
A Streamlined Approach for Fluorescence Labelling of Low-Copy-Number Plasmids for Determination of Conjugation Frequency by Flow Cytometry

Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Strains
2.2. Bacterial Growth Rate Assays
2.3. Flow Cytometry-Based Measurement of Transconjugant Frequency
2.4. Verification of Transconjugant Frequency by Selective Agar Plate Count
2.5. Flow Cytometry Data Analysis
3. Results and Discussion
3.1. Labelling of E. coli J53Az Recipient Strain with a mCherry-Bearing Non-Conjugative Small Plasmid
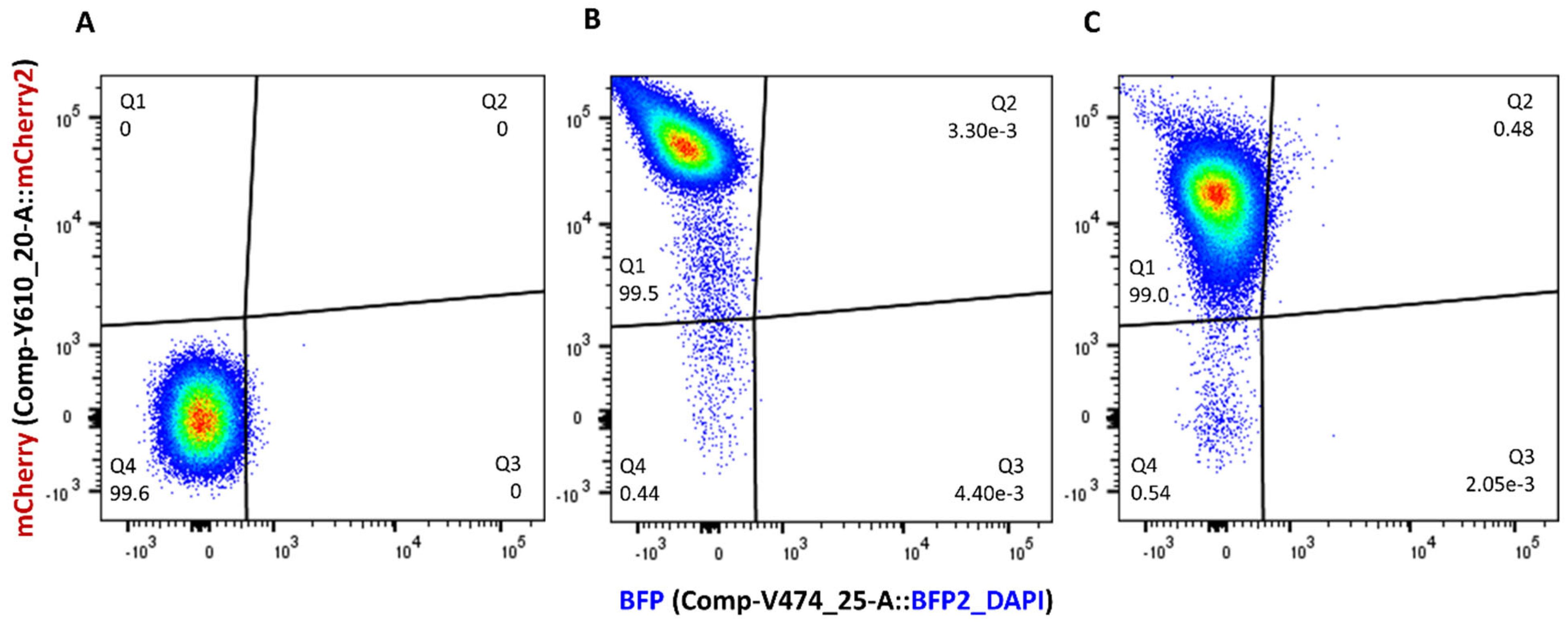
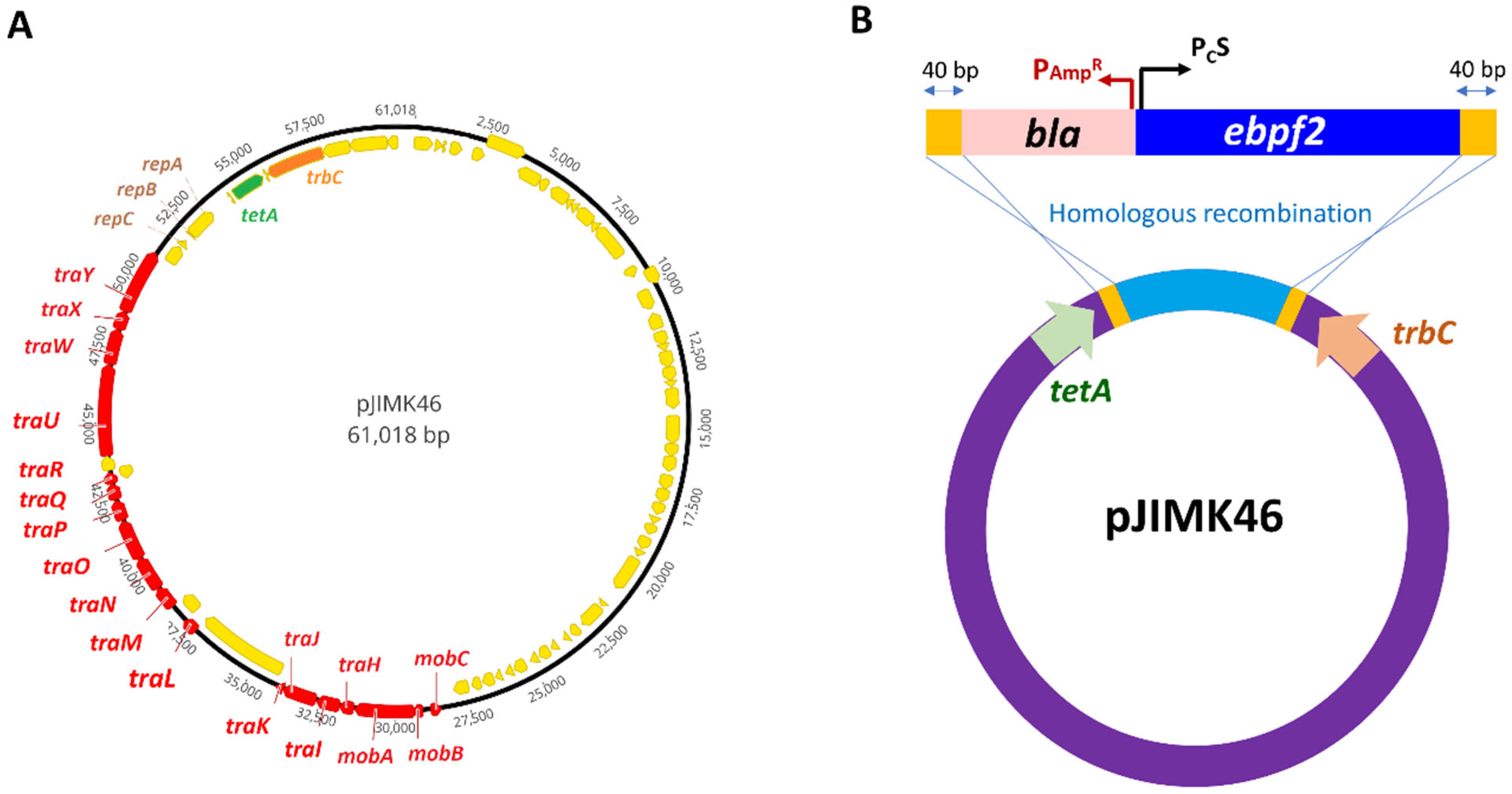
3.2. Labelling of Low-Copy-Number Plasmids with Blue Fluorescent Protein Gene
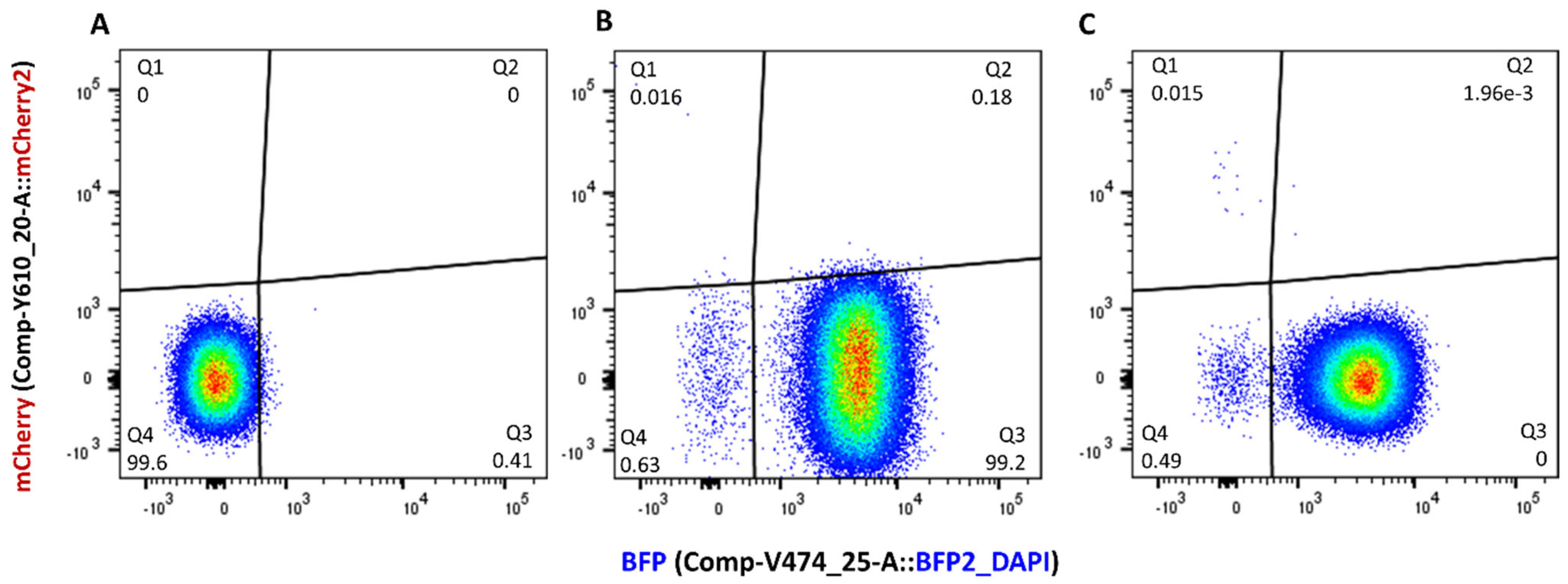
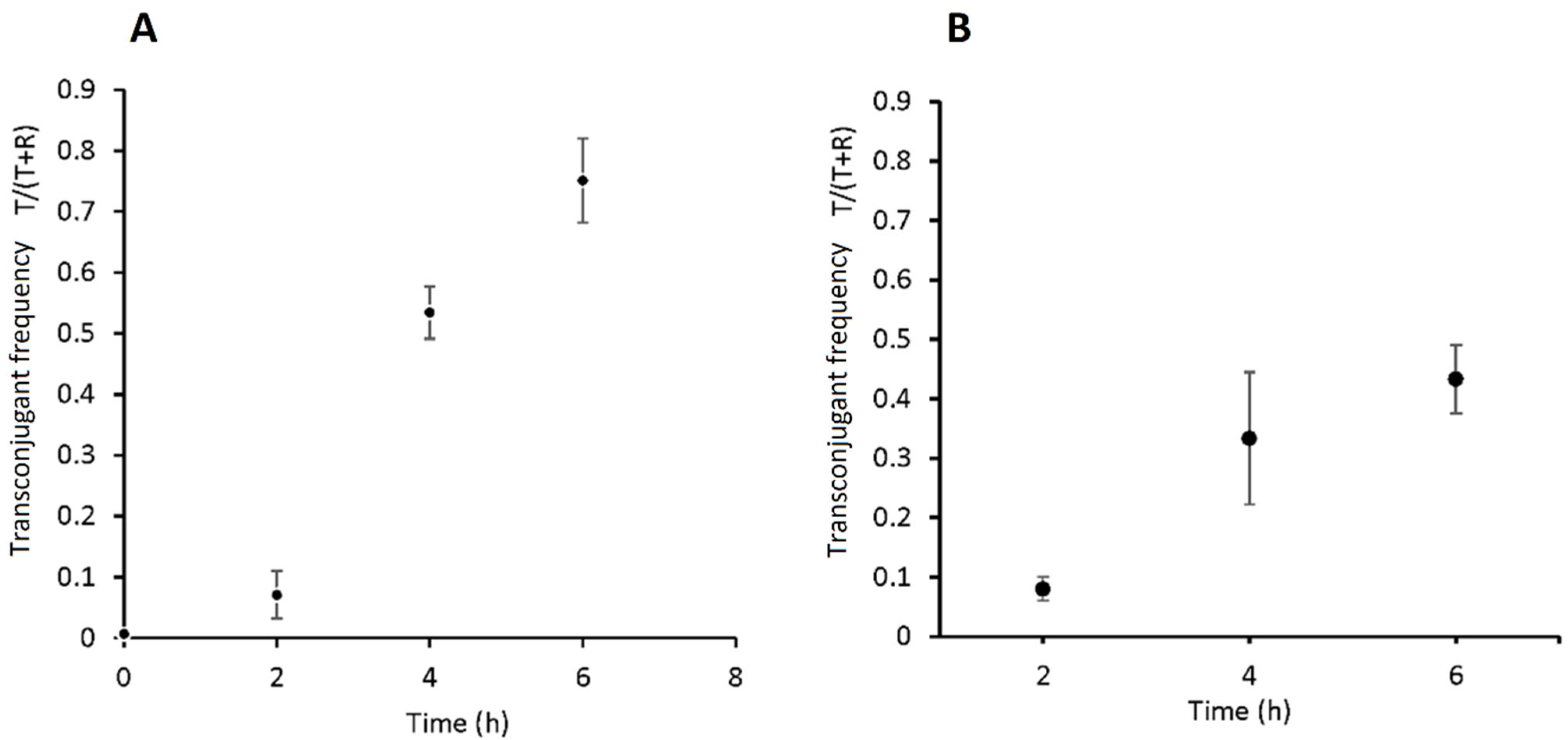
3.3. Quantifying Transconjugant Frequencies Using Flow Cytometry and Conventional Plate Count Methods
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Norman, A.; Hansen, L.H.; Sørensen, S.J. Conjugative plasmids: Vessels of the communal gene pool. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2009, 364, 2275–2289. [Google Scholar] [CrossRef] [PubMed]
- Ochman, H.; Lawrence, J.G.; Groisman, E.A. Lateral gene transfer and the nature of bacterial innovation. Nature 2000, 405, 299–304. [Google Scholar] [CrossRef]
- Virolle, C.; Goldlust, K.; Djermoun, S.; Bigot, S.; Lesterlin, C. Plasmid transfer by conjugation in Gram-negative bacteria: From the cellular to the community level. Genes 2020, 11, 1239. [Google Scholar] [CrossRef]
- Low, E.A.R.K.B. Brenner’s Encyclopedia of Genetics, 2nd ed.; Elsevier/Academic Press: Amsterdam, The Netherlands, 2013; p. 7. [Google Scholar]
- Partridge, S.R.; Kwong, S.M.; Firth, N.; Jensen, S.O. Mobile genetic elements associated with antimicrobial resistance. Clin. Microbiol. Rev. 2018, 31, e00088-17. [Google Scholar] [CrossRef]
- Zatyka, M.; Thomas, C.M. Control of genes for conjugative transfer of plasmids and other mobile elements. FEMS Microbiol. Rev. 1998, 21, 291–319. [Google Scholar] [CrossRef] [PubMed]
