
Genome-Wide Association Study of Nucleotide Variants Associated with Resistance to Nine Antimicrobials in Mycoplasma bovis

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection, Isolation, DNA Extraction, and Sequencing
2.2. Genome Assembly, Quality Control, and Data Preprocessing
2.3. Antimicrobial Resistance Phenotypes
2.4. Genome-Wide Association Study Pipeline
2.5. Visualization and Verification
3. Results and Discussion
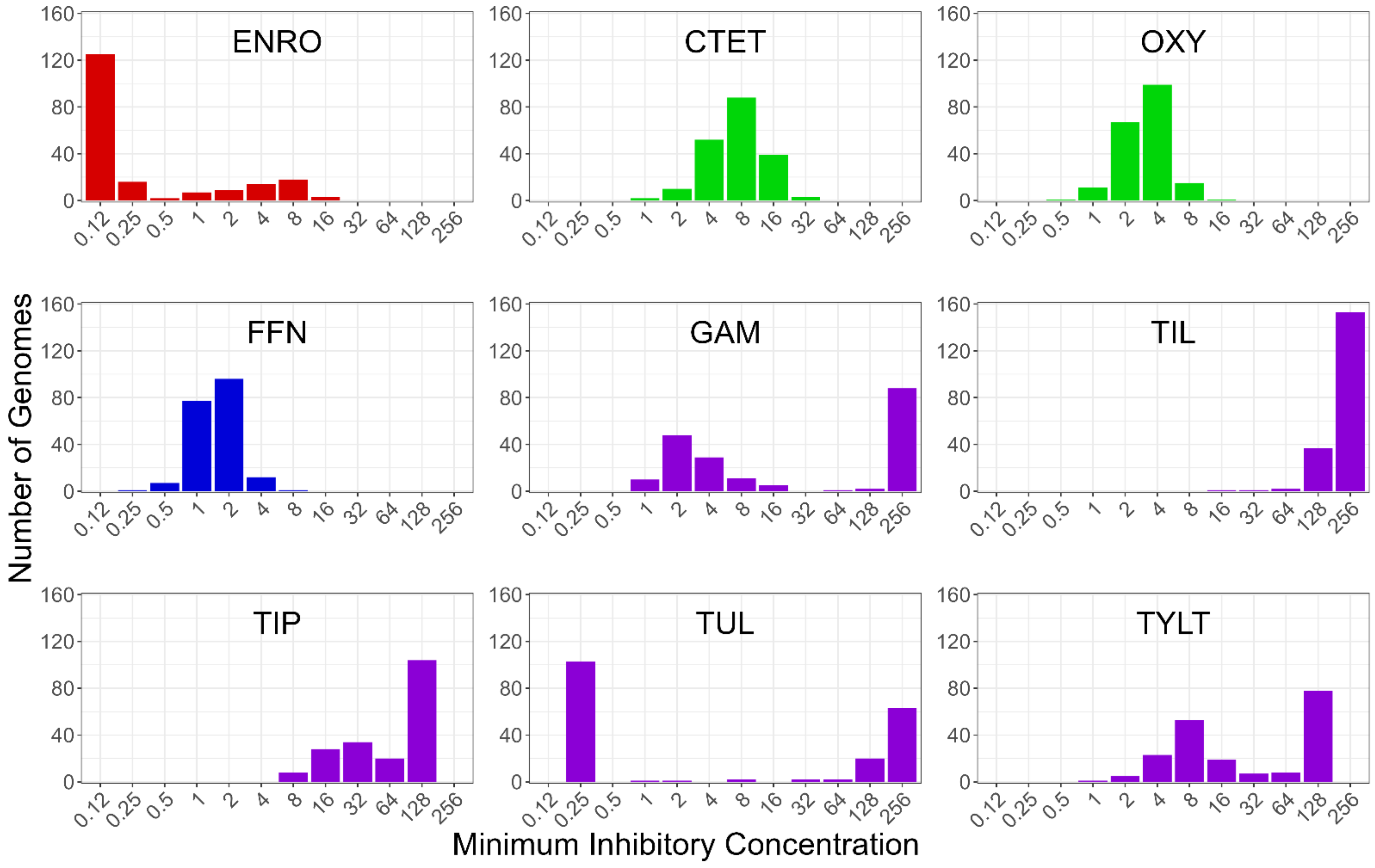
3.1. Analysis of the Genome Assembly Quality and Minimum Inhibitory Concentrations
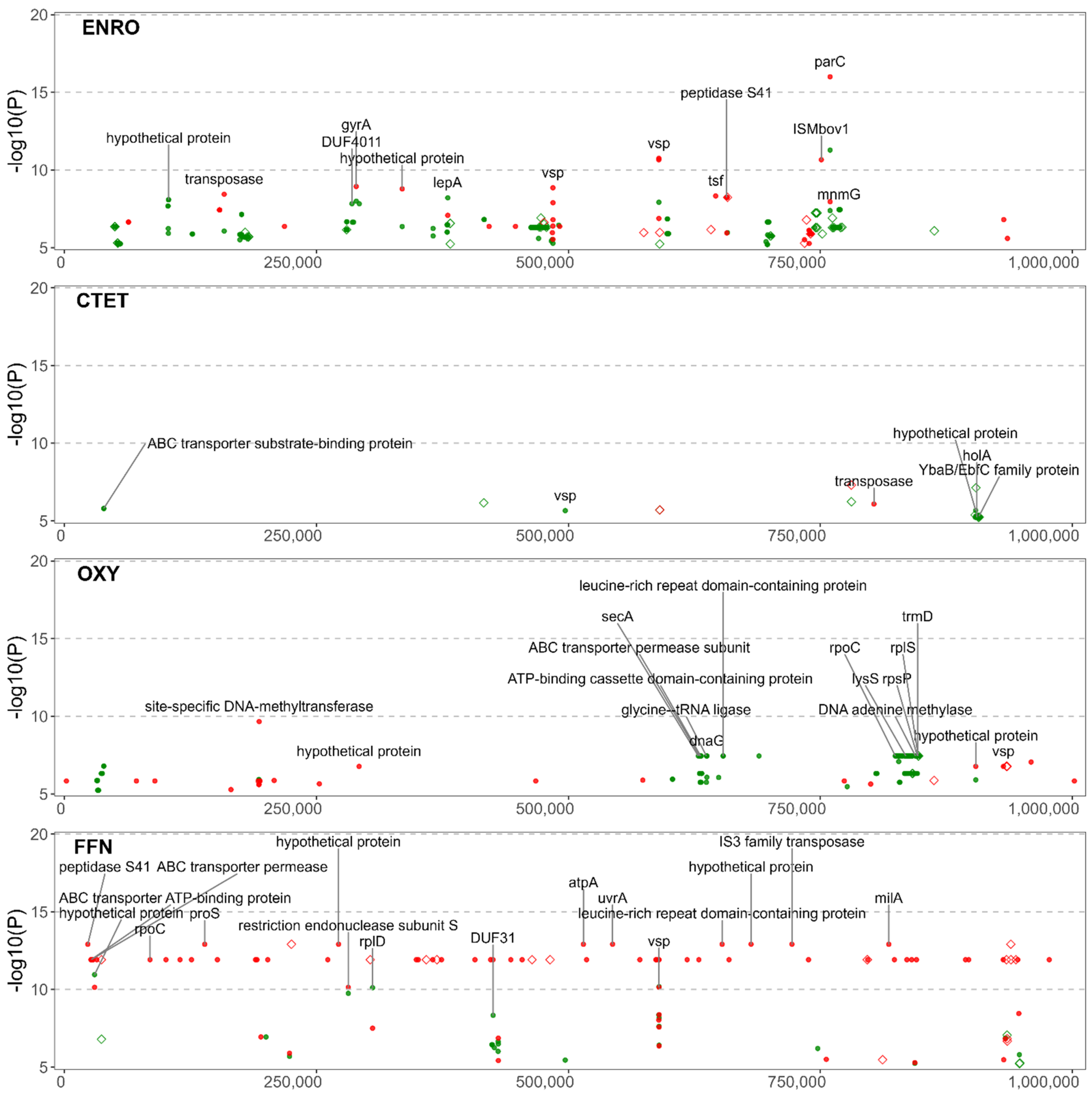
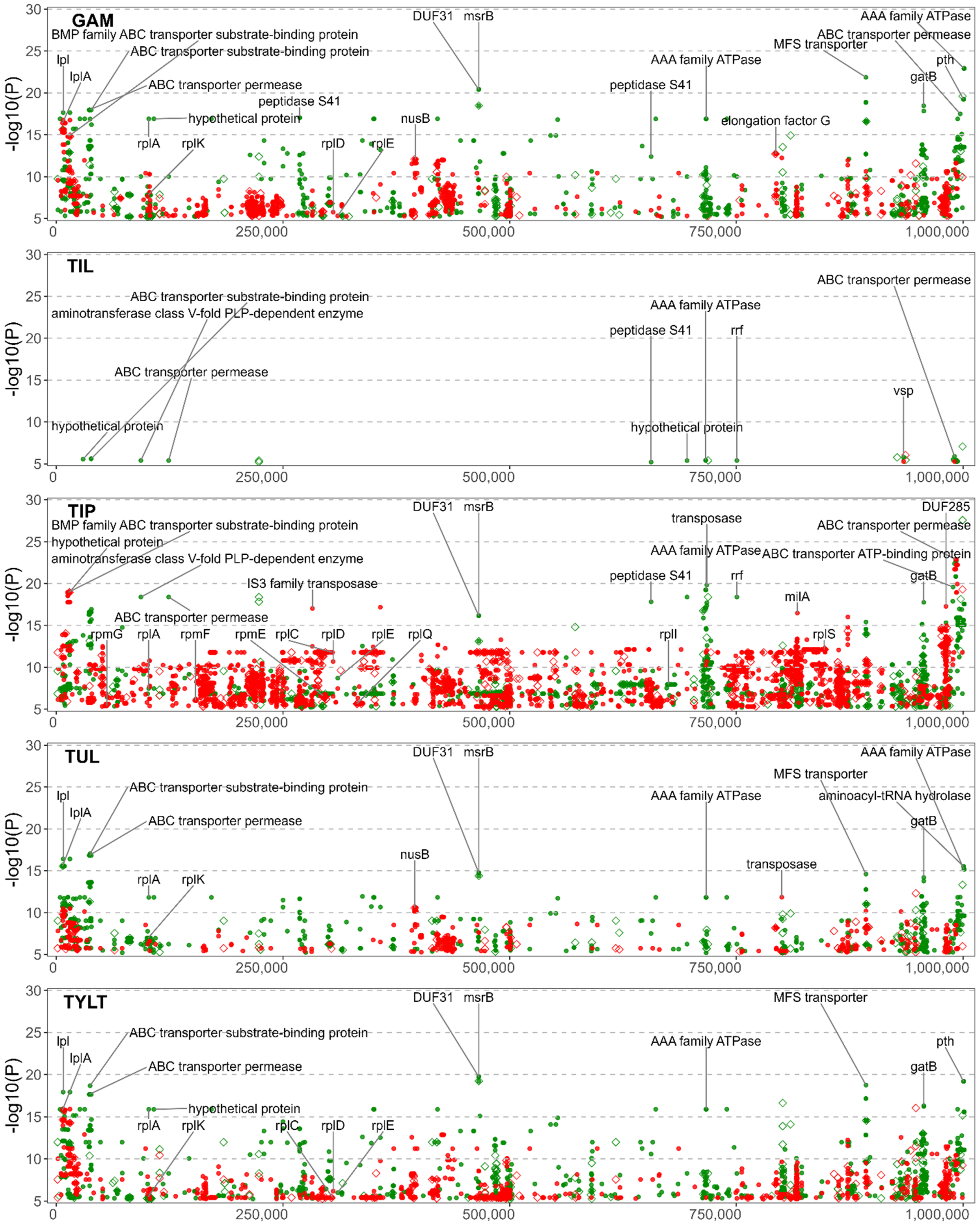
3.2. Summary Visualization of AMR-Associated Nucleotide Variants within Coding Sequences
3.3. Investigation into the p-Value Inflation Present within GWAS Results
3.4. Significant NVs Specific to Enrofloxacin Resistance
3.5. Significant NVs Specific to Chlortetracycline and Oxytetracycline Resistance
3.6. Significant NVs Specific to Florfenicol, Gamithromycin, Tilmicosin, Tildipirosin, Tulathromycin, and Tylosin Tartrate Resistance
3.7. Variants Associated with Multi-Drug Resistance
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nicholas, R.A.J.; Ayling, R.D. Mycoplasma bovis: Disease, Diagnosis, and Control. Res. Vet. Sci. 2003, 74, 105–112. [Google Scholar] [CrossRef]
- Maunsell, F.P.; Woolums, A.R.; Francoz, D.; Rosenbusch, R.F.; Step, D.L.; Wilson, D.J.; Janzen, E.D. Mycoplasma bovis Infections in Cattle. J. Vet. Intern. Med. 2011, 25, 772–783. [Google Scholar] [CrossRef] [PubMed]
- Gagea, M.I.; Bateman, K.G.; Shanahan, R.A.; van Dreumel, T.; McEwen, B.J.; Carman, S.; Archambault, M.; Caswell, J.L. Naturally Occurring Mycoplasma bovis—Associated Pneumonia and Polyarthritis in Feedlot Beef Calves. J. Vet. Diagn. Investig. 2006, 18, 29–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caswell, J.L.; Bateman, K.G.; Cai, H.Y.; Castillo-Alcala, F. Mycoplasma bovis in Respiratory Disease of Feedlot Cattle. Vet. Clin. Food Anim. Pract. 2010, 26, 365–379. [Google Scholar] [CrossRef]
- Brault, S.A.; Hannon, S.J.; Gow, S.P.; Otto, S.J.G.; Booker, C.W.; Morley, P.S. Calculation of Antimicrobial Use Indicators in Beef Feedlots—Effects of Choice of Metric and Standardized Values. Front. Vet. Sci. 2019, 6, 330. [Google Scholar] [CrossRef]
- Jelinski, M.; Kinnear, A.; Gesy, K.; Andrés-Lasheras, S.; Zaheer, R.; Weese, S.; McAllister, T.A. Antimicrobial Sensitivity Testing of Mycoplasma bovis Isolates Derived from Western Canadian Feedlot Cattle. Microorganisms 2020, 8, 124. [Google Scholar] [CrossRef] [Green Version]
- Kinnear, A.; McAllister, T.A.; Zaheer, R.; Waldner, M.; Ruzzini, A.C.; Andrés-Lasheras, S.; Parker, S.; Hill, J.E.; Jelinski, M.D. Investigation of Macrolide Resistance Genotypes in Mycoplasma bovis Isolates from Canadian Feedlot Cattle. Pathogens 2020, 9, 622. [Google Scholar] [CrossRef]
- Rosenbusch, R.F.; Kinyon, J.M.; Apley, M.; Funk, N.D.; Smith, S.; Hoffman, L.J. In Vitro Antimicrobial Inhibition Profiles of Mycoplasma bovis Isolates Recovered from Various Regions of the United States from 2002 to 2003. J. Vet. Diagn. Investig. 2005, 17, 436–441. [Google Scholar] [CrossRef] [Green Version]
