
Plant and Soil Core Mycobiomes in a Two-Year Sorghum–Legume Intercropping System of Underutilized Crops in South Africa
 , ,
, ,  , ,
, ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Field Sampling
2.2. Processing of Plant Material and Soils
2.3. DNA Extraction and Illumina Sequencing
2.4. Cluster and Data Analysis
3. Results
3.1. Illumina Sequencing and Data Analysis
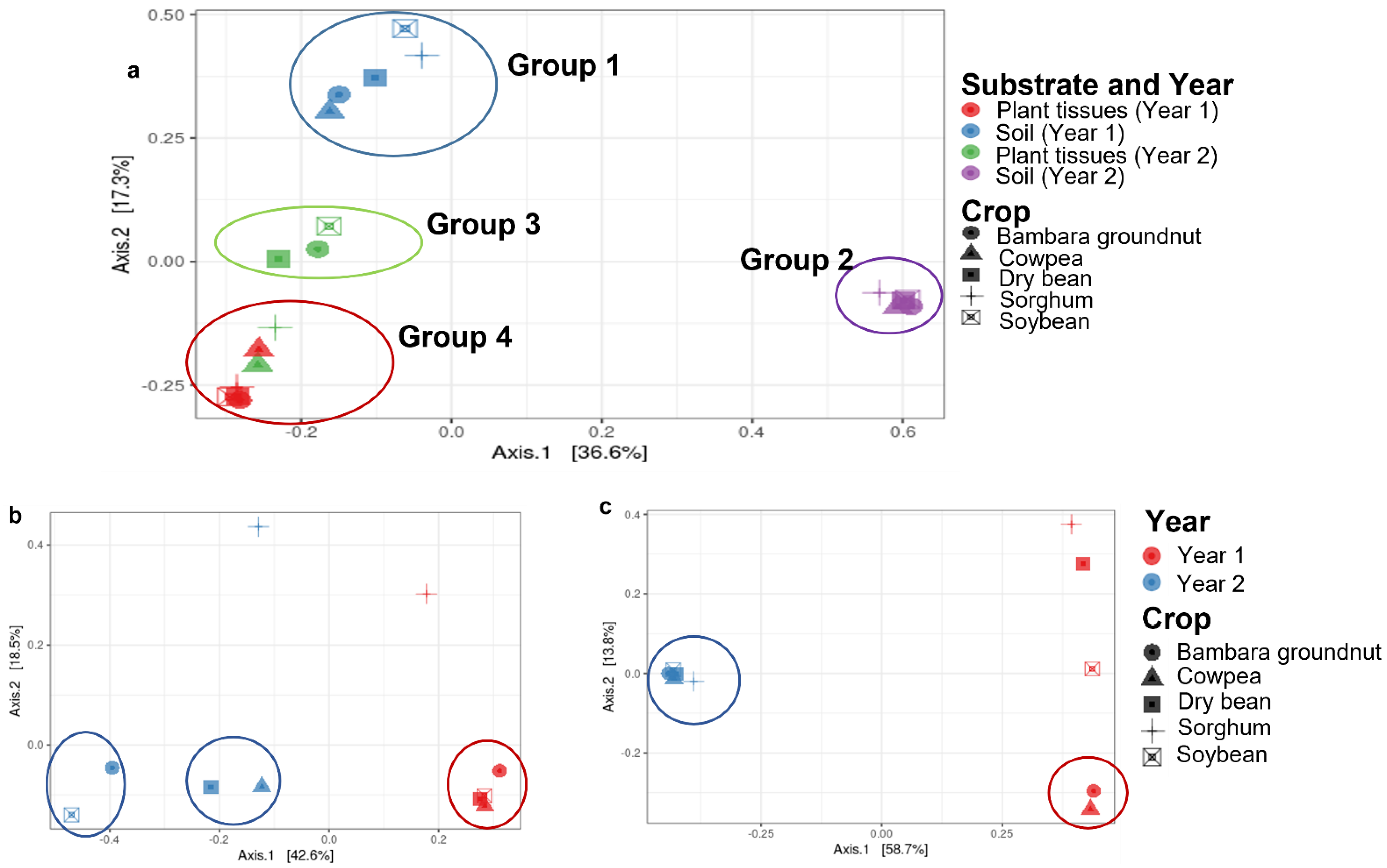
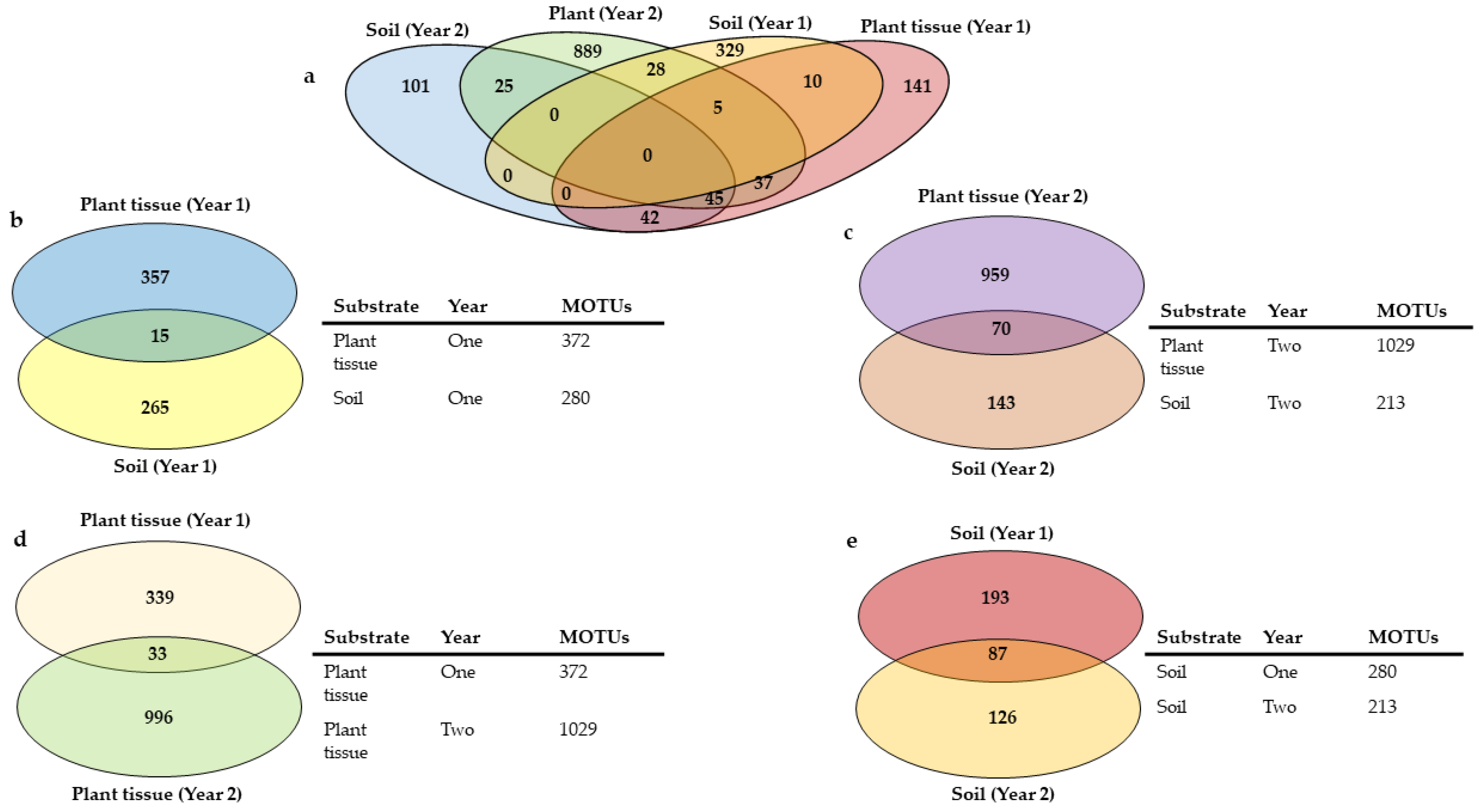
3.2. The Core Mycobiome across All Crops, Substrates, and Years
3.3. The Core Mycobiomes of Crops
3.4. Core Mycobiomes of Plant Tissues and Soils
3.5. Core Mycobiomes across the Years
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Benke, K.; Tomkins, B. Future food-production systems: Vertical farming and controlled-environment agriculture. Sustain. Sci. Pract. Policy 2017, 13, 13–26. [Google Scholar] [CrossRef] [Green Version]
- Adhikari, P.; Araya, H.; Aruna, G.; Balamatti, A.; Banerjee, S.; Baskaran, P.; Barah, B.C.; Behera, D.; Berhe, T.; Boruah, P.; et al. System of crop intensification for more productive, resource-conserving, climate-resilient, and sustainable agriculture: Experience with diverse crops in varying agroecologies. Int. J. Agric. Sustain. 2018, 16, 1–28. [Google Scholar] [CrossRef]
- Hong, Y.; Berentsen, P.; Heerink, N.; Shi, M.; van der Werf, W. The future of intercropping under growing resource scarcity and declining grain prices—A model analysis based on a case study in Northwest China. Agric. Syst. 2019, 176, 102661. [Google Scholar] [CrossRef]
- Kiwia, A.; Kimani, D.; Harawa, R.; Jama, B.; Sileshi, G.W. Sustainable Intensification with Cereal-Legume Intercropping in Eastern and Southern Africa. Sustainability 2019, 11, 2891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayes, S.; Ho, W.K.; Chai, H.H.; Gao, X.; Kundy, A.C.; Mateva, K.I.; Zahrulakmal, M.; Hahiree, M.K.I.M.; Kendabie, P.; Licea, L.C.S.; et al. Bambara groundnut: An exemplar underutilised legume for resilience under climate change. Planta 2019, 250, 803–820. [Google Scholar] [CrossRef] [Green Version]
- Mabhaudhi, T.; Chibarabada, T.; Chimonyo, V.; Murugani, V.; Pereira, L.; Sobratee, N.; Govender, L.; Slotow, R.; Modi, A. Mainstreaming Underutilized Indigenous and Traditional Crops into Food Systems: A South African Perspective. Sustainability 2018, 11, 172. [Google Scholar] [CrossRef] [Green Version]
- Chivenge, P.; Mabhaudhi, T.; Modi, A.; Mafongoya, P. The Potential Role of Neglected and Underutilised Crop Species as Future Crops under Water Scarce Conditions in Sub-Saharan Africa. Int. J. Environ. Res. Public Health 2015, 12, 5685–5711. [Google Scholar] [CrossRef] [Green Version]
- Mabhaudhi, T.; Chimonyo, V.G.P.; Modi, A.T. Status of Underutilised Crops in South Africa: Opportunities for Developing Research Capacity. Sustainability 2017, 9, 1569. [Google Scholar] [CrossRef] [Green Version]