- del Campo, I.; Ruiz, R.; Cuevas, A.; Revilla, C.; Vielva, L.; de la Cruz, F. Determination of conjugation rates on solid surfaces. Plasmid 2012, 67, 174–182. [Google Scholar] [CrossRef]
- Levin, B.R.; Lipsitch, M.; Perrot, V.; Schrag, S.; Antia, R.; Simonsen, L.; Walker, N.M.; Stewart, F.M. The population genetics of antibiotic resistance. Clin. Infect. Dis. 1997, 24 (Suppl. 1), S9–S16. [Google Scholar] [CrossRef] [PubMed]
- Simonsen, L.; Gordon, D.M.; Stewart, F.M.; Levin, B.R. Estimating the rate of plasmid transfer: An end-point method. J. Gen. Microbiol. 1990, 136, 2319–2325. [Google Scholar] [CrossRef]
- Huisman, J.S.; Benz, F.; Duxbury, S.J.N.; de Visser, J.A.G.M.; Hall, A.R.; Fischer, E.A.J.; Bonhoeffer, S. Estimating plasmid conjugation rates: A new computational tool and a critical comparison of methods. Plasmid 2022, 121, 102627. [Google Scholar] [CrossRef]
- Snyder, T.L. The relative errors of bacteriological plate counting methods. J. Bacteriol. 1947, 54, 641–654. [Google Scholar] [CrossRef]
- Mudge, C.S.; Lawler, B.M. Is the statistical method applicable to the bacterial plate count? J. Bacteriol. 1928, 15, 207–221. [Google Scholar] [CrossRef]
- Cuny, C.; Lesbats, M.; Dukan, S. Induction of a global stress response during the first step of Escherichia coli plate growth. Appl. Environ. Microbiol. 2007, 73, 885–889. [Google Scholar] [CrossRef]
- Barer, M.R.; Harwood, C.R. Bacterial viability and culturability. Adv. Microb. Physiol. 1999, 41, 93–137. [Google Scholar] [CrossRef]
- Beal, J.; Farny, N.G.; Haddock-Angelli, T.; Selvarajah, V.; Baldwin, G.S.; Buckley-Taylor, R.; Gershater, M.; Kiga, D.; Marken, J.; Sanchania, V.; et al. Robust estimation of bacterial cell count from optical density. Commun. Biol. 2020, 3, 512. [Google Scholar] [CrossRef]
- Møller, T.S.B.; Liu, G.; Boysen, A.; Thomsen, L.E.; Lüthje, F.L.; Mortensen, S.; Møller-Jensen, J.; Olsen, J.E. Treatment with cefotaxime affects expression of conjugation associated proteins and conjugation transfer frequency of an IncI1 plasmid in Escherichia coli. Front. Microbiol. 2017, 8, 2365. [Google Scholar] [CrossRef]