- Hata, E.; Harada, T.; Itoh, M. Relationship between Antimicrobial Susceptibility and Multilocus Sequence Type of Mycoplasma bovis Isolates and Development of a Method for Rapid Detection of Point Mutations Involved in Decreased Susceptibility to Macrolides, Lincosamides, Tetracyclines, and Spectinomycin. Appl. Environ. Microbiol. 2019, 85, e00575-19. [Google Scholar] [CrossRef] [Green Version]
- Ayling, R.D.; Baker, S.E.; Nicholas, R.A.; Peek, M.L.; Simon, A.J. Comparison of in Vitro Activity of Danofloxacin, Florfenicol, Oxytetracycline, Spectinomycin and Tilmicosin against Recent Field Isolates of Mycoplasma bovis. Vet. Rec. 2000, 146, 745–747. [Google Scholar] [CrossRef]
- Gerchman, I.; Levisohn, S.; Mikula, I.; Lysnyansky, I. In Vitro Antimicrobial Susceptibility of Mycoplasma bovis Isolated in Israel from Local and Imported Cattle. Vet. Microbiol. 2009, 137, 268–275. [Google Scholar] [CrossRef] [PubMed]
- Gautier-Bouchardon, A.V.; Ferré, S.; Grand, D.L.; Paoli, A.; Gay, E.; Poumarat, F. Overall Decrease in the Susceptibility of Mycoplasma bovis to Antimicrobials over the Past 30 Years in France. PLoS ONE 2014, 9, e87672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sulyok, K.M.; Kreizinger, Z.; Fekete, L.; Hrivnák, V.; Magyar, T.; Jánosi, S.; Schweitzer, N.; Turcsányi, I.; Makrai, L.; Erdélyi, K.; et al. Antibiotic Susceptibility Profiles of Mycoplasma bovis Strains Isolated from Cattle in Hungary, Central Europe. BMC Vet. Res. 2014, 10, 256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heuvelink, A.; Reugebrink, C.; Mars, J. Antimicrobial Susceptibility of Mycoplasma bovis Isolates from Veal Calves and Dairy Cattle in the Netherlands. Vet. Microbiol. 2016, 189, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Klein, U.; de Jong, A.; Moyaert, H.; El Garch, F.; Leon, R.; Richard-Mazet, A.; Rose, M.; Maes, D.; Pridmore, A.; Thomson, J.R.; et al. Antimicrobial Susceptibility Monitoring of Mycoplasma hyopneumoniae and Mycoplasma bovis Isolated in Europe. Vet. Microbiol. 2017, 204, 188–193. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, B.P. Resistance to Rifampicin: A Review. J. Antibiot. 2014, 67, 625–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor-Robinson, D.; Bébéar, C. Antibiotic Susceptibilities of Mycoplasmas and Treatment of Mycoplasmal Infections. J. Antimicrob. Chemother. 1997, 40, 622–630. [Google Scholar] [CrossRef] [Green Version]
- Lerner, U.; Amram, E.; Ayling, R.D.; Mikula, I.; Gerchman, I.; Harrus, S.; Teff, D.; Yogev, D.; Lysnyansky, I. Acquired Resistance to the 16-Membered Macrolides Tylosin and Tilmicosin by Mycoplasma bovis. Vet. Microbiol. 2014, 168, 365–371. [Google Scholar] [CrossRef]
- Sulyok, K.M.; Kreizinger, Z.; Wehmann, E.; Lysnyansky, I.; Bányai, K.; Marton, S.; Jerzsele, Á.; Rónai, Z.; Turcsányi, I.; Makrai, L.; et al. Mutations Associated with Decreased Susceptibility to Seven Antimicrobial Families in Field and Laboratory-Derived Mycoplasma bovis Strains. Antimicrob. Agents Chemother. 2017, 61, e01983-16. [Google Scholar] [CrossRef] [Green Version]