- Hadebe, S.T.; Modi, A.T.; Mabhaudhi, T. Drought Tolerance and Water Use of Cereal Crops: A Focus on Sorghum as a Food Security Crop in Sub-Saharan Africa. J. Agron. Crop Sci. 2017, 203, 177–191. [Google Scholar] [CrossRef]
- Khojely, D.M.; Ibrahim, S.E.; Sapey, E.; Han, T. History, current status, and prospects of soybean production and research in sub-Saharan Africa. Crop J. 2018, 6, 226–235. [Google Scholar] [CrossRef]
- Jackson, J.; Kinabo, J.; Lekalake, R.; Mogotsi, K. Processing and Utilization of Dry Beans and Pulses in Africa. Dry Beans Pulses 2022, 409–430. [Google Scholar] [CrossRef]
- Mfeka, N.; Mulidzi, R.A.; Lewu, F.B. Growth and yield parameters of three cowpea (Vigna unguiculata L. Walp) lines as affected by planting date and zinc application rate. S. Afr. J. Sci. 2019, 115. [Google Scholar] [CrossRef] [Green Version]
- Chimonyo, V.G.P.; Modi, A.T.; Mabhaudhi, T. Water use and productivity of a sorghum-cowpea-bottle gourd intercrop system. Agric. Water Manag. 2016, 165, 82–96. [Google Scholar] [CrossRef]
- Tsujimoto, Y.; Antonio Pedro, J.; Boina, G.; Murracama, M.V.; Ito, O.; Tobita, S.; Oya, T.; Estevao Cuambe, C.; Martinho, C. Plant Production Science Performance of Maize-Soybean Intercropping under Various N Application Rates and Soil Moisture Conditions in Northern Mozambique. Plant Prod. Sci. 2015, 18, 365–376. [Google Scholar] [CrossRef]
- Da Silva, L.L.; Veloso, T.G.R.; Manhães, J.H.C.; da Silva, C.C.; de Queiroz, M.V. The plant organs and rhizosphere determine the common bean mycobiome. Brazilian J. Microbiol. 2020, 51, 765–772. [Google Scholar] [CrossRef] [PubMed]
- Snoeijenbos, M.; Cárdenas, M.; Guevara-Suarez, M.; Bernal, A.; Jiménez, P.; Restrepo, S. Phytobiomes, the Reason Why Microbiologists and Botanists Should Work Together. In Annual Plant Reviews online; Wiley: New York, NY, USA, 2019; pp. 585–618. [Google Scholar]
- Schlaeppi, K.; Bulgarelli, D. The Plant Microbiome at Work. Mol. Plant-Microbe Interact. 2015, 28, 212–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shade, A.; Stopnisek, N. Abundance-occupancy distributions to prioritize plant core microbiome membership. Curr. Opin. Microbiol. 2019, 49, 50–58. [Google Scholar] [CrossRef]
- Risely, A. Applying the core microbiome to understand host–microbe systems. J. Anim. Ecol. 2020, 89, 1549–1558. [Google Scholar] [CrossRef] [Green Version]
- Shade, A.; Handelsman, J. Beyond the Venn diagram: The hunt for a core microbiome. Environ. Microbiol. 2012, 14, 4–12. [Google Scholar] [CrossRef]
- Huse, S.M.; Ye, Y.; Zhou, Y.; Fodor, A.A. A core human microbiome as viewed through 16S rRNA sequence clusters. PLoS ONE 2012, 7, e34242. [Google Scholar] [CrossRef]
- Eyre, A.W.; Wang, M.; Oh, Y.; Dean, R.A. Identification and Characterization of the Core Rice Seed Microbiome. Phytobiomes J. 2019, 3, 148–157. [Google Scholar] [CrossRef] [Green Version]