- Liu, G.; Bogaj, K.; Bortolaia, V.; Olsen, J.E.; Thomsen, L.E. Antibiotic-induced, increased conjugative transfer is common to diverse naturally occurring ESBL plasmids in Escherichia coli. Front. Microbiol. 2019, 10, 2119. [Google Scholar] [CrossRef]
- Benz, F.; Huisman, J.S.; Bakkeren, E.; Herter, J.A.; Stadler, T.; Ackermann, M.; Diard, M.; Egli, A.; Hall, A.R.; Hardt, W.D.; et al. Plasmid- and strain-specific factors drive variation in ESBL-plasmid spread in vitro and in vivo. ISME J. 2020, 15, 862–878. [Google Scholar] [CrossRef]
- Bakkeren, E.; Huisman, J.S.; Fattinger, S.A.; Hausmann, A.; Furter, M.; Egli, A.; Slack, E.; Sellin, M.E.; Bonhoeffer, S.; Regoes, R.R.; et al. Salmonella persisters promote the spread of antibiotic resistance plasmids in the gut. Nature 2019, 573, 276–280. [Google Scholar] [CrossRef]
- Stevenson, C.; Hall, J.P.J.; Brockhurst, M.A.; Harrison, E. Plasmid stability is enhanced by higher-frequency pulses of positive selection. Proc. Biol. Sci. 2018, 285, 20172497. [Google Scholar] [CrossRef]
- Pinedo, C.A.; Smets, B.F. Conjugal TOL transfer from Pseudomonas putida to Pseudomonas aeruginosa: Effects of restriction proficiency, toxicant exposure, cell density ratios, and conjugation detection method on observed transfer efficiencies. Appl. Environ. Microbiol. 2005, 71, 51–57. [Google Scholar] [CrossRef]
- Flett, F.; Mersinias, V.; Smith, C.P. High efficiency intergeneric conjugal transfer of plasmid DNA from Escherichia coli to methyl DNA-restricting streptomycetes. FEMS Microbiol. Lett. 1997, 155, 223–229. [Google Scholar] [CrossRef]
- Nikodinovic, J.; Barrow, K.D.; Chuck, J.A. High frequency transformation of the amphotericin-producing bacterium Streptomyces nodosus. J. Microbiol. Methods 2003, 55, 273–277. [Google Scholar] [CrossRef] [PubMed]
- Kamruzzaman, M.; Shoma, S.; Thomas, C.M.; Partridge, S.R.; Iredell, J.R. Plasmid interference for curing antibiotic resistance plasmids in vivo. PLoS ONE 2017, 12, e0172913. [Google Scholar] [CrossRef] [PubMed]
- Phornphisutthimas, S.; Thamchaipenet, A.; Panijpan, B. Conjugation in Escherichia coli: A laboratory exercise. Biochem. Mol. Biol. Educ. 2007, 35, 440–445. [Google Scholar] [CrossRef] [PubMed]