- Amram, E.; Mikula, I.; Schnee, C.; Ayling, R.D.; Nicholas, R.A.J.; Rosales, R.S.; Harrus, S.; Lysnyansky, I. 16S RRNA Gene Mutations Associated with Decreased Susceptibility to Tetracycline in Mycoplasma bovis. Antimicrob. Agents Chemother. 2015, 59, 796–802. [Google Scholar] [CrossRef] [Green Version]
- Sato, T.; Okubo, T.; Usui, M.; Higuchi, H.; Tamura, Y. Amino Acid Substitutions in GyrA and ParC Are Associated with Fluoroquinolone Resistance in Mycoplasma bovis Isolates from Japanese Dairy Calves. J. Vet. Med. Sci. 2013, 75, 1063–1065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khalil, D.; Becker, C.A.M.; Tardy, F. Alterations in the Quinolone Resistance-Determining Regions and Fluoroquinolone Resistance in Clinical Isolates and Laboratory-Derived Mutants of Mycoplasma bovis: Not All Genotypes May Be Equal. Appl. Environ. Microbiol. 2016, 82, 1060–1068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lysnyansky, I.; Mikula, I.; Gerchman, I.; Levisohn, S. Rapid Detection of a Point Mutation in the ParC Gene Associated with Decreased Susceptibility to Fluoroquinolones in Mycoplasma bovis. Antimicrob. Agents Chemother. 2009, 53, 4911–4914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fàbrega, A.; Madurga, S.; Giralt, E.; Vila, J. Mechanism of Action of and Resistance to Quinolones. Microb. Biotechnol. 2009, 2, 40–61. [Google Scholar] [CrossRef] [Green Version]
- Calcutt, M.J.; Lysnyansky, I.; Sachse, K.; Fox, L.K.; Nicholas, R.A.J.; Ayling, R.D. Gap Analysis of Mycoplasma bovis Disease, Diagnosis and Control: An Aid to Identify Future Development Requirements. Transbound. Emerg. Dis. 2018, 65, 91–109. [Google Scholar] [CrossRef] [Green Version]
- Ledger, L.; Eidt, J.; Cai, H.Y. Identification of Antimicrobial Resistance-Associated Genes through Whole Genome Sequencing of Mycoplasma bovis Isolates with Different Antimicrobial Resistances. Pathogens 2020, 9, 588. [Google Scholar] [CrossRef]
- Bokma, J.; Vereecke, N.; Nauwynck, H.; Haesebrouck, F.; Theuns, S.; Pardon, B.; Boyen, F. Genome-Wide Association Study Reveals Genetic Markers for Antimicrobial Resistance in Mycoplasma bovis. Microbiol. Spectr. 2021, 9, e00262-21. [Google Scholar] [CrossRef]
- Tam, V.; Patel, N.; Turcotte, M.; Bossé, Y.; Paré, G.; Meyre, D. Benefits and Limitations of Genome-Wide Association Studies. Nat. Rev. Genet. 2019, 20, 467–484. [Google Scholar] [CrossRef]
- Vilhjálmsson, B.J.; Nordborg, M. The Nature of Confounding in Genome-Wide Association Studies. Nat. Rev. Genet. 2013, 14, 1–2. [Google Scholar] [CrossRef]
- Falush, D.; Bowden, R. Genome-Wide Association Mapping in Bacteria? Trends Microbiol. 2006, 14, 353–355. [Google Scholar] [CrossRef]
- Yang, J.; Weedon, M.N.; Purcell, S.; Lettre, G.; Estrada, K.; Willer, C.J.; Smith, A.V.; Ingelsson, E.; O’connell, J.R.; Mangino, M.; et al. Genomic inflation factors under polygenic inheritance. Eur. J. Hum. Genet. 2011, 19, 807–812. [Google Scholar] [CrossRef] [PubMed]
