
The Gut Microbiota of Broilers Reared with and without Antibiotic Treatment

 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Description of Samples Used in Study
2.2. Sample Processing
2.3. 16S Amplicon Metagenomic Sequencing of Samples
2.4. Statistical Analysis
2.4.1. Processing of Sequence Data
2.4.2. OTU Cluster and Taxonomic Annotation
2.4.3. Alpha Diversity, Beta Diversity and LEfSe Analysis
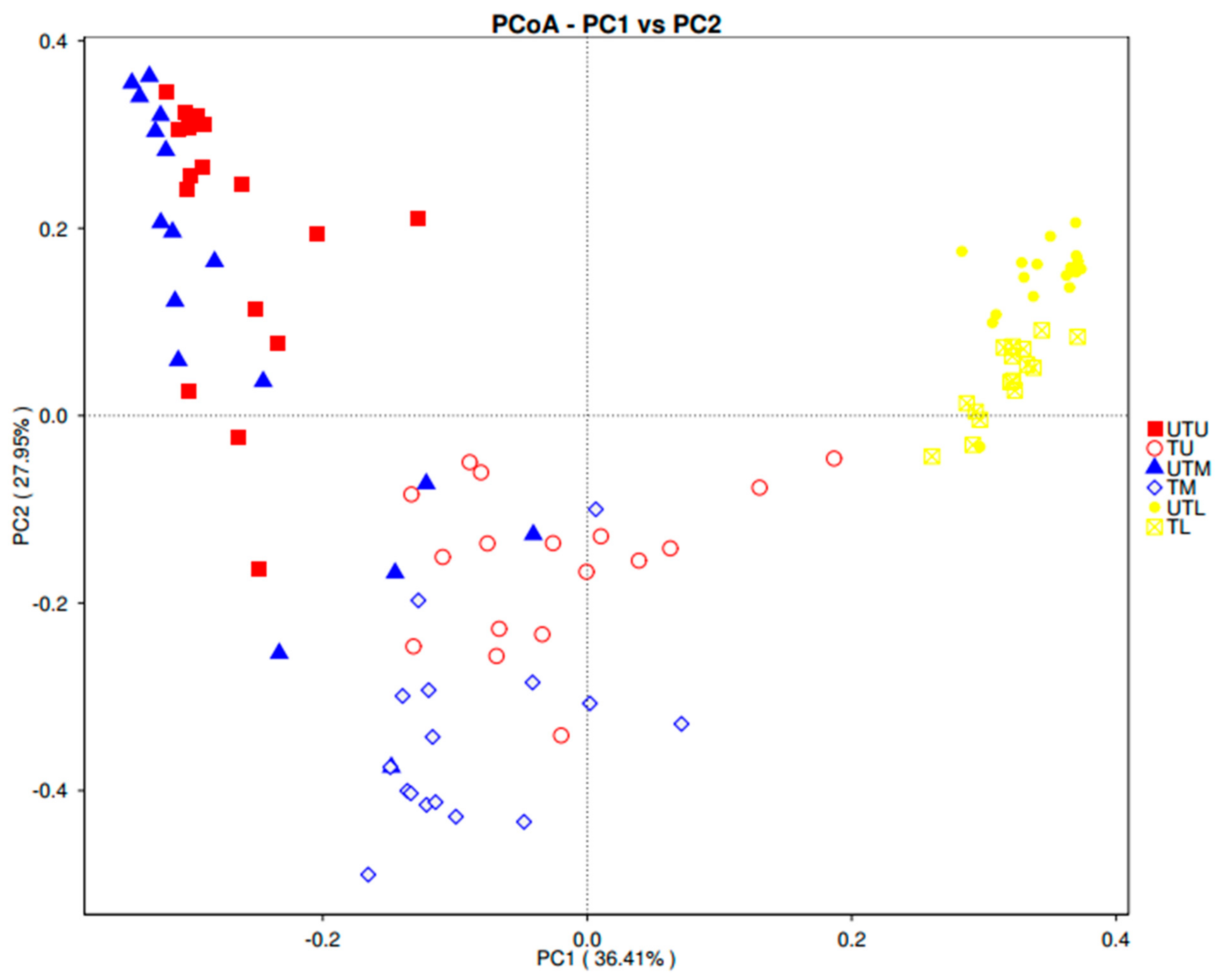
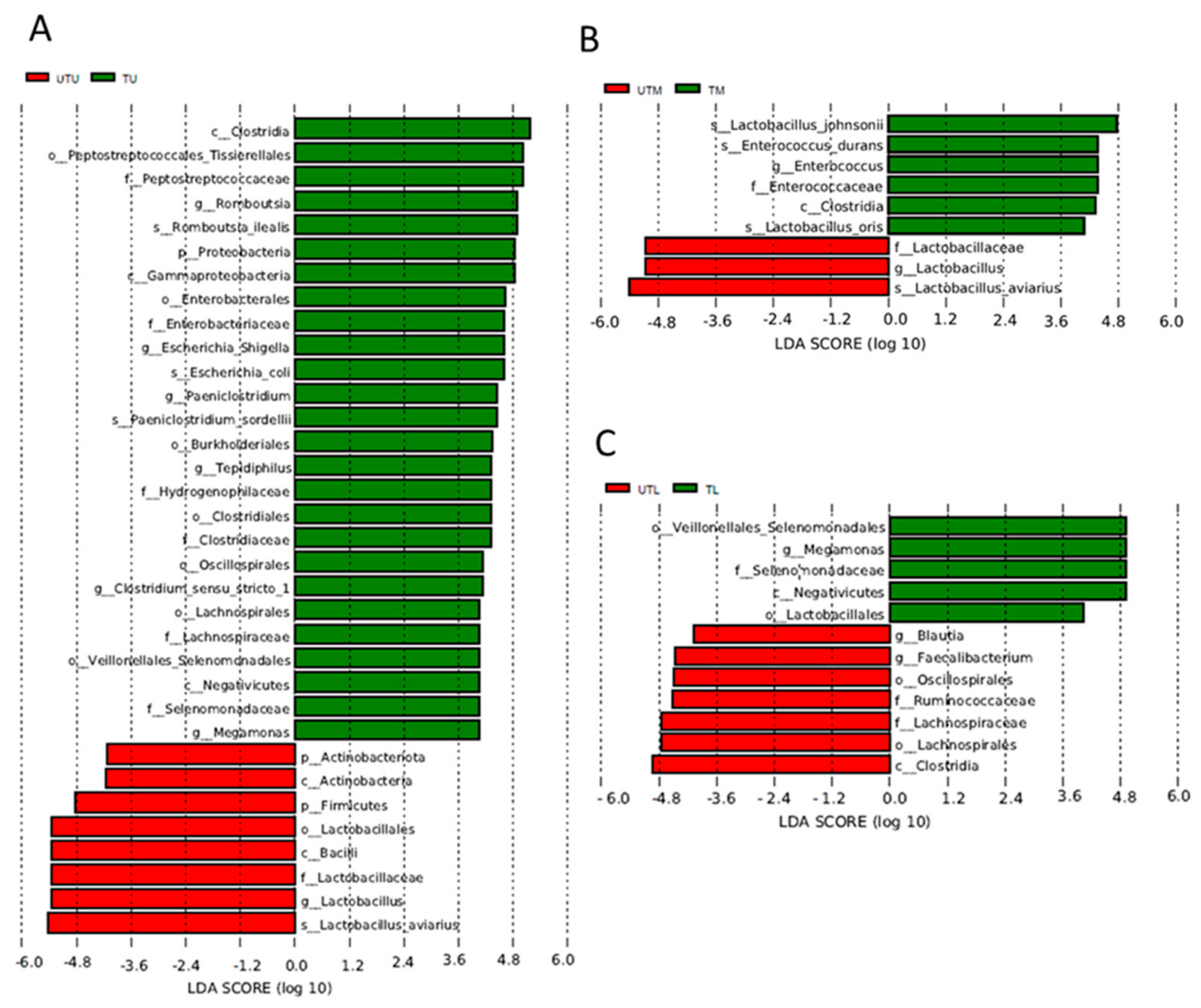
3. Results
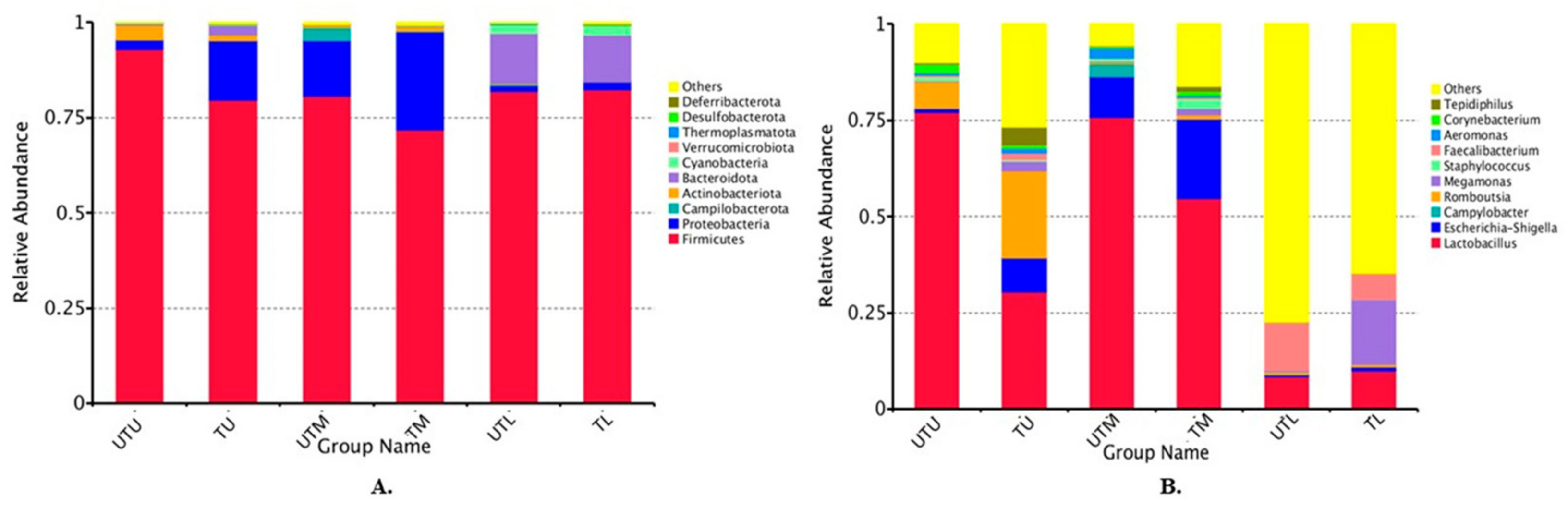
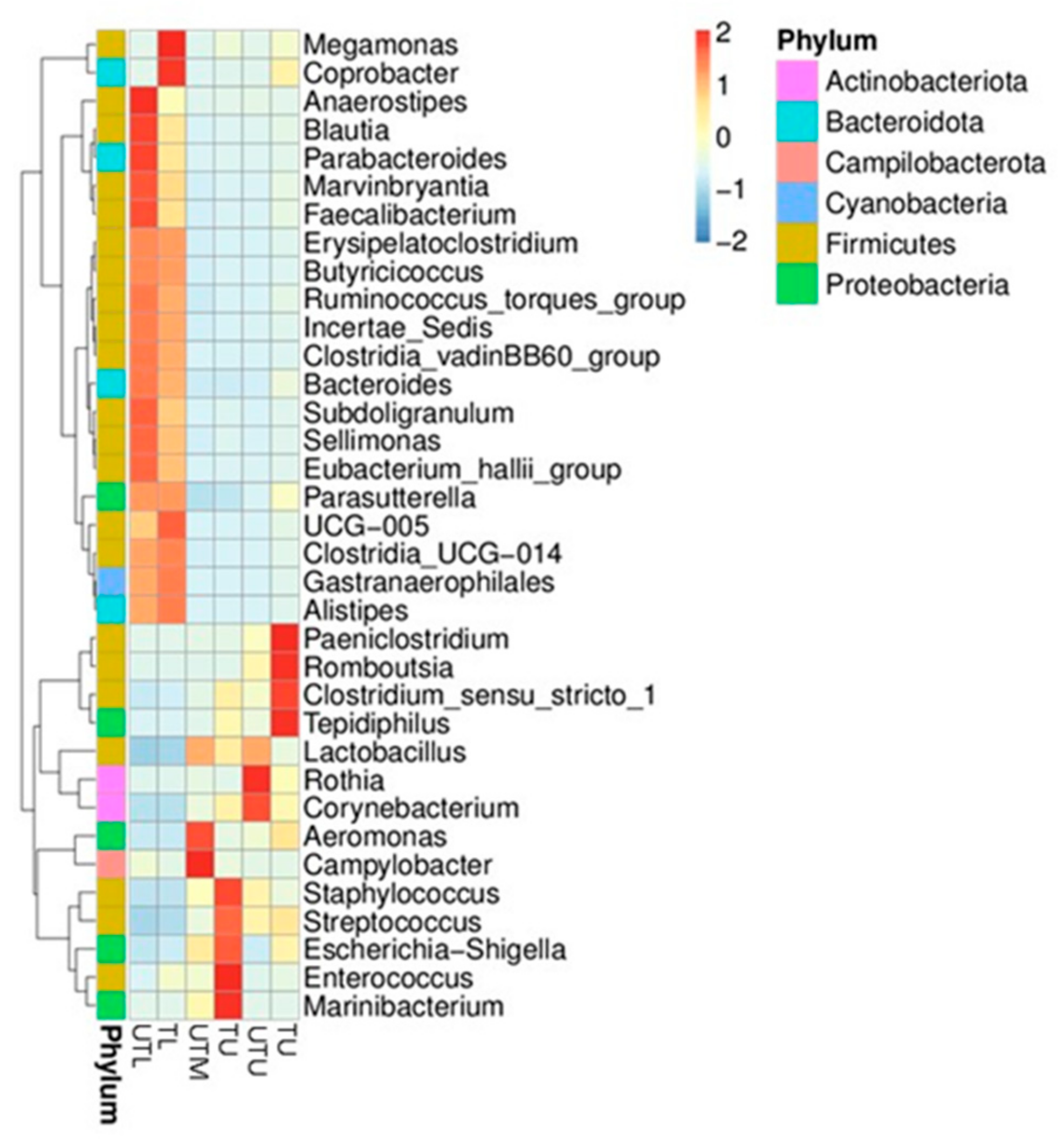