- Shintani, M.; Ohkuma, M.; Kimbara, K. High-resolution comparison of bacterial conjugation frequencies. J. Vis. Exp. 2019, 143, e57812. [Google Scholar] [CrossRef]
- Pinilla-Redondo, R.; Riber, L.; Sørensen, S.J. Fluorescence recovery allows the implementation of fluorescence reporter gene platform applicable for the detection and quantification of horizontal gene transfer in anoxic environments. Appl. Environ. Microbiol. 2018, 84, e02507-17. [Google Scholar] [CrossRef]
- Buckner, M.M.C.; Ciusa, M.L.; Meek, R.W.; Moorey, A.R.; McCallum, G.E.; Prentice, E.L.; Reid, J.P.; Alderwick, L.J.; Di Maio, A.; Piddock, L.J.V. HIV drugs inhibit transfer of plasmids carrying extended-spectrum β-lactamase and carbapenemase genes. mBio 2020, 11, e03355-19. [Google Scholar] [CrossRef]
- Klümper, U.; Riber, L.; Dechesne, A.; Sannazzarro, A.; Hansen, L.H.; Sørensen, S.J.; Smets, B.F. Broad host range plasmids can invade an unexpectedly diverse fraction of a soil bacterial community. ISME J. 2015, 9, 934–945. [Google Scholar] [CrossRef]
- Klümper, U.; Dechesne, A.; Riber, L.; Brandt, K.K.; Gülay, A.; Sørensen, S.J.; Smets, B.F. Metal stressors consistently modulate bacterial conjugal plasmid uptake potential in a phylogenetically conserved manner. ISME J. 2017, 11, 152–165. [Google Scholar] [CrossRef] [PubMed]
- Malwade, A.; Nguyen, A.; Sadat-Mousavi, P.; Ingalls, B.P. Predictive modeling of a batch filter mating process. Front. Microbiol. 2017, 8, 461. [Google Scholar] [CrossRef]
- Wu, A.Y.; Kamruzzaman, M.; Iredell, J.R. Specialised functions of two common plasmid mediated toxin-antitoxin systems, ccdAB and pemIK, in Enterobacteriaceae. PLoS ONE 2020, 15, e0230652. [Google Scholar] [CrossRef]
- Chang, A.C.; Cohen, S.N. Construction and characterization of amplifiable multicopy DNA cloning vehicles derived from the P15A cryptic miniplasmid. J. Bacteriol. 1978, 134, 1141–1156. [Google Scholar] [CrossRef]
- Kim, K.H.; Choi, K.; Bartley, B.; Sauro, H.M. Controlling E. coli gene expression noise. IEEE Trans. Biomed. Circuits Syst. 2015, 9, 497–504. [Google Scholar] [CrossRef]