- Hong, E.P.; Park, J.W. Sample Size and Statistical Power Calculation in Genetic Association Studies. Genomics Inf. 2012, 10, 117. [Google Scholar] [CrossRef] [PubMed]
- Brynildsrud, O.; Bohlin, J.; Scheffer, L.; Eldholm, V. Rapid Scoring of Genes in Microbial Pan-Genome-Wide Association Studies with Scoary. Genome Biol. 2016, 17, 238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Springer. Theoretical and Applied Genetics: Submission Guidelines. Available online: https://www.springer.com/journal/122/submission-guidelines (accessed on 6 June 2022).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Li, H. Aligning Sequence Reads, Clone Sequences and Assembly Contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map Format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Garrison, E.; Marth, G. Haplotype-Based Variant Detection from Short-Read Sequencing. arXiv 2012, arXiv:1207.3907. [Google Scholar]
- Lees, J.A.; Galardini, M.; Bentley, S.D.; Weiser, J.N.; Corander, J. Pyseer: A Comprehensive Tool for Microbial Pangenome-Wide Association Studies. Bioinformatics 2018, 34, 4310–4312. [Google Scholar] [CrossRef]
- Lees, J.A.; Vehkala, M.; Välimäki, N.; Harris, S.R.; Chewapreecha, C.; Croucher, N.J.; Marttinen, P.; Davies, M.R.; Steer, A.C.; Tong, S.Y.C.; et al. Sequence Element Enrichment Analysis to Determine the Genetic Basis of Bacterial Phenotypes. Nat. Commun. 2016, 7, 12797. [Google Scholar] [CrossRef]
- Ondov, B.D.; Treangen, T.J.; Melsted, P.; Mallonee, A.B.; Bergman, N.H.; Koren, S.; Phillippy, A.M. Mash: Fast Genome and Metagenome Distance Estimation Using MinHash. Genome Biol. 2016, 17, 132. [Google Scholar] [CrossRef] [Green Version]
- Seemann, T. Prokka: Rapid Prokaryotic Genome Annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-Generation PLINK: Rising to the Challenge of Larger and Richer Datasets. GigaScience 2015, 4, s13742-015. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H. Ggplot2. WIREs Comput. Stat. 2011, 3, 180–185. [Google Scholar] [CrossRef]
- Devlin, B.; Roeder, K. Genomic Control for Association Studies. Biometrics 1999, 55, 997–1004. [Google Scholar] [CrossRef]
- Cardon, L.R.; Palmer, L.J. Population Stratification and Spurious Allelic Association. Lancet 2003, 361, 598–604. [Google Scholar] [CrossRef]
- Hinrichs, A.L.; Larkin, E.K.; Suarez, B.K. Population Stratification and Patterns of Linkage Disequilibrium. Genet. Epidemiol. 2009, 33, S88–S92. [Google Scholar] [CrossRef] [Green Version]
- Gustafsson, C.; Govindarajan, S.; Minshull, J. Codon Bias and Heterologous Protein Expression. Trends Biotechnol. 2004, 22, 346–353. [Google Scholar] [CrossRef]
- Brandis, G.; Hughes, D. The Selective Advantage of Synonymous Codon Usage Bias in Salmonella. PLoS Genet. 2016, 12, e1005926. [Google Scholar] [CrossRef] [Green Version]
- Zhao, C.; Hartke, A.; La Sorda, M.; Posteraro, B.; Laplace, J.-M.; Auffray, Y.; Sanguinetti, M. Role of Methionine Sulfoxide Reductases A and B of Enterococcus Faecalis in Oxidative Stress and Virulence. Infect Immun. 2010, 78, 3889–3897. [Google Scholar] [CrossRef] [Green Version]