- Simonin, M.; Dasilva, C.; Terzi, V.; Ngonkeu, E.L.M.; DIouf, D.; Kane, A.; Béna, G.; Moulin, L. Influence of plant genotype and soil on the wheat rhizosphere microbiome: Evidences for a core microbiome across eight African and European soils. FEMS Microbiol. Ecol. 2020, 96. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Wu, H.; Yan, B.; Zhao, H.; Liu, F.; Zhang, H.; Sheng, Q.; Miao, F.; Liang, Z. Core microbiome of medicinal plant salvia miltiorrhiza seed: A rich reservoir of beneficial microbes for secondary metabolism? Int. J. Mol. Sci. 2018, 19, 672. [Google Scholar] [CrossRef] [Green Version]
- Lundberg, D.S.; Lebeis, S.L.; Paredes, S.H.; Yourstone, S.; Gehring, J.; Malfatti, S.; Tremblay, J.; Engelbrektson, A.; Kunin, V.; Del Rio, T.G.; et al. Defining the core Arabidopsis thaliana root microbiome. Nature 2012, 488, 86–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Eyre, A.W.; Thon, M.R.; Oh, Y.; Dean, R.A. Dynamic Changes in the Microbiome of Rice During Shoot and Root Growth Derived From Seeds. Front. Microbiol. 2020, 11, 559728. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Hussain, M.; Zhang, W.; Stadler, M.; Liu, X.; Xiang, M. Current insights into fungal species diversity and perspective on naming the environmental DNA sequences of fungi. Mycology 2019, 10, 127–140. [Google Scholar] [CrossRef] [Green Version]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Huntley, J.; Fierer, N.; Owens, S.M.; Betley, J.; Fraser, L.; Bauer, M.; et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 2012, 6, 1621–1624. [Google Scholar] [CrossRef] [Green Version]
- Kinge, T.; Cason, E.; Valverde, A.; Nyaga, M.; Gryzenhout, M. Endophytic seed mycobiome of six sorghum (Sorghum bicolor) cultivars from commercial seedlots using an Illumina sequencing approach. Mycsophere 2019, 10, 739–756. [Google Scholar] [CrossRef]
- Andrews, S. FastQC A Quality Control tool for High Throughput Sequence Data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 12 November 2018).
- Schmieder, R.; Edwards, R. Quality control and preprocessing of metagenomic datasets. Bioinformatics 2011, 27, 863–864. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Kobert, K.; Flouri, T.; Stamatakis, A. PEAR: A fast and accurate Illumina Paired-End reAd mergeR. Bioinformatics 2014, 30, 614–620. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [Green Version]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 2016, 2016, e2584. [Google Scholar] [CrossRef] [Green Version]
- Nilsson, R.H.; Larsson, K.-H.; Taylor, A.F.S.; Bengtsson-Palme, J.; Jeppesen, T.S.; Schigel, D.; Kennedy, P.; Picard, K.; Gï Ockner, F.O., 10th; Tedersoo, L.; et al. The UNITE database for molecular identification of fungi: Handling dark taxa and parallel taxonomic classifications. Nucleic Acids Res. 2018, 47, 259–264. [Google Scholar] [CrossRef]
- Solanki, M.K.; Abdelfattah, A.; Britzi, M.; Zakin, V.; Wisniewski, M.; Droby, S.; Sionov, E. Shifts in the composition of the microbiota of stored wheat grains in response to fumigation. Front. Microbiol. 2019, 10, 1098. [Google Scholar] [CrossRef] [Green Version]