- Reynolds, M.G. Compensatory evolution in rifampin-resistant Escherichia coli. Genetics 2000, 156, 1471–1481. [Google Scholar] [CrossRef]
- Matsumura, Y.; Peirano, G.; Pitout, J.D.D. Complete genome sequence of Escherichia coli J53, an azide-resistant laboratory strain used for conjugation experiments. Genome Announc. 2018, 6, e00433-18. [Google Scholar] [CrossRef]
- Yi, H.; Cho, Y.J.; Yong, D.; Chun, J. Genome sequence of Escherichia coli J53, a reference strain for genetic studies. J. Bacteriol. 2012, 194, 3742–3743. [Google Scholar] [CrossRef]
- de la Cruz, F.; Grinsted, J. Genetic and molecular characterization of Tn21, a multiple resistance transposon from R100.1. J. Bacteriol. 1982, 151, 222–228. [Google Scholar] [CrossRef]
- Bassalo, M.C.; Garst, A.D.; Halweg-Edwards, A.L.; Grau, W.C.; Domaille, D.W.; Mutalik, V.K.; Arkin, A.P.; Gill, R.T. Rapid and efficient one-step metabolic pathway integration in E. coli. ACS Synth. Biol. 2016, 5, 561–568. [Google Scholar] [CrossRef]
- Guzman, L.M.; Belin, D.; Carson, M.J.; Beckwith, J. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J. Bacteriol. 1995, 177, 4121–4130. [Google Scholar] [CrossRef]
- Lev, S.; Kaufman-Francis, K.; Desmarini, D.; Juillard, P.G.; Li, C.; Stifter, S.A.; Feng, C.G.; Sorrell, T.C.; Grau, G.E.; Bahn, Y.S.; et al. Pho4 is essential for dissemination of Cryptococcus neoformans to the host brain by promoting phosphate uptake and growth at alkaline pH. mSphere 2017, 2, e00381-16. [Google Scholar] [CrossRef]
- Kamruzzaman, M.; Patterson, J.D.; Shoma, S.; Ginn, A.N.; Partridge, S.R.; Iredell, J.R. Relative strengths of promoters provided by common mobile genetic elements associated with resistance gene expression in Gram-negative bacteria. Antimicrob. Agents Chemother. 2015, 59, 5088–5091. [Google Scholar] [CrossRef] [PubMed]
- Espedido, B.A.; Partridge, S.R.; Iredell, J.R. blaIMP-4 in different genetic contexts in Enterobacteriaceae isolates from Australia. Antimicrob. Agents Chemother. 2008, 52, 2984–2987. [Google Scholar] [CrossRef]