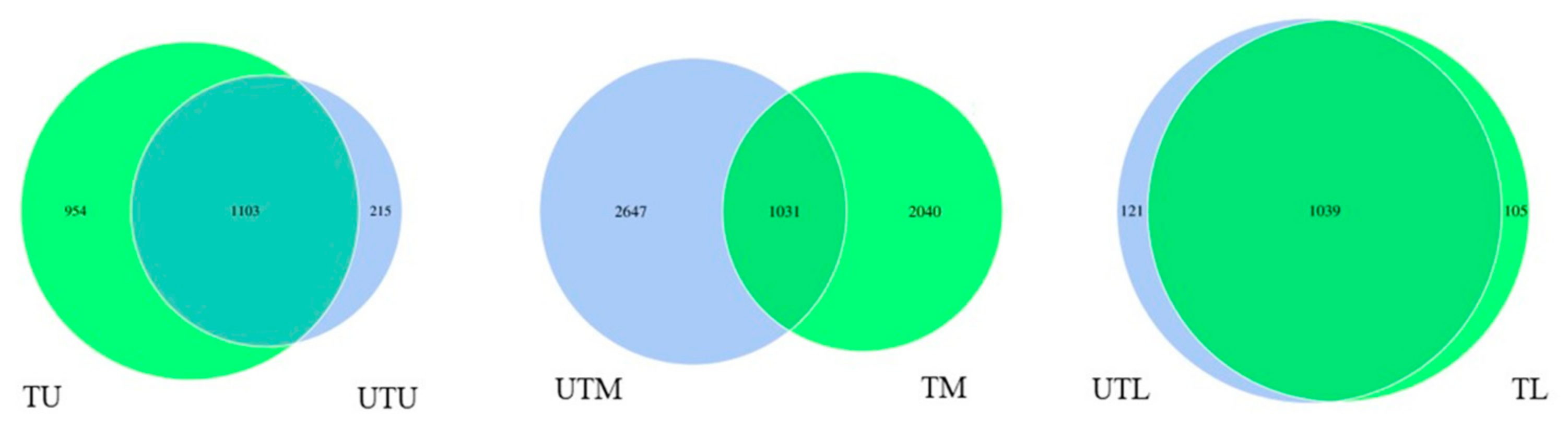
3.1. OTU Identification and Taxonomic Annotation
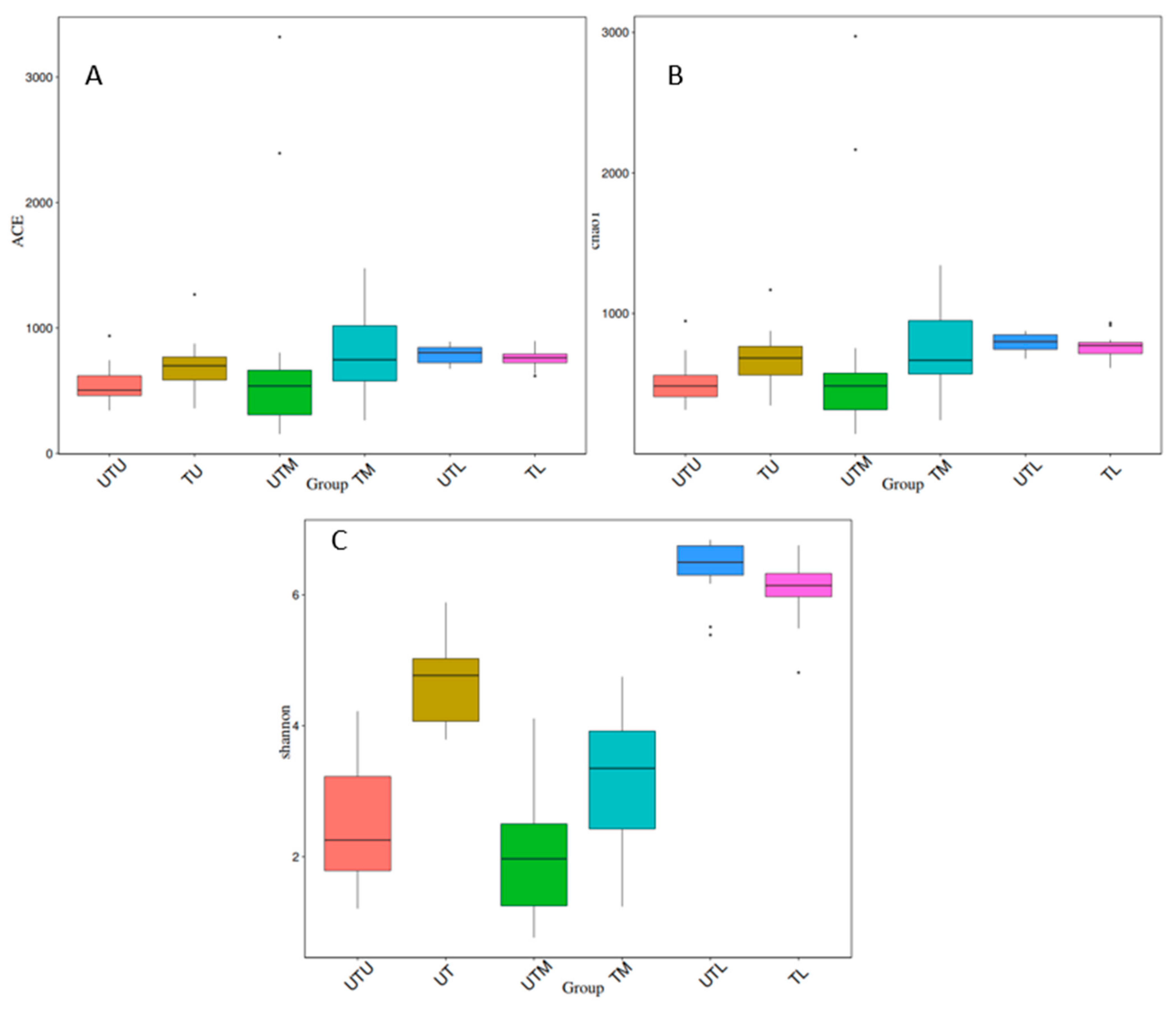
3.2. Alpha Diversity Analysis
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Roth, N.; Käsbohrer, A.; Mayrhofer, S.; Zitz, U.; Hofacre, C.; Domig, K.J. The application of antibiotics in broiler production and the resulting antibiotic resistance in Escherichia coli: A global overview. Poult. Sci. 2019, 98, 1791–1804. [Google Scholar] [CrossRef] [PubMed]
- Mehdi, Y.; Létourneau-Montminy, M.P.; Gaucher, M.; Chorfi, Y.; Suresh, G.; Rouissi, T.; Brar, S.K.; Côté, C.; Ramirez, A.A.; Godbout, S. Use