- Romsang, A.; Atichartpongkul, S.; Trinachartvanit, W.; Vattanaviboon, P.; Mongkolsuk, S. Gene Expression and Physiological Role of Pseudomonas Aeruginosa Methionine Sulfoxide Reductases during Oxidative Stress. J. Bacteriol. 2013, 195, 3299–3308. [Google Scholar] [CrossRef] [Green Version]
- Woodhead, J.L.; Yang, K.; Oldach, D.; MacLauchlin, C.; Fernandes, P.; Watkins, P.B.; Siler, S.Q.; Howell, B.A. Analyzing the Mechanisms Behind Macrolide Antibiotic-Induced Liver Injury Using Quantitative Systems Toxicology Modeling. Pharm. Res. 2019, 36, 48. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Huang, X.; Xie, Y.; Song, M.; Zhu, K.; Ding, S. Macrolides Induce Severe Cardiotoxicity and Developmental Toxicity in Zebrafish Embryos. Sci. Total Environ. 2019, 649, 1414–1421. [Google Scholar] [CrossRef] [PubMed]
- Maiden, M.C.; Davis, E.O.; Baldwin, S.A.; Moore, D.C.; Henderson, P.J. Mammalian and Bacterial Sugar Transport Proteins Are Homologous. Nature 1987, 325, 641–643. [Google Scholar] [CrossRef] [PubMed]
- Hurdle, J.G.; O’Neill, A.J.; Chopra, I. Prospects for Aminoacyl-TRNA Synthetase Inhibitors as New Antimicrobial Agents. Antimicrob. Agents Chemother. 2005, 49, 4821–4833. [Google Scholar] [CrossRef] [Green Version]
- Chernov, V.M.; Chernova, O.A.; Mouzykantov, A.A.; Medvedeva, E.S.; Baranova, N.B.; Malygina, T.Y.; Aminov, R.I.; Trushin, M.V. Antimicrobial Resistance in Mollicutes: Known and Newly Emerging Mechanisms. FEMS Microbiol. Lett. 2018, 365, fny185. [Google Scholar] [CrossRef]
- Bielecki, P.; Lukat, P.; Hüsecken, K.; Dötsch, A.; Steinmetz, H.; Hartmann, R.W.; Müller, R.; Häussler, S. Mutation in Elongation Factor G Confers Resistance to the Antibiotic Argyrin in the Opportunistic Pathogen Pseudomonas aeruginosa. Chembiochem 2012, 13, 2339–2345. [Google Scholar] [CrossRef]
- Weisblum, B. Erythromycin Resistance by Ribosome Modification. Antimicrob. Agents Chemother. 1995, 39, 577–585. [Google Scholar]
- Siguier, P.; Gourbeyre, E.; Chandler, M. Bacterial Insertion Sequences: Their Genomic Impact and Diversity. FEMS Microbiol. Rev. 2014, 38, 865–891. [Google Scholar] [CrossRef] [Green Version]
- Higgins, C.F. ABC Transporters: From Microorganisms to Man. Annu. Rev. Cell Biol. 1992, 8, 67–113. [Google Scholar] [CrossRef]
- Marquez, B. Bacterial Efflux Systems and Efflux Pumps Inhibitors. Biochimie 2005, 87, 1137–1147. [Google Scholar] [CrossRef]
- Fyfe, C.; Grossman, T.H.; Kerstein, K.; Sutcliffe, J. Resistance to Macrolide Antibiotics in Public Health Pathogens. Cold Spring Harb. Perspect. Med. 2016, 6, a025395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.S.; Mei, S.H.; Li, Z.B.; Fei, L.I.U.; Qing, Z.H. Whole Genome Analysis Reveals New Insights into Macrolide Resistance in Mycoplasma pneumoniae. Biomed. Environ. Sci. 2017, 30, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Paulsen, I.T.; Brown, M.H.; Skurray, R.A. Proton-Dependent Multidrug Efflux Systems. Microbiol. Rev. 1996, 60, 575–608. [Google Scholar] [CrossRef] [PubMed]