- Amábile-Cuevas, C.F. Antibiotic resistance: From Darwin to Lederberg to Keynes. Microb. Drug Resist. 2013, 19, 73–87. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.M.; Nielsen, K.M. Mechanisms of, and barriers to, horizontal gene transfer between bacteria. Nat. Rev. Microbiol. 2005, 3, 711–721. [Google Scholar] [CrossRef]
- Dimitriu, T.; Marchant, L.; Buckling, A.; Raymond, B. Bacteria from natural populations transfer plasmids mostly towards their kin. Proc. Biol. Sci. 2019, 286, 20191110. [Google Scholar] [CrossRef]
- Cavaliere, R.; Ball, J.L.; Turnbull, L.; Whitchurch, C.B. The biofilm matrix destabilizers, EDTA and DNaseI, enhance the susceptibility of nontypeable Hemophilus influenzae biofilms to treatment with ampicillin and ciprofloxacin. Microbiologyopen 2014, 3, 557–567. [Google Scholar] [CrossRef]
- Finnegan, S.; Percival, S.L. EDTA: An antimicrobial and antibiofilm agent for use in wound care. Adv. Wound Care 2015, 4, 415–421. [Google Scholar] [CrossRef]
- Roostalu, J.; Jõers, A.; Luidalepp, H.; Kaldalu, N.; Tenson, T. Cell division in Escherichia coli cultures monitored at single cell resolution. BMC Microbiol. 2008, 8, 68. [Google Scholar] [CrossRef]
- Pletnev, P.; Osterman, I.; Sergiev, P.; Bogdanov, A.; Dontsova, O. Survival guide: Escherichia coli in the stationary phase. Acta Nat. 2015, 7, 22–33. [Google Scholar] [CrossRef]
- Gaforio, J.J.; Serrano, M.J.; Ortega, E.; Algarra, I.; Alvarez de Cienfuegos, G. Use of SYTOX green dye in the flow cytometric analysis of bacterial phagocytosis. Cytometry 2002, 48, 93–96. [Google Scholar] [CrossRef] [PubMed]
- Lina, W.; Shuo, W.; Yiyi, S.; Xu, W.; Xiaomei, Y. Applications and challenges for single-bacteria analysis by flow cytometry. Sci. China Chem. 2016, 59, 30–39. [Google Scholar] [CrossRef]
- Bhowmick, D.; Sheridan, R.T.C.; Bushnell, T.P.; Spalding, K.L. Practical guidelines for optimization and characterization of the Beckman Coulter CytoFLEX™ platform. Cytometry A 2020, 97, 800–810. [Google Scholar] [CrossRef] [PubMed]
- Lister, A. Detecting and Counting Bacteria with the CytoFLEX Research Flow Cytometer; CytoFlex flow Cytometer Application Notes; Beckman Coulter Life Sciences: Indianapolis, IN, USA, 2015. [Google Scholar]