of antibiotics in broiler production: Global impacts and alternatives. Anim. Nutr. 2018, 4, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Pereira, R.; Bortoluzzi, C.; Durrer, A.; Fagundes, N.S.; Pedroso, A.A.; Rafael, J.M.; de Lima Perim, J.E.; Zavarize, K.C.; Napty, G.S.; Andreote, F.D.; et al. Performance and intestinal microbiota of chickens receiving probiotic in the feed and submitted to antibiotic therapy. J. Anim. Physiol. Ani. Nutr. 2019, 103, 72–86. [Google Scholar] [CrossRef] [PubMed]
- Schokker, D.; de Klerk, B.; Borg, R.; Bossers, A.; Rebel, J.M.J. Factors Influencing the Succession of the Fecal Microbiome in Broilers. Livest. Sci. 2021, 247, 104486. [Google Scholar] [CrossRef]
- Clavijo, V.; Flórez, M.J.V. The gastrointestinal microbiome and its association with the control of pathogens in broiler chicken production: A review. Poult. Sci. 2018, 97, 1006–1021. [Google Scholar] [CrossRef]
- Rychlik, I. Composition and function of chicken gut microbiota. Animals 2020, 10, 103. [Google Scholar] [CrossRef] [Green Version]
- Stanley, D.; Geier, M.S.; Denman, S.E.; Haring, V.R.; Crowley, T.M.; Hughes, R.J.; Moore, R.J. Identification of chicken intestinal microbiota correlated with the efficiency of energy extraction from feed. Vet. Microbiol. 2013, 164, 85–92. [Google Scholar] [CrossRef]
- Feye, K.M.; Baxter, M.F.; Tellez-Isaias, G.; Kogut, M.H.; Ricke, S.C. Influential factors on the composition of the conventionally raised broiler gastrointestinal microbiomes. Poult. Sci. 2020, 99, 653–659. [Google Scholar] [CrossRef] [PubMed]
- Rebollar Serrano, M.E.; Serrano, M.E.R. Evaluación de Indicadores Productivos en Pollos de Engorda al Incluir Maíz y Pasta de Soya Extruidos y Malta de Cebada. Master’s Thesis, Universidad de Colima, Colima, Mexico, 2002; p. 134. [Google Scholar]
- Xiong, W.; Wang, Y.; Sun, Y.; Ma, L.; Zeng, Q.; Jiang, X.; Li, A.; Zeng, Z.; Zhang, T. Antibiotic-mediated changes in the fecal microbiome of broiler chickens define the incidence of antibiotic resistance genes. Microbiome 2018, 6, 34. [Google Scholar] [CrossRef] [Green Version]
- Irish Farmers Association. Poultry Council Report May 2020. Available online: https://www.ifa.ie/policy-areas/poultry-council-report-may-2020/ (accessed on 22 November 2022).