- Pambuka, G.T.; Kinge, T.R.; Ghosh, S.; Cason, E.D.; Nyaga, M.M.; Gryzenhout, M. Baseline Data of the Fungal Phytobiome of Three Sorghum (Sorghum bicolor) Cultivars in South Africa using Targeted Environmental Sequencing. J. Fungi 2021, 7, 978. [Google Scholar] [CrossRef]
- Paulson, J.N.; Stine, O.C.; Bravo, H.C.; Pop, M. Differential abundance analysis for microbial marker-gene surveys. Nat. Methods 2013, 10, 1200–1202. [Google Scholar] [CrossRef] [Green Version]
- Layeghifard, M.; Hwang, D.M.; Guttman, D.S. Constructing and Analyzing Microbiome Networks in R. In Methods in Molecular Biology; Humana Press Inc.: New York, NY, USA, 2018; Volume 1849, pp. 243–266. [Google Scholar]
- McMurdie, P.J.; Holmes, S. phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duchene, O.; Vian, J.F.; Celette, F. Intercropping with legume for agroecological cropping systems: Complementarity and facilitation processes and the importance of soil microorganisms. A review. Agric. Ecosyst. Environ. 2017, 240, 148–161. [Google Scholar] [CrossRef]
- Li, L.; Tilman, D.; Lambers, H.; Zhang, F.S. Plant diversity and overyielding: Insights from belowground facilitation of intercropping in agriculture. New Phytol. 2014, 203, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Rachid, C.T.C.C.; Balieiro, F.C.; Fonseca, E.S.; Peixoto, R.S.; Chaer, G.M.; Tiedje, J.M.; Rosado, A.S. Intercropped Silviculture Systems, a Key to Achieving Soil Fungal Community Management in Eucalyptus Plantations. PLoS ONE 2015, 10, e0118515. [Google Scholar] [CrossRef] [PubMed]
- Lian, T.; Mu, Y.; Ma, Q.; Cheng, Y.; Gao, R.; Cai, Z.; Jiang, B.; Nian, H. Use of sugarcane–soybean intercropping in acid soil impacts the structure of the soil fungal community. Sci. Rep. 2018, 8, 14488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, X.L.; Wang, C.B.; Bao, X.G.; Wang, P.; Li, X.F.; Yang, S.C.; Ding, G.c.; Christie, P.; Li, L. Crop diversity facilitates soil aggregation in relation to soil microbial community composition driven by intercropping. Plant Soil 2019, 436, 173–192. [Google Scholar] [CrossRef]
- Zhou, X.; Yu, G.; Wu, F. Effects of intercropping cucumber with onion or garlic on soil enzyme activities, microbial communities and cucumber yield. Eur. J. Soil Biol. 2011, 47, 279–287. [Google Scholar] [CrossRef]
- Chen, Y.L.; Xu, T.L.; Veresoglou, S.D.; Hu, H.W.; Hao, Z.P.; Hu, Y.J.; Liu, L.; Deng, Y.; Rillig, M.C.; Chen, B.D. Plant diversity represents the prevalent determinant of soil fungal community structure across temperate grasslands in northern China. Soil Biol. Biochem. 2017, 110, 12–21. [Google Scholar] [CrossRef]
- Li, N.; Gao, D.; Zhou, X.; Chen, S.; Li, C.; Wu, F. Intercropping with Potato-Onion Enhanced the Soil Microbial Diversity of Tomato. Microorganisms 2020, 8, 834. [Google Scholar] [CrossRef]
- Granzow, S.; Kaiser, K.; Wemheuer, B.; Pfeiffer, B.; Daniel, R.; Vidal, S.; Wemheuer, F. The Effects of Cropping Regimes on Fungal and Bacterial Communities of Wheat and Faba Bean in a Greenhouse Pot Experiment Differ between Plant Species and Compartment. Front. Microbiol. 2017, 8, 902. [Google Scholar] [CrossRef] [Green Version]