- Saidijam, M.; Benedetti, G.; Ren, Q.; Xu, Z.; Hoyle, C.J.; Palmer, S.L.; Ward, A.; Bettaney, K.E.; Szakonyi, G.; Meuller, J.; et al. Microbial Drug Efflux Proteins of the Major Facilitator Superfamily. Curr. Drug Targets 2006, 7, 793–811. [Google Scholar] [CrossRef]
- Kumar, S.; Mukherjee, M.M.; Varela, M.F. Modulation of Bacterial Multidrug Resistance Efflux Pumps of the Major Facilitator Superfamily. Int. J. Bacteriol. 2013, 2013, 204141. [Google Scholar] [CrossRef] [Green Version]
- Kapoor, G.; Saigal, S.; Elongavan, A. Action and Resistance Mechanisms of Antibiotics: A Guide for Clinicians. J. Anaesthesiol. Clin. Pharmacol. 2017, 33, 300–305. [Google Scholar] [CrossRef]
- Magalhães, S.; Aroso, M.; Roxo, I.; Ferreira, S.; Cerveira, F.; Ramalheira, E.; Ferreira, R.; Vitorino, R. Proteomic Profile of Susceptible and Multidrug-Resistant Clinical Isolates of Escherichia coli and Klebsiella pneumoniae Using Label-Free and Immunoproteomic Strategies. Res. Microbiol. 2017, 168, 222–233. [Google Scholar] [CrossRef]
- Szczuka, E.; Kaznowski, A.; Bosacka, K.; Strzemieczna, E. Antimicrobial Resistance and Presence of IleS-2 Gene Encoding Mupirocin Resistance in Clinical Isolates of Methicillin-Resistant Staphylococcus sp. Folia Microbiol. 2009, 54, 153–156. [Google Scholar] [CrossRef]
- Pang, L.; Weeks, S.D.; Van Aerschot, A. Aminoacyl-tRNA Synthetases as Valuable Targets for Antimicrobial Drug Discovery. Int. J. Mol. Sci. 2021, 22, 1750. [Google Scholar] [CrossRef]
- Harvey, K.L.; Jarocki, V.M.; Charles, I.G.; Djordjevic, S.P. The Diverse Functional Roles of Elongation Factor Tu (EF-Tu) in Microbial Pathogenesis. Front. Microbiol. 2019, 10, 2351. [Google Scholar] [CrossRef]
- Kraal, B.; Zeef, L.A.; Mesters, J.R.; Boon, K.; Vorstenbosch, E.L.; Bosch, L.; Anborgh, P.H.; Parmeggiani, A.; Hilgenfeld, R. Antibiotic Resistance Mechanisms of Mutant EF-Tu Species in Escherichia coli. Biochem. Cell Biol. 1995, 73, 1167–1177. [Google Scholar] [CrossRef] [PubMed]
- Pech, M.; Karim, Z.; Yamamoto, H.; Kitakawa, M.; Qin, Y.; Nierhaus, K.H. Elongation Factor 4 (EF4/LepA) Accelerates Protein Synthesis at Increased Mg2+ Concentrations. Proc. Natl. Acad. Sci. USA 2011, 108, 3199–3203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.-H.; Hilal, T.; Loll, B.; Bürger, J.; Mielke, T.; Böttcher, C.; Said, N.; Wahl, M.C. Structure-Based Mechanisms of a Molecular RNA Polymerase/Chaperone Machine Required for Ribosome Biosynthesis. Mol. Cell 2020, 79, 1024–1036.e5. [Google Scholar] [CrossRef]
- Stagno, J.R.; Altieri, A.S.; Bubunenko, M.; Tarasov, S.G.; Li, J.; Court, D.L.; Byrd, R.A.; Ji, X. Structural Basis for RNA Recognition by NusB and NusE in the Initiation of Transcription Antitermination. Nucleic Acids Res. 2011, 39, 7803–7815. [Google Scholar] [CrossRef]