- Martin, H.; Manzanilla, E.G.; More, S.J.; O’Neill, L.; Bradford, L.; Carty, C.I.; Collins, Á.B.; McAloon, C.G. Current antimicrobial use in farm animals in the Republic of Ireland. Ir. Vet. J. 2020, 73, 11. [Google Scholar] [CrossRef]
- Yu, Y.; Lee, C.; Kim, J.; Hwang, S. Group-specific primer and probe sets to detect methanogenic communities using quantitative real-time polymerase chain reaction. Biotechnol. Bioeng. 2005, 89, 670–679. [Google Scholar] [CrossRef]
- Magoč, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bokulich, N.A.; Subramanian, S.; Faith, J.J.; Gevers, D.; Gordon, J.I.; Knight, R.; Mills, D.A.; Caporaso, J.G. Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat. Meth. 2013, 10, 57–59. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinnger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Gonzalez Peña, A.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Meth. 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haas, B.J.; Gevers, D.; Earl, A.M.; Feldgarden, M.; Ward, D.V.; Giannoukos, G.; Ciulla, D.; Tabbaa, D.; Highlander, S.K.; Sodergren, E.; et al. Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome Res. 2011, 21, 494–504. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Meth. 2013, 10, 996–998. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, 590–596. [Google Scholar] [CrossRef]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naïve Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, J.R.; Nagarajan, N.; Pop, M. Statistical Methods for Detecting Differentially Abundant Features in Clinical Metagenomic Samples. PLoS Comput. Biol. 2009, 5, e1000352. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.; Morrison, M.; Yu, Z. Bacterial census of poultry intestinal microbiome. Poult. Sci. 2012, 92, 671–683. [Google Scholar] [CrossRef] [PubMed]
- Elokil, A.A.; Abouelezz, K.F.M.; Ahmad, H.I.; Pan, Y.; Li, S. Investigation of the Impacts of Antibiotic Exposure on the Diversity of the Gut Microbiota in Chicks. Animals 2020, 10, 896. [Google Scholar] [CrossRef] [PubMed]
- Videnska, P.; Faldynova, M.; Juricova, H.; Babak, V.; Sisak, F.; Havlickova, H.; Rychlik, I. Chicken faecal microbiota and disturbances induced by single or repeated therapy with tetracycline and streptomycin. BMC Vet. Res. 2013, 9, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, D.; Yu, Z. Intestinal microbiome of poultry and its interaction with host and diet. Gut Microbes 2013, 5, 108–119. [Google Scholar] [CrossRef] [Green Version]
- Fernández-Bravo, A.; Figueras, M.J. An update on the genus Aeromonas: Taxonomy, epidemiology, and pathogenicity. Microorganisms 2020, 8, 129. [Google Scholar] [CrossRef] [Green Version]
- Praveen, P.K.; Debnath, C.; Shekhar, S.; Dalai, N.; Ganguly, S. Incidence of Aeromonas spp. infection in fish and chicken meat and its related public health hazards: A review. Vet. World 2016, 9, 6–11. [Google Scholar] [CrossRef] [Green Version]