- Zhong, X.; Droesch, J.; Fox, R.; Top, E.M.; Krone, S.M. On the meaning and estimation of plasmid transfer rates for surface-associated and well-mixed bacterial populations. J. Theor. Biol. 2012, 294, 144–152. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Measured Quantity | Unit | Descriptions | Reference |
|---|---|---|---|
| Transconjugant frequency; T/(T + R) | Dimensionless | Ratio of the number of transconjugants to the combined populations of transconjugants and recipients at the time of measurement; also known as transfer frequency, proportion (or fraction) of transconjugants in literature | [7,18,19,20] |
| Transconjugant frequency; T/(DR) | mL CFU−1 | Ratio of the number of transconjugants to the product of the number of donors multiplied by the number of recipients at the time of measurement | [21] |
| Exconjugant frequency; T/R0 | Dimensionless | The number of transconjugants at the end of the conjugation per recipient cell at the start of conjugation (R0) | [22,23] |
| Conjugation efficiency; T/D0 | Dimensionless | The number of transconjugants at the end of the conjugation per donor cell at the start of conjugation (D0) | [24,25] |
| Bacterial Strain | Characteristics | Reference |
|---|---|---|
| E. coli J53Az | A sodium azide-resistant derivative of the E. coli K-12 reference strain | [36] |
| E. coli UB5201Rf | Rifampicin-resistant derivative of the E. coli UB5201 wild-type strain with amino acid substitution S512P in the β-subunit of RNA polymerase encoded by rpoB | [38] |
| Primer Name | Oligonucleotide Sequence (5′->3′) a |
|---|---|
| mCherry_XbaI-F | CCATCTAGAGTGAGCAAGGGCGAGGAGGAT |
| mCherry_SacI-R | GCTGAGCTCACGATTCTTTTACTTGTAC |
| ccdAB_EC_HpaI-F | CGGTTAACACGAAACGGGAATGCGGTAA |
| ccdAB_EC_SacI-R | GCGAGCTCATGACTGCAGACTGGCTGTGT |
| PcS_XbaI-F | CACTCTAGAAAACGGATGAAGGCACGAAC |
| PcS_XbaI-RBS-R | GACTCTAGATCCTCCTTTGCTGCTCCATAACATCAA |
| pJIMK46::PcS-ebfp2-F | AGGCCTATGCCATGCGGGTCAAGGCGACTTCCGGCTACTTGAGTAAACTTGGTCTGACAG |