- Court, D.L.; Patterson, T.A.; Baker, T.; Costantino, N.; Mao, X.; Friedman, D.I. Structural and Functional Analyses of the Transcription-Translation Proteins NusB and NusE. J. Bacteriol. 1995, 177, 2589–2591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, Y.; Chan, S.T.; Lin, L.; Shek, T.L.; Tsang, T.F.; Zhang, Y.; Ip, M.; Chan, P.K.; Blanchard, N.; Hanquet, G.; et al. Nusbiarylins, a New Class of Antimicrobial Agents: Rational Design of Bacterial Transcription Inhibitors Targeting the Interaction between the NusB and NusE Proteins. Bioorganic Chem. 2019, 92, 103203. [Google Scholar] [CrossRef]
- Chu, A.J.; Qiu, Y.; Harper, R.; Lin, L.; Ma, C.; Yang, X. Nusbiarylins Inhibit Transcription and Target Virulence Factors in Bacterial Pathogen Staphylococcus aureus. Int. J. Mol. Sci. 2020, 21, 5772. [Google Scholar] [CrossRef]
- Rawlings, N.D.; Barrett, A.J.; Bateman, A. MEROPS: The Peptidase Database. Nucleic Acids Res. 2010, 38, D227–D233. [Google Scholar] [CrossRef]
- Allam, A.B.; Brown, M.B.; Reyes, L. Disruption of the S41 Peptidase Gene in Mycoplasma mycoides capri Impacts Proteome Profile, H2O2 Production, and Sensitivity to Heat Shock. PLoS ONE 2012, 7, e51345. [Google Scholar] [CrossRef] [Green Version]
- Ganter, S.; Miotello, G.; Manso-Silván, L.; Armengaud, J.; Tardy, F.; Gaurivaud, P.; Thiaucourt, F. Proteases as Secreted Exoproteins in Mycoplasmas from Ruminant Lungs and Their Impact on Surface-Exposed Proteins. Appl. Environ. Microbiol. 2019, 85, e01439-19. [Google Scholar] [CrossRef]
- Razin, A.; Razin, S. Methylated Bases in Mycoplasmal DNA. Nucleic Acids Res. 1980, 8, 1383–1390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wojciechowski, M.; Czapinska, H.; Bochtler, M. CpG Underrepresentation and the Bacterial CpG-Specific DNA Methyltransferase, M.MpeI. Proc. Natl. Acad. Sci. USA 2013, 110, 105–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnes, M.H.; Tarantino, P.M.; Spacciapoli, P.; Brown, N.C.; Yu, H.; Dybvig, K. DNA Polymerase III of Mycoplasma pulmonis: Isolation and Characterization of the Enzyme and Its Structural Gene, PolC. Mol. Microbiol. 1994, 13, 843–854. [Google Scholar] [CrossRef] [PubMed]
- Michel, G.; Sauvé, V.; Larocque, R.; Li, Y.; Matte, A.; Cygler, M. The Structure of the RlmB 23S RRNA Methyltransferase Reveals a New Methyltransferase Fold with a Unique Knot. Structure 2002, 10, 1303–1315. [Google Scholar] [CrossRef]
- Bachar, A.; Itzhaki, E.; Gleizer, S.; Shamshoom, M.; Milo, R.; Antonovsky, N. Point Mutations in Topoisomerase I Alter the Mutation Spectrum in E. Coli and Impact the Emergence of Drug Resistance Genotypes. Nucleic Acids Res. 2020, 48, 761–769. [Google Scholar] [CrossRef]
- Faucher, M.; Nouvel, L.-X.; Dordet-Frisoni, E.; Sagné, E.; Baranowski, E.; Hygonenq, M.-C.; Marenda, M.-S.; Tardy, F.; Citti, C. Mycoplasmas under Experimental Antimicrobial Selection: The Unpredicted Contribution of Horizontal Chromosomal Transfer. PLoS Genet. 2019, 15, e1007910. [Google Scholar] [CrossRef] [Green Version]
- Citti, C.; Dordet-Frisoni, E.; Nouvel, L.X.; Kuo, C.H.; Baranowski, E. Horizontal Gene Transfers in Mycoplasmas (Mollicutes). Curr. Issues Mol. Biol. 2018, 29, 3–22. [Google Scholar] [CrossRef] [Green Version]
- Blötz, C.; Singh, N.; Dumke, R.; Stülke, J. Characterization of an Immunoglobulin Binding Protein (IbpM) From Mycoplasma pneumoniae. Front. Microbiol. 2020, 11, 685. [Google Scholar] [CrossRef] [Green Version]