- Igbinosa, I.H. Antibiogram profiling and pathogenic status of Aeromonas species recovered from Chicken. Saudi J. Biol. Sci. 2014, 21, 481–485. [Google Scholar] [CrossRef] [Green Version]
- Greene, G.; Koolman, L.; Whyte, P.; Lynch, H.; Coffey, A.; Lucey, B.; Egan, J.; O’Connor, L.; Bolton, D. An in vitro investigation of the survival and/or growth of Campylobacter jejuni in broiler digestate from different feed types. Lett. Appl. Microbiol. 2020, 72, 36–40. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, S.; Chaloner, G.; Kemmett, K.; Davidson, N.; Williams, N.; Kipar, A.; Humphrey, T.; Wigley, P. Campylobacter jejuni is not merely a commensal in commercial broiler chickens and affects bird welfare. mBio 2014, 5, e01364-14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oakley, B.B.; Lillehoj, H.S.; Kogut, M.H.; Kim, W.K.; Maurer, J.J.; Pedroso, A.; Lee, M.D.; Collett, S.R.; Johnson, T.J.; Cox, N.A. The chicken gastrointestinal microbiome. FEMS Microbiol. Lett. 2014, 360, 100–112. [Google Scholar] [CrossRef] [PubMed]
- Koolman, L.; Whyte, P.; Bolton, D.J. An investigation of broiler caecal Campylobacter counts at first and second thinning. J. Appl. Microbiol. 2014, 117, 876–881. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples/p-Value | ACE | Chao1 | Shannon | |||
|---|---|---|---|---|---|---|
| Tukey | Wilcox | Tukey | Wilcox | Tukey | Wilcox | |
| UTU v TU | 0.79264 | 0.0074 1 | 0.65193 | 0.0045 1 | 0 1 | 0 1 |
| UTM v TM | 0.98912 | 0.0011 1 | 0.98309 | 0.002 1 | 0 1 | 0 1 |
| UTL v TL | 0.99994 | 0.5193 | 0.99987 | 0.4221 | 0.76 | 0.0266 1 |
| UTU v UTM | 0.65501 | 0.7621 | 0.75718 | 0.9435 | 0.41 | 0.1065 |
| UTM v UTL | 0.99915 | 0 1 | 0.91642 | 0 1 | 0 1 | 0 1 |
| UTU v UTL | 0.4216 | 0 1 | 0.18507 | 0 1 | 0 1 | 0 1 |
| TU v TM | 0.9571 | 0.3333 | 0.99541 | 0.7022 | 0 1 | 0 1 |
| TM v TL | 0.9971 | 0.6757 | 1 | 0.2472 | 0 1 | 0 1 |
| TU v TL | 0.99901 | 0.1652 | 0.99277 | 0.1208 | 0 1 | 0 1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Greene, G.; Koolman, L.; Whyte, P.; Burgess, C.; Bolton, D. The Gut Microbiota of Broilers Reared with and without Antibiotic Treatment. Microorganisms 2023, 11, 876. https://doi.org/10.3390/microorganisms11040876
Greene G, Koolman L, Whyte P, Burgess C, Bolton D. The Gut Microbiota of Broilers Reared with and without Antibiotic Treatment. Microorganisms. 2023; 11(4):876. https://doi.org/10.3390/microorganisms11040876
Chicago/Turabian StyleGreene, Genevieve, Leonard Koolman, Paul Whyte, Catherine Burgess, and Declan Bolton. 2023. "The Gut Microbiota of Broilers Reared with and without Antibiotic Treatment" Microorganisms 11, no. 4: 876. https://doi.org/10.3390/microorganisms11040876