- Egidi, E.; Delgado-Baquerizo, M.; Plett, J.M.; Wang, J.; Eldridge, D.J.; Bardgett, R.D.; Maestre, F.T.; Singh, B.K. A few Ascomycota taxa dominate soil fungal communities worldwide. Nat. Commun. 2019, 10, 2369. [Google Scholar] [CrossRef] [Green Version]
- De Souza, R.S.C.; Okura, V.K.; Armanhi, J.S.L.; Jorrín, B.; Lozano, N.; da Silva, M.J.; González-Guerrero, M.; de Araújo, L.M.; Verza, N.C.; Bagheri, H.C.; et al. Unlocking the bacterial and fungal communities assemblages of sugarcane microbiome. Sci. Rep. 2016, 6, 28774. [Google Scholar] [CrossRef] [Green Version]
- Fulthorpe, R.; Martin, A.R.; Isaac, M.E. Root Endophytes of Coffee (Coffea arabica): Variation Across Climatic Gradients and Relationships with Functional Traits. Phytobiomes J. 2020, 4, 27–39. [Google Scholar] [CrossRef] [Green Version]
- Toju, H.; Peay, K.G.; Yamamichi, M.; Narisawa, K.; Hiruma, K.; Naito, K.; Fukuda, S.; Ushio, M.; Nakaoka, S.; Onoda, Y.; et al. Core microbiomes for sustainable agroecosystems. Nat. Plants 2018, 4, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Busby, P.E.; Soman, C.; Wagner, M.R.; Friesen, M.L.; Kremer, J.; Bennett, A.; Morsy, M.; Eisen, J.A.; Leach, J.E.; Dangl, J.L. Research priorities for harnessing plant microbiomes in sustainable agriculture. PLoS Biol. 2017, 15, e2001793. [Google Scholar] [CrossRef] [PubMed]
- Geiser, D.M.; Aoki, T.; Bacon, C.W.; Baker, S.E.; Bhattacharyya, M.K.; Brandt, M.E.; Brown, D.W.; Burgess, L.W.; Chulze, S.; Coleman, J.J.; et al. One Fungus, One Name: Defining the Genus Fusarium in a Scientifically Robust Way That Preserves Longstanding Use. Phytopathology 2013, 103, 400–408. [Google Scholar] [CrossRef] [Green Version]
- O’Donnell, K.; Ward, T.J.; Robert, V.A.R.G.; Crous, P.W.; Geiser, D.M.; Kang, S. DNA sequence-based identification of Fusarium: Current status and future directions. Phytoparasitica 2015, 43, 583–595. [Google Scholar] [CrossRef] [Green Version]
- Summerell, B.A. Resolving Fusarium: Current Status of the Genus. Annu. Rev. Phytopathol. 2019, 57, 323–339. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Wu, F. Diversity and Co-occurrence Patterns of Soil Bacterial and Fungal Communities in Seven Intercropping Systems. Front. Microbiol. 2018, 9, 1521. [Google Scholar] [CrossRef]
- Bensch, K.; Braun, U.; Groenewald, J.Z.; Crous, P.W. The genus Cladosporium. Stud. Mycol. 2012, 72, 1–401. [Google Scholar] [CrossRef] [Green Version]
- Thomma, B.P.H.J.; Van Esse, H.P.; Crous, P.W.; De Wit, P.J.G.M. Cladosporium fulvum (syn. Passalora fulva), a highly specialized plant pathogen as a model for functional studies on plant pathogenic Mycosphaerellaceae. Mol. Plant Pathol. 2005, 6, 379–393. [Google Scholar] [CrossRef]
- De Wit, P.J.G.M.; van der Burgt, A.; Ökmen, B.; Stergiopoulos, I.; Abd-Elsalam, K.A.; Aerts, A.L.; Bahkali, A.H.; Beenen, H.G.; Chettri, P.; Cox, M.P.; et al. The Genomes of the Fungal Plant Pathogens Cladosporium fulvum and Dothistroma septosporum Reveal Adaptation to Different Hosts and Lifestyles But Also Signatures of Common Ancestry. PLoS Genet. 2012, 8, e1003088. [Google Scholar] [CrossRef]
