobial Resistance and Virulence Determinants in Pseudomonas aeruginosa Associated with Fresh Vegetables. Int. J. Microbiol. 2012, 2012, 426241. [Google Scholar] [CrossRef] [PubMed]
- Go Eun, B.; Chung, I.-Y.; Kim, H.; Seok, K.-S.; Kim, B.; Yoo, Y.-J.; Jang, Y.; Chae, J.-C. Diversity of ampicillin resistant bacteria in domestic streams. Korean J. Microbiol. 2015, 51, 440–443. [Google Scholar] [CrossRef]
- Pang, Z.; Raudonis, R.; Glick, B.R.; Lin, T.-J.; Cheng, Z. Antibiotic resistance in Pseudomonas aeruginosa: Mechanisms and alternative therapeutic strategies. Biotechnol. Adv. 2019, 37, 177–192. [Google Scholar] [CrossRef]
- Noguchi, T.; Matsumura, Y.; Yamamoto, M.; Nagao, M.; Takakura, S.; Ichiyama, S. Clinical and microbiologic characteristics of cefotaxime-non-susceptible Enterobacteriaceae bacteremia: A case control study. BMC Infect. Dis 2017, 17, 44. [Google Scholar] [CrossRef]
- EFSA. Scientific Opinion on Chloramphenicol in food and feed. EFSA J. 2014, 12, 3907. [Google Scholar] [CrossRef]
- Schwaiger, K.; Helmke, K.; Hölzel, C.; Bauer, J. Antibiotic resistance in bacteria isolated from vegetables with regards to the marketing stage (farm vs. supermarket). Int. J. Food Microbiol. 2011, 148, 191–196. [Google Scholar] [CrossRef]


| Fertilizer Source | Surrounding Soils | Lettuce Leaves |
|---|---|---|
| Non-treated soil | 20 | 44 |
| Cow manure | 5 | 26 |
| Pig manure | 3 | 25 |
| Poultry manure | 3 | 18 |
| Chemical fertilizer | 10 | 25 |
| Chemical fertilizer + cow and poultry manure | 12 | 23 |
| Chemical fertilizer + cow + poultry + pig manure | 13 | 21 |
| Total number of isolates | 66 | 182 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jeong, S.; Kim, I.; Kim, B.-E.; Jeong, M.-I.; Oh, K.-K.; Cho, G.-S.; Franz, C.M.A.P. Identification and Characterization of Antibiotic-Resistant, Gram-Negative Bacteria Isolated from Korean Fresh Produce and Agricultural Environment. Microorganisms 2023, 11, 1241. https://doi.org/10.3390/microorganisms11051241
Jeong S, Kim I, Kim B-E, Jeong M-I, Oh K-K, Cho G-S, Franz CMAP. Identification and Characterization of Antibiotic-Resistant, Gram-Negative Bacteria Isolated from Korean Fresh Produce and Agricultural Environment. Microorganisms. 2023; 11(5):1241. https://doi.org/10.3390/microorganisms11051241
Chicago/Turabian StyleJeong, Sunyoung, Ile Kim, Bo-Eun Kim, Myeong-In Jeong, Kwang-Kyo Oh, Gyu-Sung Cho, and Charles M. A. P. Franz. 2023. "Identification and Characterization of Antibiotic-Resistant, Gram-Negative Bacteria Isolated from Korean Fresh Produce and Agricultural Environment" Microorganisms 11, no. 5: 1241. https://doi.org/10.3390/microorganisms11051241