- Qin, L.; Chen, Y.; You, X. Subversion of the Immune Response by Human Pathogenic Mycoplasmas. Front Microbiol 2019, 10, 1934. [Google Scholar] [CrossRef]
- Waters, L.S.; Storz, G. Regulatory RNAs in Bacteria. Cell 2009, 136, 615–628. [Google Scholar] [CrossRef] [Green Version]
- Maeda, T.; Iwasawa, J.; Kotani, H.; Sakata, N.; Kawada, M.; Horinouchi, T.; Sakai, A.; Tanabe, K.; Furusawa, C. High-Throughput Laboratory Evolution Reveals Evolutionary Constraints in Escherichia coli. Nat. Commun. 2020, 11, 5970. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Category | ENRO | CTET | OXY | FFN | GAM | TIL | TIP | TUL | TYLT |
|---|---|---|---|---|---|---|---|---|---|
| 30S rRNA and proteins | 0 | 0 | 1 | 0 | 1 | 0 | 6 | 1 | 3 |
| 50S rRNA and proteins | 0 | 0 | 1 | 1 | 5 | 0 | 11 | 3 | 6 |
| ABC transporter | 2 | 1 | 8 | 5 | 24 | 4 | 32 | 25 | 27 |
| ATPase | 1 | 0 | 2 | 1 | 4 | 1 | 9 | 3 | 4 |
| Elongation factor | 1 | 0 | 0 | 0 | 1 | 0 | 2 | 1 | 2 |
| Hypothetical protein | 12 | 1 | 5 | 14 | 68 | 2 | 107 | 45 | 74 |
| Membrane protein | 1 | 0 | 0 | 3 | 6 | 0 | 9 | 3 | 8 |
| Methyltransferase | 2 | 0 | 6 | 4 | 8 | 0 | 21 | 6 | 10 |
| Nuclease | 4 | 0 | 2 | 3 | 12 | 0 | 22 | 9 | 18 |
| Peptidase | 3 | 0 | 1 | 3 | 13 | 1 | 22 | 12 | 16 |
| Polymerase | 1 | 2 | 4 | 2 | 4 | 0 | 11 | 2 | 6 |
| Topoisomerase | 2 | 1 | 0 | 1 | 2 | 0 | 6 | 1 | 4 |
| Transmembrane protein | 2 | 0 | 0 | 0 | 4 | 0 | 9 | 2 | 6 |
| Transmem. transport protein | 0 | 0 | 1 | 0 | 3 | 0 | 9 | 3 | 6 |
| Transposase | 5 | 1 | 3 | 3 | 20 | 0 | 31 | 14 | 23 |
| tRNA Ligase | 2 | 0 | 3 | 2 | 2 | 0 | 13 | 2 | 4 |
| Uncharacterized protein | 4 | 0 | 3 | 1 | 14 | 0 | 20 | 10 | 12 |
| Variable surface lipoproteins | 5 | 1 | 1 | 5 | 16 | 1 | 19 | 14 | 17 |
| GWAS Model | ENRO | CTET | OXY | FFN | GAM | TIL | TIP | TUL | TYLT |
|---|---|---|---|---|---|---|---|---|---|
| Fixed Effects Model | 1.06 | 1 | 1 | 1 | 3.1 | 1 | 2.85 | 2.27 | 2.45 |
| Linear Mixed Model | 1 | 1 | 1.02 | 1.17 | 3.87 | 1.89 | 4.84 | 3.63 | 2.93 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Waldner, M.; Kinnear, A.; Yacoub, E.; McAllister, T.; Register, K.; Li, C.; Jelinski, M. Genome-Wide Association Study of Nucleotide Variants Associated with Resistance to Nine Antimicrobials in Mycoplasma bovis. Microorganisms 2022, 10, 1366. https://doi.org/10.3390/microorganisms10071366
Waldner M, Kinnear A, Yacoub E, McAllister T, Register K, Li C, Jelinski M. Genome-Wide Association Study of Nucleotide Variants Associated with Resistance to Nine Antimicrobials in Mycoplasma bovis. Microorganisms. 2022; 10(7):1366. https://doi.org/10.3390/microorganisms10071366
Chicago/Turabian StyleWaldner, Matthew, Andrea Kinnear, Elhem Yacoub, Tim McAllister, Karen Register, Changxi Li, and Murray Jelinski. 2022. "Genome-Wide Association Study of Nucleotide Variants Associated with Resistance to Nine Antimicrobials in Mycoplasma bovis" Microorganisms 10, no. 7: 1366. https://doi.org/10.3390/microorganisms10071366