- Robles-Yerena, L.; Ayala-Escobar, V.; Leyva-Mir, S.G.; Lima, N.B.; Camacho-Tapia, M.; Tovar-Pedraza, J.M. First report of Cladosporium cladosporioides causing leaf spot on tomato in Mexico. J. Plant Pathol. 2019, 101, 759. [Google Scholar] [CrossRef] [Green Version]
- Torres, D.E.; Rojas-Martínez, R.I.; Zavaleta-Mejía, E.; Guevara-Fefer, P.; Márquez-Guzmán, G.J.; Pérez-Martínez, C. Cladosporium cladosporioides and Cladosporium pseudocladosporioides as potential new fungal antagonists of Puccinia horiana Henn., the causal agent of chrysanthemum white rust. PLoS ONE 2017, 12, e0170782. [Google Scholar] [CrossRef]
- Rai, M.K.; Tiwari, V.V.; Irinyi, L.; Kövics, G.J. Advances in Taxonomy of Genus Phoma: Polyphyletic Nature and Role of Phenotypic Traits and Molecular Systematics. Indian J. Microbiol. 2014, 54, 123–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bennett, A.; Ponder, M.; Garcia-Diaz, J. Phoma Infections: Classification, Potential Food Sources, and Their Clinical Impact. Microorganisms 2018, 6, 58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rai, M.; Agarkar, G. Plant-fungal interactions: What triggers the fungi to switch among lifestyles? Crit. Rev. Microbiol. 2016, 42, 428–438. [Google Scholar] [CrossRef] [PubMed]
- Deb, D.; Khan, A.; Dey, N. Phoma diseases: Epidemiology and control. Plant Pathol. 2020, 69, 1203–1217. [Google Scholar] [CrossRef]
- Abdelfattah, A.; Wisniewski, M.; Li Destri Nicosia, M.G.; Cacciola, S.O.; Schena, L. Metagenomic Analysis of Fungal Diversity on Strawberry Plants and the Effect of Management Practices on the Fungal Community Structure of Aerial Organs. PLoS ONE 2016, 11, e0160470. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Yao, Y.-F. Endophytic Fungal Communities Associated with Vascular Plants in the High Arctic Zone Are Highly Diverse and Host-Plant Specific. PLoS ONE 2015, 10, e0130051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aljohani, R.; Samarasinghe, H.; Ashu, T.; Xu, J. Diversity and relationships among strains of culturable yeasts in agricultural soils in Cameroon. Sci. Rep. 2018, 8, 15687. [Google Scholar] [CrossRef] [Green Version]
- Yurkov, A. Yeasts in forest soils. In Yeasts in Natural Ecosystems: Diversity; Springer International Publishing: Berlin/Heidelberg, Germany, 2017; pp. 87–116. ISBN 9783319626833. [Google Scholar]
- Moller, L.; Kessler, K.D.; Steyn, A.; Valentine, A.J.; Botha, A. The role of Cryptococcus laurentii and mycorrhizal fungi in the nutritional physiology of Lupinus angustifolius L. hosting N2-fixing nodules. Plant Soil 2016, 409, 345–360. [Google Scholar] [CrossRef]
- May, R.C.; Stone, N.R.H.; Wiesner, D.L.; Bicanic, T.; Nielsen, K. Cryptococcus: From environmental saprophyte to global pathogen. Nat. Rev. Microbiol. 2016, 14, 106–117. [Google Scholar] [CrossRef]