| pJIMK46::PcS- ebfp2-R | TTTGCCGGCGTTGTTAATCAGGAGGCCAAACGATGGCTGAGGGATCCGAATTCAGGAGGT |
| Plasmid | Characteristics b | Source |
|---|---|---|
| pSS9 | A pBR322-based vector containing a green fluorescent protein gene (gfpuv); TcR | [39] |
| pSS9_mCherry | pSS9 with an mCherry red fluorescent protein gene and the ccdABEC toxin–antitoxin system of an E. coli IncF plasmid cloned into the XbaI/SacI and SacI/HpaI sites, respectively; TcR | This study |
| pBAD33-Gm | A low–medium-copy-number vector with the p15A origin of replication and an L-arabinose inducible promoter; GmR | [33,34,40] |
| p_mCherry-stable | pBAD33 with mCherry-ccdABEC cloned into the XbaI/HindIII site, followed by single-site cloning of the PcS promoter into XbaI; GmR | This study |
| pJIMK46 | IncM interference plasmid derived from pJIBE401 clinical plasmid by deleting the multi-drug resistance (MDR) region and the toxin gene pemK of the pemIK TA system | [24] |
| pMini_blue-strong | Codon-optimized enhanced blue fluorescent protein gene (ebfp2) was cloned into the multiple cloning site of the NEB cloning vector pMiniT downstream of a strong constitutive promoter PcS; AmpR | This study |
| pConj_blue-strong | pJIMK46 was tagged with PcS-ebfp2-bla between the trbC and tetA genes in pJIMK46; AmpR |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qi, Q.; Kamruzzaman, M.; Iredell, J.R. A Streamlined Approach for Fluorescence Labelling of Low-Copy-Number Plasmids for Determination of Conjugation Frequency by Flow Cytometry. Microorganisms 2023, 11, 878. https://doi.org/10.3390/microorganisms11040878
Qi Q, Kamruzzaman M, Iredell JR. A Streamlined Approach for Fluorescence Labelling of Low-Copy-Number Plasmids for Determination of Conjugation Frequency by Flow Cytometry. Microorganisms. 2023; 11(4):878. https://doi.org/10.3390/microorganisms11040878
Chicago/Turabian StyleQi, Qin, Muhammad Kamruzzaman, and Jonathan R. Iredell. 2023. "A Streamlined Approach for Fluorescence Labelling of Low-Copy-Number Plasmids for Determination of Conjugation Frequency by Flow Cytometry" Microorganisms 11, no. 4: 878. https://doi.org/10.3390/microorganisms11040878