- Afkhami, M.E.; Rudgers, J.A. Symbiosis lost: Imperfect vertical transmission of fungal endophytes in grasses. Am. Nat. 2008, 172, 405–416. [Google Scholar] [CrossRef]
- Sánchez Márquez, S.; Bills, G.F.; Herrero, N.; Zabalgogeazcoa, Í. Non-systemic fungal endophytes of grasses. Fungal Ecol. 2012, 5, 289–297. [Google Scholar] [CrossRef]
- Hibbett, D.; Glotzer, D. Where are all the undocumented fungal species? A study of Mortierella demonstrates the need for sequence-based classification. New Phytol. 2011, 191, 592–596. [Google Scholar] [CrossRef]
- Tedersoo, L.; Lindahl, B. Fungal identification biases in microbiome projects. Environ. Microbiol. Rep. 2016, 8, 774–779. [Google Scholar] [CrossRef]
- Senanayake, I.C.; Crous, P.W.; Groenewald, J.Z.; Maharachchikumbura, S.S.N.; Jeewon, R.; Phillips, A.J.L.; Bhat, J.D.; Perera, R.H.; Li, Q.R.; Li, W.J.; et al. Families of Diaporthales based on morphological and phylogenetic evidence. Stud. Mycol. 2017, 86, 217–296. [Google Scholar] [CrossRef]
- Ariyawansa, H.A.; Tsai, I.; Thambugala, K.M.; Chuang, W.Y.; Lin, S.R.; Hozzein, W.N.; Cheewangkoon, R. Species diversity of Pleosporalean taxa associated with Camellia sinensis (L.) Kuntze in Taiwan. Sci. Rep. 2020, 10, 12762. [Google Scholar] [CrossRef]
- Mohamed Nor, N.M.I.; Salleh, B.; Leslie, J.F. Fusarium species from sorghum in thailand. Plant Pathol. J. 2019, 35, 301–312. [Google Scholar] [CrossRef]
- Petrovic, T.; Walsh, J.L.; Burgess, L.W.; Summerell, B.A. Fusarium species associated with stalk rot of grain sorghum in the northern grain belt of eastern Australia. In Proceedings of the Australasian Plant Pathology; Springer: Berlin/Heidelberg, Germany, 2009; Volume 38, pp. 373–379. [Google Scholar]
- Mclaren, N.W. Evaluation of sorghum hybrids for resistance to the root rot complex. S. Afr. J. Plant Soil 2002, 19, 37–42. [Google Scholar] [CrossRef] [Green Version]
- Idris, H.A.; Labuschagne, N.; Korsten, L. Suppression of Pythium ultimum root rot of sorghum by rhizobacterial isolates from Ethiopia and South Africa. Biol. Control 2008, 45, 72–84. [Google Scholar] [CrossRef]
- Arias, M.M.D.; Leandro, L.F.; Munkvold, G.P. Aggressiveness of Fusarium species and impact of root infection on growth and yield of soybeans. Phytopathology 2013, 103, 822–832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.P.; You, M.P.; Barbetti, M.J. Species of Pythium Associated with Seedling Root and Hypocotyl Disease on Common Bean (Phaseolus vulgaris) in Western Australia. Plant Dis. 2014, 98, 1241–1247. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Crop | Year | Number of Reads | Total MOTUs | |
|---|---|---|---|---|
| Plant tissues | Bambara groundnut | One | 9470 | 77 |
| Dry bean | One | 8923 | 87 | |
| Soybean | One | 7720 | 72 | |
| Cowpea | One | 8019 | 66 | |
| Sorghum | One | 2098 | 60 | |
| Bambara groundnut | Two | 357,819 | 185 | |
| Dry bean | Two | 679,123 | 225 | |
| Soybean | Two | 845,135 | 179 | |
| Cowpea | Two | 214,228 | 212 | |
| Sorghum | Two | 165,487 | 229 | |
| Soils | Bambara groundnut | One | 77,709 | 747 |
| Dry bean | One | 66,207 | 364 | |
| Soybean | One | 120,151 | 277 | |
| Cowpea | One | 50,611 | 791 | |
| Sorghum | One | 139,545 | 277 | |
| Bambara groundnut | Two | 77,709 | 155 | |
| Dry bean | Two | 76,769 | 229 | |
| Soybean | Two | 74,069 | 203 | |
| Cowpea | Two | 57,655 | 148 | |
| Sorghum | Two | 75,901 | 272 |
| Sorghum | Bambara Groundnut | Cowpea | Dry Bean | Soybean | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Niche | Phylum | Core MOTUs | Year 1 | Year 2 | Year 1 | Year 2 | Year 1 | Year 2 | Year 1 | Year 2 | Year 1 | Year 2 |
| Plant tissues | Ascomycota | Davidiellaceae sp. (Cladosporium) | 4.9 | 4.8 | 4.5 | 0.1 | 3.7 | 0.6 | 9.5 | 6.2 | 3.0 | 0.7 |
| Didymellaceae sp. 1 (Phoma) | 29.8 | 8.6 | 31.8 | 12.1 | 24.2 | 31.3 | 28.4 | 14.3 | 37.4 | 3.9 | ||
| Didymellaceae sp. 2 (Epicoccum) | 4.8 | 3.8 | 3.3 | 0.3 | 0.8 | 0.1 | 0.8 | 0.1 | 1.7 | 0.2 | ||
| Fusarium 2 | 5.6 | 7.7 | 2.7 | 11.3 | 2.6 | 3.0 | 5.8 | 33.1 | 7.6 | 15.3 | ||
| Unidentified | 12.9 | 9.0 | 20.9 | 2.4 | 25 | 11.4 | 51.1 | 7.4 | 45.7 | 4.9 | ||
| Basidiomycota | Cryptococcus | 3.5 | 4.9 | 1.5 | 6.2 | 0.2 | 3.7 | 0.4 | 13.9 | 0.2 | 2.3 | |
| Unidentified | Unidentified | 2.5 | 4.0 | 1.0 | 0.6 | 0.6 | 1.7 | 1.6 | 2.0 | 0.9 | 12.6 | |
| Soil | Ascomycota | Davidiellaceae sp. (Cladosporium) | 0.3 | 3.2 | 0.2 | 0.2 | 0.1 | 0.1 | 1.0 | 0.5 | 0.3 | 0.1 |
| Didymellaceae sp. 1 (Phoma) | 4.3 | 2.4 | 15.6 | 2.3 | 16.7 | 1.6 | 7.6 | 5.4 | 3.1 | 1.6 | ||
| Didymellaceae sp. 2 (Epicoccum) | 0.2 | 0.1 | 0.1 | 0.1 | 0.2 | 0.1 | 0.5 | 0.2 | 0.1 | 0.1 | ||
| Fusarium 2 | 2.4 | 0.8 | 26.3 | 1.2 | 2.6 | 4.8 | 8.9 | 1.3 | 26.0 | 6.8 | ||
| Unidentified | 1.6 | 0.7 | 3.0 | 0.1 | 3.5 | 0.1 | 2.2 | 0.5 | 0.8 | 0.2 | ||
| Basidiomycota | Cryptococcus | 0.1 | 1.5 | 0.3 | 7.0 | 0.2 | 8.5 | 1.8 | 6.9 | 0.1 | 2.0 | |
| Unidentified | Unidentified | 61.2 | 17.3 | 7.3 | 7.9 | 6.7 | 14.5 | 23.9 | 16.7 | 12.3 | 14.5 | |
| Compared Categories | D.F | Sum of Sqs | R2 | p-Value |
|---|---|---|---|---|
| Plant and soil | 3 | 4.626 | 0.60156 | 0.001 |
| Years | 1 | 1.6955 | 0.22048 | 0.001 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pambuka, G.T.; Kinge, T.R.; Ghosh, S.; Cason, E.D.; Nyaga, M.M.; Gryzenhout, M. Plant and Soil Core Mycobiomes in a Two-Year Sorghum–Legume Intercropping System of Underutilized Crops in South Africa. Microorganisms 2022, 10, 2079. https://doi.org/10.3390/microorganisms10102079
Pambuka GT, Kinge TR, Ghosh S, Cason ED, Nyaga MM, Gryzenhout M. Plant and Soil Core Mycobiomes in a Two-Year Sorghum–Legume Intercropping System of Underutilized Crops in South Africa. Microorganisms. 2022; 10(10):2079. https://doi.org/10.3390/microorganisms10102079
Chicago/Turabian StylePambuka, Gilmore T., Tonjock Rosemary Kinge, Soumya Ghosh, Errol D. Cason, Martin M. Nyaga, and Marieka Gryzenhout. 2022. "Plant and Soil Core Mycobiomes in a Two-Year Sorghum–Legume Intercropping System of Underutilized Crops in South Africa" Microorganisms 10, no. 10: 2079. https://doi.org/10.3390/microorganisms10102079